
Integrated Hair Follicle Profiles of microRNAs and mRNAs to Reveal the Pattern Formation of Hu Sheep Lambskin

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Library Preparation for Sequencing
2.3. Quality Control, Reference Genome Alignment, and Assembly
2.4. Quantification and Differential Expression of miRNA and mRNA
2.5. Target Gene Prediction and Functional Enrichment Analysis of miRNAs
2.6. Integrated Analysis of miRNA and mRNA Expression Profiles
2.7. Verification of RNA-seq Results
3. Results
3.1. The Expression Profile of miRNA and mRNA
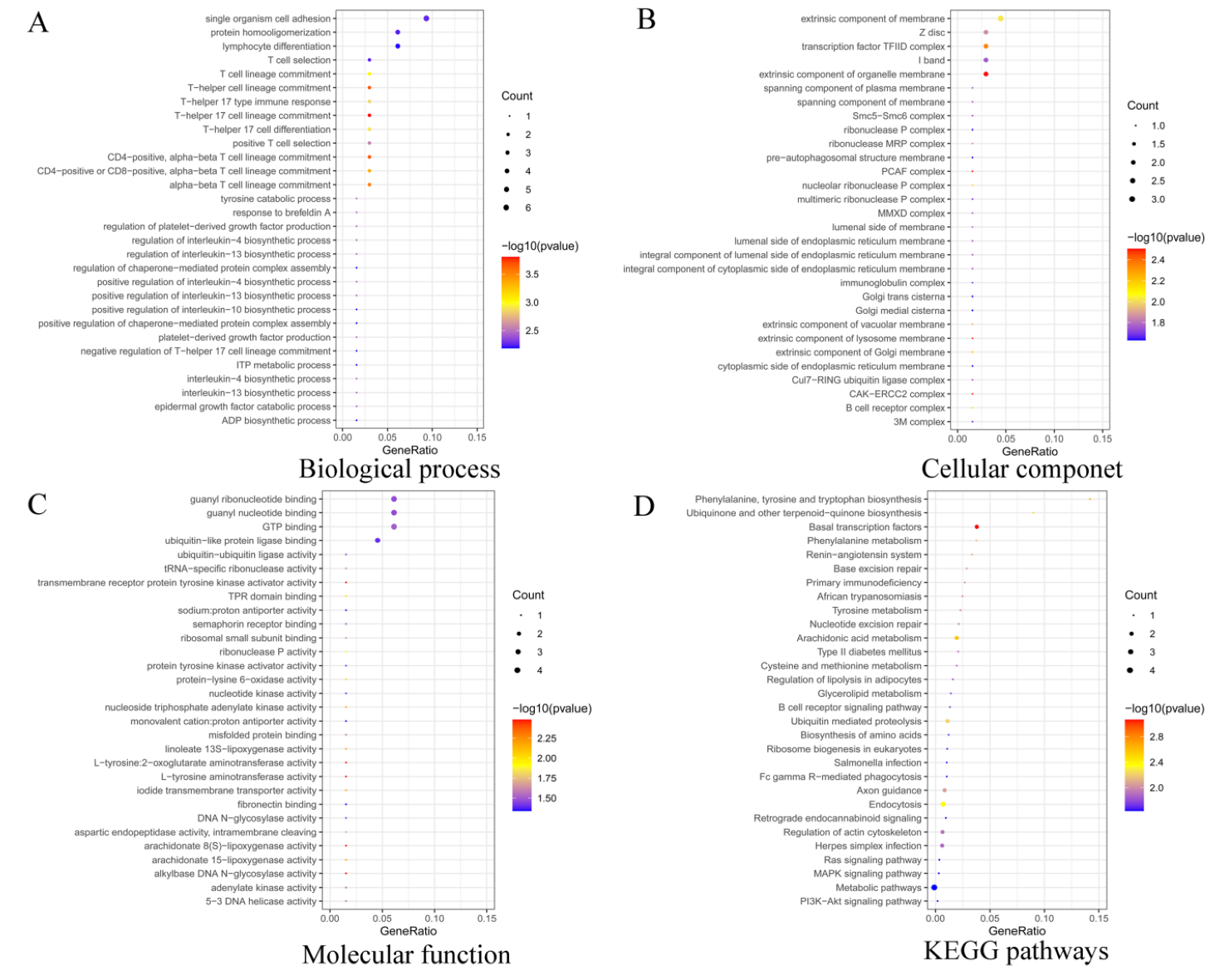
3.2. Functional Enrichment Analysis of Target Genes
3.3. Co-Expression Analysis of Integrated DE miRNA-mRNA
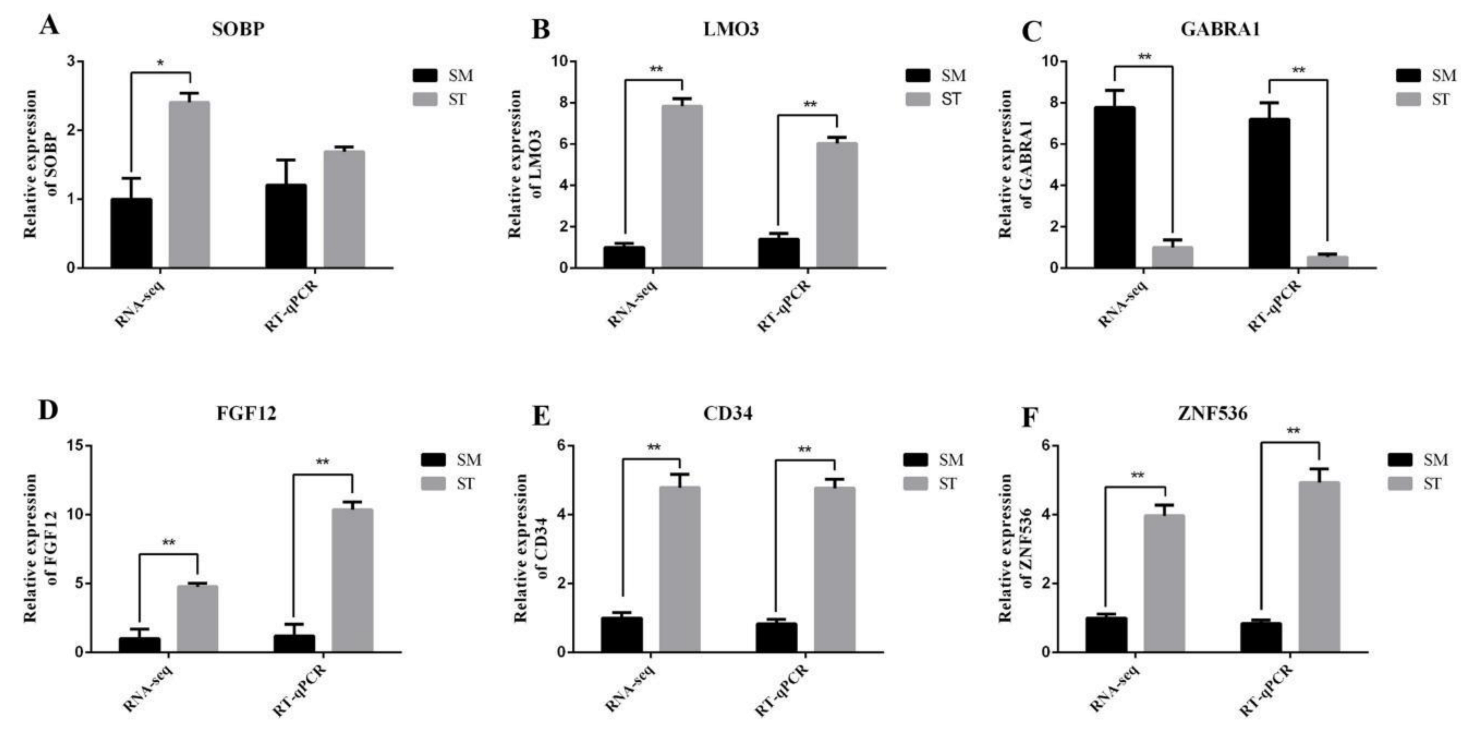
3.4. Data Validation
4. Discussion
4.1. miRNAs and Hair Follicle Development
4.2. DE miRNAs Analysis in Different Degree of Curliness
4.3. Functional Enrichment Analysis of DE miRNAs between SM vs. ST
4.4. Co-Expression Analysis of miRNA-mRNA
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jiang, L.; Schlesinger, F.; Davis, C.A.; Zhang, Y.; Li, R.; Salit, M.; Gingeras, T.R.; Oliver, B. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011, 21, 1543–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demars, J.; Cano, M.; Drouilhet, L.; Plisson-Petit, F.; Bardou, P.; Fabre, S.; Servin, B.; Sarry, J.; Woloszyn, F.; Mulsant, P.; et al. Genome-Wide Identification of the Mutation Underlying Fleece Variation and Discriminating Ancestral Hairy Species from Modern Woolly Sheep. Mol. Biol. Evol. 2017, 34, 1722–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, S.; Andl, T.; Bagasra, A.; Lu, M.M.; Epstein, D.J.; Morrisey, E.E.; Millar, S.E. Characterization of Wnt gene expression in developing and postnatal hair follicles and identification of Wnt5a as a target of Sonic hedgehog in hair follicle morphogenesis. Mech. Dev. 2001, 107, 69–82. [Google Scholar] [CrossRef]
- Zhou, P.; Byrne, C.; Jacobs, J.; Fuchs, E. Lymphoid enhancer factor 1 directs hair follicle patterning and epithelial cell fate. Genes Dev. 1995, 9, 700–713. [Google Scholar] [CrossRef] [Green Version]
- Ouspenskaia, T.; Matos, I.; Mertz, A.F.; Fiore, V.F.; Fuchs, E. WNT-SHH Antagonism Specifies and Expands Stem Cells prior to Niche Formation. Cell 2016, 164, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.M.; Jarrell, A.; Guo, C.T.; Lang, R.; Atit, R. Dermal β-catenin activity in response to epidermal Wnt ligands is required for fibroblast proliferation and hair follicle initiation. Development 2012, 139, 1522–1533. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Lai, Y.C.; Figueroa, S.; Yang, T.; Widelitz, R.B.; Kobielak, K.; Nie, Q.; Chuong, C.M. Deciphering principles of morphogenesis from temporal and spatial patterns on the integument. Dev. Dyn. 2015, 244, 905–920. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tomann, P.; Andl, T.; Gallant, N.M.; Huelsken, J.; Jerchow, B.; Birchmeier, W.; Paus, R.; Piccolo, S.; Mikkola, M.L.; et al. Reciprocal requirements for EDA/EDAR/NF-kappaB and Wnt/β-catenin signaling pathways in hair follicle induction. Dev. Cell 2009, 17, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Cetera, M.; Leybova, L.; Joyce, B.; Devenport, D. Counter-rotational cell flows drive morphological and cell fate asymmetries in mammalian hair follicles. Nat. Cell Biol. 2018, 20, 541–552. [Google Scholar] [CrossRef]
- Qiu, W.; Li, X.; Tang, H.; Huang, A.S.; Panteleyev, A.A.; Owens, D.M.; Su, G.H. Conditional activin receptor type 1B (Acvr1b) knockout mice reveal hair loss abnormality. J. Invest. Derm. 2011, 131, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.H.; Zhang, Y.J.; Zhang, J.X.; Chang, Z.L.; Li, J.Q.; Yan, Z.W.; Zhang, W.G. Hoxc13/β-catenin Correlation with Hair Follicle Activity in Cashmere Goat. J. Integr. Agric. 2012, 11, 1159–1166. [Google Scholar] [CrossRef]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A brief review on the mechanisms of miRNA regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Vishnoi, A.; Rani, S. MiRNA Biogenesis and Regulation of Diseases: An Overview. Methods Mol. Biol 2017, 1509, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Andl, T.; Botchkareva, N.V. MicroRNAs (miRNAs) in the control of HF development and cycling: The next frontiers in hair research. Exp. Derm. 2015, 24, 821–826. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Stokes, N.; Polak, L.; Fuchs, E. Specific microRNAs are preferentially expressed by skin stem cells to balance self-renewal and early lineage commitment. Cell Stem Cell 2011, 8, 294–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Zhang, Z.; O’Loughlin, E.; Wang, L.; Fan, X.; Lai, E.C.; Yi, R. MicroRNA-205 controls neonatal expansion of skin stem cells by modulating the PI(3)K pathway. Nat. Cell Biol. 2013, 15, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.I.; Alam, M.; Emelianov, V.U.; Poterlowicz, K.; Patel, A.; Sharov, A.A.; Mardaryev, A.N.; Botchkareva, N.V. MicroRNA-214 controls skin and hair follicle development by modulating the activity of the Wnt pathway. J. Cell Biol. 2014, 207, 549–567. [Google Scholar] [CrossRef] [Green Version]
- Mardaryev, A.N.; Ahmed, M.I.; Vlahov, N.V.; Fessing, M.Y.; Gill, J.H.; Sharov, A.A.; Botchkareva, N.V. Micro-RNA-31 controls hair cycle-associated changes in gene expression programs of the skin and hair follicle. FASEB J. 2010, 24, 3869–3881. [Google Scholar] [CrossRef] [Green Version]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Stark, A.; Brennecke, J.; Russell, R.B.; Cohen, S.M. Identification of Drosophila MicroRNA targets. PLoS Biol. 2003, 1, E60. [Google Scholar] [CrossRef]
- Yu, Z.; Gordon, S.W.; Nixon, A.J.; Bawden, C.S.; Rogers, M.A.; Wildermoth, J.E.; Maqbool, N.J.; Pearson, A.J. Expression patterns of keratin intermediate filament and keratin associated protein genes in wool follicles. Differentiation 2009, 77, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Liu, R.; Tang, X.; Cao, J.; Zhao, S.; Yu, M. Expression profiling reveals genes involved in the regulation of wool follicle bulb regression and regeneration in sheep. Int. J. Mol. Sci. 2015, 16, 9152–9166. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Chen, W.; Sun, W.; Hussain, Z.; Chen, L.; Wang, S.; Wang, J. Expression profile analysis to identify circular RNA expression signatures in hair follicle of Hu sheep lambskin. Genomics 2020, 112, 4454–4462. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Chen, W.; Sun, W.; Hussain, Z.; Chen, L.; Wang, S.; Wang, J. Analysis of lncRNAs Expression Profiles in Hair Follicle of Hu Sheep Lambskin. Animals 2020, 10, 1035. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Ni, R.; Yin, J.F.; Musa, H.H.; Ding, J.T.; Chen, L. Genome array of hair follicle genes in lambskin with different patterns. PLoS ONE 2013, 8, e68840. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Sun, W.; Yin, J.; Lv, X.; Bao, J.; Yu, J.; Wang, L.; Jin, C.; Hu, L. Screening candidate microRNAs (miRNAs) in different lambskin hair follicles in Hu sheep. PLoS ONE 2017, 12, e0176532. [Google Scholar] [CrossRef]
- Erlich, Y.; Mitra, P.P.; delaBastide, M.; McCombie, W.R.; Hannon, G.J. Alta-Cyclic: A self-optimizing base caller for next-generation sequencing. Nat. Methods 2008, 5, 679–682. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef] [Green Version]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, J.; Li, Z.; Li, X.; Hu, X.; Huang, Y.; Zhao, X.; Liang, C.; Wang, Y.; Sun, L.; et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS ONE 2010, 5, e15224. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 4, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.M.; Lu, Y.L.; Sio, C.P.; Wu, G.C.; Tzou, W.S.; Pai, T.W. Gene Ontology based housekeeping gene selection for RNA-seq normalization. Methods 2014, 67, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Krishnaswami, S.R.; Cowing-Zitron, C.; Hung, N.J.; Reilly-Rhoten, H.; Burns, J.; Yu, B.D. Negative regulation of Shh levels by Kras and Fgfr2 during hair follicle development. Dev. Biol. 2013, 373, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.; Lei, M.; Zhou, L.; Bai, X.; Lai, X.; Yu, Y.; Yang, T.; Lian, X. Hair follicle stem cell proliferation, Akt and Wnt signaling activation in TPA-induced hair regeneration. Histochem. Cell Biol. 2017, 147, 749–758. [Google Scholar] [CrossRef]
- Xiao, S.; Wang, J.; Chen, Q.; Miao, Y.; Hu, Z. The mechanism of activated platelet-rich plasma supernatant promotion of hair growth by cultured dermal papilla cells. J. Cosmet. Derm. 2019, 18, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Nissimov, J.N.; Das Chaudhuri, A.B. Hair curvature: A natural dialectic and review. Biol. Rev. Camb. Philos. Soc. 2014, 89, 723–766. [Google Scholar] [CrossRef]
- Yi, R.; Pasolli, H.A.; Landthaler, M.; Hafner, M.; Ojo, T.; Sheridan, R.; Sander, C.; O’Carroll, D.; Stoffel, M.; Tuschl, T.; et al. DGCR8-dependent microRNA biogenesis is essential for skin development. Proc. Natl. Acad. Sci. USA 2009, 106, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Sun, W.; Yin, J.; Ni, R.; Su, R.; Wang, Q.; Gao, W.; Bao, J.; Yu, J.; Wang, L.; et al. An Integrated Analysis of MicroRNA and mRNA Expression Profiles to Identify RNA Expression Signatures in Lambskin Hair Follicles in Hu Sheep. PLoS ONE 2016, 11, e0157463. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, J.; Xu, Q.; Kang, X.; Wang, K.; Wu, K.; Fang, M. Integrated miRNA-mRNA analysis reveals regulatory pathways underlying the curly fleece trait in Chinese tan sheep. BMC Genom. 2018, 19, 360. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qu, H.; Jiang, H.; Zhao, Z.; Zhang, Q. Transcriptome-Wide Comparative Analysis of microRNA Profiles in the Telogen Skins of Liaoning Cashmere Goats (Capra hircus) and Fine-Wool Sheep (Ovis aries) by Solexa Deep Sequencing. DNA Cell Biol. 2016, 35, 696–705. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Li, X.; Han, W.; Yang, K.; Wang, H.; Zhang, Y.; Su, R.; Liu, Z.; Wang, R.; et al. High-throughput sequencing of hair follicle development-related micrornas in cashmere goat at various fetal periods. Saudi J. Biol. Sci. 2018, 25, 1494–1508. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.; Mok, K.W.; Rendl, M. An updated classification of hair follicle morphogenesis. Exp. Dermatol. 2019, 28, 332–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, K.M.; Jeong, K.H.; Kim, J.E.; Park, Y.M.; Kang, H. Hair growth-promotion effects of different alternating current parameter settings are mediated by the activation of Wnt/β-catenin and MAPK pathway. Exp. Dermatol. 2015, 24, 958–963. [Google Scholar] [CrossRef]
- Sawada, A.; Shinya, M.; Jiang, Y.J.; Kawakami, A.; Kuroiwa, A.; Takeda, H. Fgf/MAPK signalling is a crucial positional cue in somite boundary formation. Development 2001, 128, 4873–4880. [Google Scholar] [CrossRef]
- Jiang, L.; Xu, J.; Jin, R.; Bai, H.; Zhang, M.; Yang, S.; Zhang, X.; Zhang, X.; Han, Z.; Zeng, S. Transcriptomic analysis of chicken cochleae after gentamicin damage and the involvement of four signaling pathways (Notch, FGF, Wnt and BMP) in hair cell regeneration. Hear. Res. 2018, 361, 66–79. [Google Scholar] [CrossRef]
- Zhu, H.L.; Gao, Y.H.; Yang, J.Q.; Li, J.B.; Gao, J. Serenoa repens extracts promote hair regeneration and repair of hair loss mouse models by activating TGF-β and mitochondrial signaling pathway. Eur. Rev. Med. Pharm. Sci. 2018, 22, 4000–4008. [Google Scholar] [CrossRef]
- Battista, M.; Musto, A.; Navarra, A.; Minopoli, G.; Russo, T.; Parisi, S. miR-125b Regulates the Early Steps of ESC Differentiation through Dies1 in a TGF-Independent Manner. Int. J. Mol. Sci. 2013, 14, 13482–13496. [Google Scholar] [CrossRef] [Green Version]
- Emmrich, S.; Rasche, M.; Schoning, J.; Reimer, C.; Keihani, S.; Maroz, A.; Xie, Y.; Li, Z.; Schambach, A.; Reinhardt, D.; et al. miR-99a/100 similar to 125b tricistrons regulate hematopoietic stem and progenitor cell homeostasis by shifting the balance between TGF β and Wnt signaling. Gene Dev. 2014, 28, 858–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, M.T.; Shyh-Chang, N.; Khaw, S.L.; Chin, L.; Teh, C.; Tay, J.; O’Day, E.; Korzh, V.; Yang, H.; Lal, A.; et al. Conserved regulation of p53 network dosage by microRNA-125b occurs through evolving miRNA-target gene pairs. PLoS Genet. 2011, 7, e1002242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, A.G.; Sahoo, D.; Adorno, M.; Wang, Y.; Weissman, I.L.; Park, C.Y. MicroRNA-125b expands hematopoietic stem cells and enriches for the lymphoid-balanced and lymphoid-biased subsets. Proc. Natl. Acad. Sci. USA 2010, 107, 21505–21510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoh, Y.; Hoffman, R.M. Hair follicle-associated-pluripotent (HAP) stem cells. Cell Cycle 2017, 16, 2169–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantauzzo, K.A.; Bazzi, H.; Jahoda, C.A.; Christiano, A.M. Dynamic expression of the zinc-finger transcription factor Trps1 during hair follicle morphogenesis and cycling. Gene Expr. Patterns 2008, 8, 51–57. [Google Scholar] [CrossRef]
- Oliver, C.H.; Nichols, J.; Watson, C.J. The KRAB domain zinc finger protein, Zfp157, is expressed in multiple tissues during mouse embryogenesis and in specific cells in adult mammary gland and skin. Genesis 2013, 51, 179–186. [Google Scholar] [CrossRef]
- Su, R.; Fan, Y.; Qiao, X.; Li, X.; Zhang, L.; Li, C.; Li, J. Transcriptomic analysis reveals critical genes for the hair follicle of Inner Mongolia cashmere goat from catagen to telogen. PLoS ONE 2018, 13, e0204404. [Google Scholar] [CrossRef] [Green Version]
- Hoefert, J.E.; Bjerke, G.A.; Wang, D.; Yi, R. The microRNA-200 family coordinately regulates cell adhesion and proliferation in hair morphogenesis. J. Cell Biol. 2018, 217, 2185–2204. [Google Scholar] [CrossRef] [Green Version]
- Ge, M.; Liu, C.; Li, L.; Lan, M.; Yu, Y.; Gu, L.; Su, Y.; Zhang, K.; Zhang, Y.; Wang, T.; et al. miR-29a/b1 Inhibits Hair Follicle Stem Cell Lineage Progression by Spatiotemporally Suppressing WNT and BMP Signaling. Cell Rep. 2019, 29, 2489–2504. [Google Scholar] [CrossRef] [Green Version]
- Hillege, M.M.G.; Galli Caro, R.A.; Offringa, C.; de Wit, G.M.J.; Jaspers, R.T.; Hoogaars, W.M.H. TGF-β Regulates Collagen Type I Expression in Myoblasts and Myotubes via Transient Ctgf and Fgf-2 Expression. Cells 2020, 9, 375. [Google Scholar] [CrossRef] [Green Version]
- Mossahebi-Mohammadi, M.; Quan, M.; Zhang, J.S.; Li, X. FGF Signaling Pathway: A Key Regulator of Stem Cell Pluripotency. Front. Cell Dev. Biol. 2020, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, X.; Chen, W.; Wang, S.; Cao, X.; Yuan, Z.; Getachew, T.; Mwacharo, J.M.; Haile, A.; Sun, W. Integrated Hair Follicle Profiles of microRNAs and mRNAs to Reveal the Pattern Formation of Hu Sheep Lambskin. Genes 2022, 13, 342. https://doi.org/10.3390/genes13020342
Lv X, Chen W, Wang S, Cao X, Yuan Z, Getachew T, Mwacharo JM, Haile A, Sun W. Integrated Hair Follicle Profiles of microRNAs and mRNAs to Reveal the Pattern Formation of Hu Sheep Lambskin. Genes. 2022; 13(2):342. https://doi.org/10.3390/genes13020342
Chicago/Turabian StyleLv, Xiaoyang, Weihao Chen, Shanhe Wang, Xiukai Cao, Zehu Yuan, Tesfaye Getachew, Joram M. Mwacharo, Aynalem Haile, and Wei Sun. 2022. "Integrated Hair Follicle Profiles of microRNAs and mRNAs to Reveal the Pattern Formation of Hu Sheep Lambskin" Genes 13, no. 2: 342. https://doi.org/10.3390/genes13020342
APA StyleLv, X., Chen, W., Wang, S., Cao, X., Yuan, Z., Getachew, T., Mwacharo, J. M., Haile, A., & Sun, W. (2022). Integrated Hair Follicle Profiles of microRNAs and mRNAs to Reveal the Pattern Formation of Hu Sheep Lambskin. Genes, 13(2), 342. https://doi.org/10.3390/genes13020342