
Multi-Level Analysis and Identification of Tumor Mutational Burden Genes across Cancer Types

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Gene Source and Collection Method
- The number of genes in the panel is below 500;
- The gene mutation frequency is over 0.01;
- Cancer-associated genes in the published literature are preferentially selected;
- Immune regulatory genes are preferentially selected;
- Drug-target genes, prognosis-related genes, or genes in clinical research are preferentially selected;
- Synonymous mutations are included in the TMB assessment.
2.3. TMB Calculation
2.4. DEGs and Functional Enrichment Analysis
2.5. TMB Differentially Expressed Genes and Immune Cell Infiltration Process
2.6. Prognosis Prediction Model Construction
3. Results
3.1. Establishment of Screening Criteria for TMB Gene Panel Collection
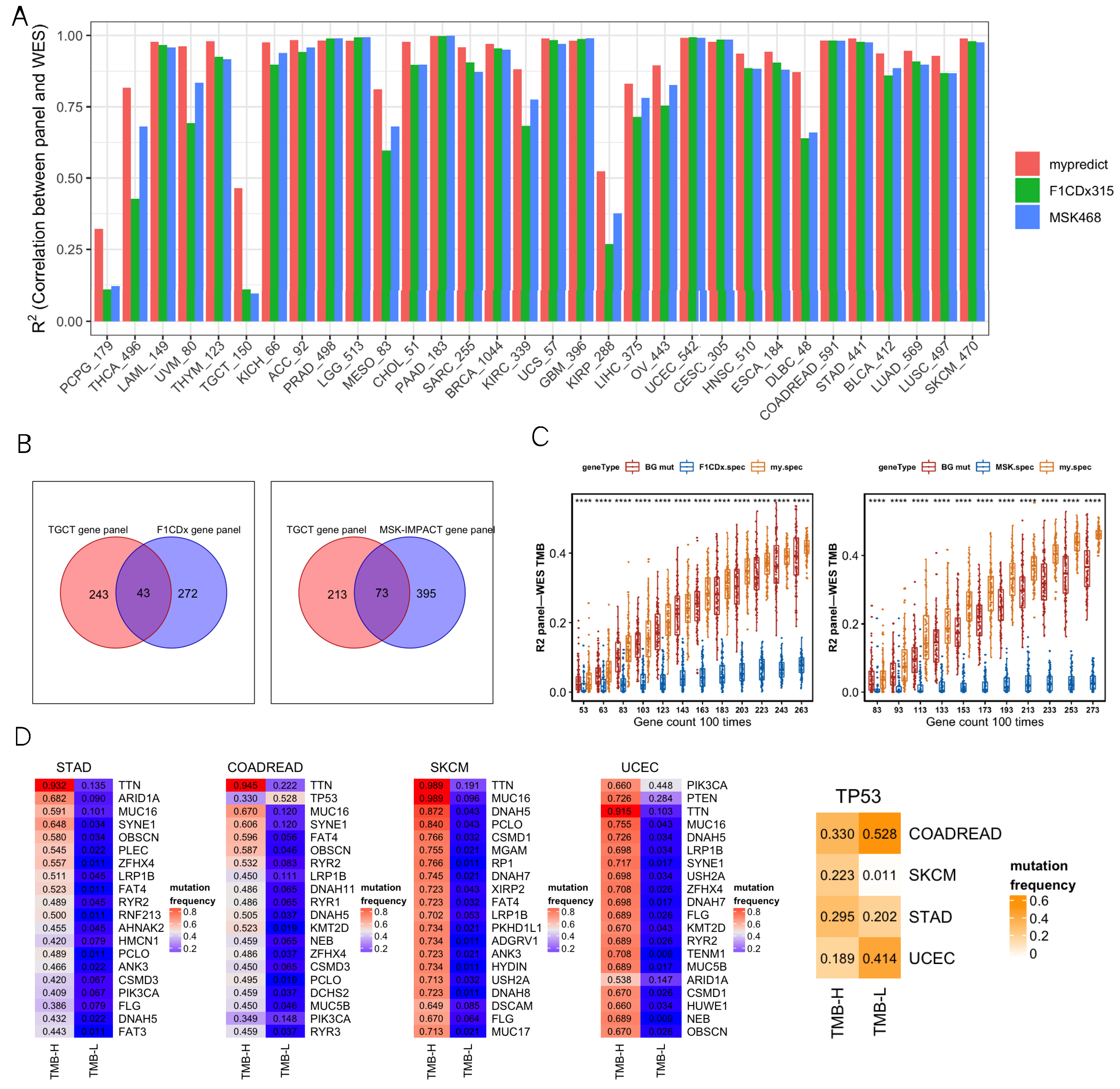
3.2. Comprehensive Analysis of SepPanel in R2, GO Enrichment, and Mutation Frequency
3.3. TMB-Related Differentially Expressed Genes
3.4. Analysis of TMB-Related DEGs and CD8+ T Cell Infiltration
3.5. Prognostic-Related Genes of TMB-Differentially Expressed Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Addeo, A.; Friedlaender, A.; Banna, G.L.; Weiss, G.J. TMB or not TMB as a biomarker: That is the question. Crit. Rev. Oncol. Hematol. 2021, 163, 103374. [Google Scholar] [CrossRef]
- Buder-Bakhaya, K.; Hassel, J.C. Biomarkers for clinical benefit of immune checkpoint inhibitor treatment-a review from the melanoma perspective and beyond. Front. Immunol. 2018, 9, 1474. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.; Abu-Akeel, M.; et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 2018, 33, 843–852.e844. [Google Scholar] [CrossRef] [Green Version]
- Jia, Q.; Chu, Q.; Zhang, A.; Yu, J.; Liu, F.; Qian, K.; Xiao, Y.; Wang, X.; Yang, Y.; Zhao, Y.; et al. Mutational burden and chromosomal aneuploidy synergistically predict survival from radiotherapy in non-small cell lung cancer. Commun. Biol. 2021, 4, 131. [Google Scholar] [CrossRef]
- Tian, Y.; Zhai, X.; Yan, W.; Zhu, H.; Yu, J. Clinical outcomes of immune checkpoint blockades and the underlying immune escape mechanisms in squamous and adenocarcinoma NSCLC. Cancer Med. 2021, 10, 3–14. [Google Scholar] [CrossRef]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Schwarze, K.; Buchanan, J.; Taylor, J.C.; Wordsworth, S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 2018, 20, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- FDA. FDA Announces Approval, CMS Proposes Coverage of First Breakthrough-Designated Test to Detect Extensive Number of Cancer Biomarkers. Available online: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm587273.htm (accessed on 18 June 2019).
- Wang, Z.; Duan, J.; Cai, S.; Han, M.; Dong, H.; Zhao, J.; Zhu, B.; Wang, S.; Zhuo, M.; Sun, J.; et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non-small cell lung cancer with use of a next-generation sequencing cancer gene panel. JAMA Oncol. 2019, 5, 696–702. [Google Scholar] [CrossRef]
- Roszik, J.; Haydu, L.E.; Hess, K.R.; Oba, J.; Joon, A.Y.; Siroy, A.E.; Karpinets, T.V.; Stingo, F.C.; Baladandayuthapani, V.; Tetzlaff, M.T.; et al. Novel algorithmic approach predicts tumor mutation load and correlates with immunotherapy clinical outcomes using a defined gene mutation set. BMC Med. 2016, 14, 168. [Google Scholar] [CrossRef] [Green Version]
- Campesato, L.F.; Barroso-Sousa, R.; Jimenez, L.; Correa, B.R.; Sabbaga, J.; Hoff, P.M.; Reis, L.F.; Galante, P.A.; Camargo, A.A. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to PD-1 blockade in clinical practice. Oncotarget 2015, 6, 34221–34227. [Google Scholar] [CrossRef] [Green Version]
- Lyu, G.Y.; Yeh, Y.H.; Yeh, Y.C.; Wang, Y.C. Mutation load estimation model as a predictor of the response to cancer immunotherapy. NPJ Genom. Med. 2018, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Spizzo, G.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; Arora, S.P.; Khushman, M.; Salem, M.E.; Battaglin, F.; et al. Molecular profile of BRCA-mutated biliary tract cancers. ESMO Open 2020, 5, e000682. [Google Scholar] [CrossRef]
- Klein, S.; Quaas, A.; Noh, K.W.; Cartolano, M.; Abedpour, N.; Mauch, C.; Quantius, J.; Reinhardt, H.C.; Buettner, R.; Peifer, M.; et al. Integrative Analysis of pleomorphic dermal sarcomas reveals fibroblastic differentiation and susceptibility to immunotherapy. Clin. Cancer Res. 2020, 26, 5638–5645. [Google Scholar] [CrossRef]
- Huang, T.; Liang, Y.; Zhang, H.; Chen, X.; Wei, H.; Sun, W.; Wang, Y. CSMD1 mutations are associated with increased mutational burden, favorable prognosis, and anti-tumor immunity in gastric cancer. Genes 2021, 12, 1715. [Google Scholar] [CrossRef]
- Sesma, A.; Pardo, J.; Cruellas, M.; Galvez, E.M.; Gascon, M.; Isla, D.; Martinez-Lostao, L.; Ocariz, M.; Pano, J.R.; Quilez, E.; et al. From tumor mutational burden to blood t cell receptor: Looking for the best predictive biomarker in lung cancer treated with immunotherapy. Cancers 2020, 12, 2974. [Google Scholar] [CrossRef]
- Liu, T.; Tan, J.; Wu, M.; Fan, W.; Wei, J.; Zhu, B.; Guo, J.; Wang, S.; Zhou, P.; Zhang, H.; et al. High-affinity neoantigens correlate with better prognosis and trigger potent antihepatocellular carcinoma (HCC) activity by activating CD39(+)CD8(+) T cells. Gut 2021, 70, 1965–1977. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Stadler, Z.K.; Battaglin, F.; Middha, S.; Hechtman, J.F.; Tran, C.; Cercek, A.; Yaeger, R.; Segal, N.H.; Varghese, A.M.; Reidy-Lagunes, D.L.; et al. Reliable detection of mismatch repair deficiency in colorectal cancers using mutational load in next-generation sequencing panels. J. Clin. Oncol. 2016, 34, 2141–2147. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, M. Correlate tumor mutation burden with immune signatures in human cancers. BMC Immunol. 2019, 20, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varn, F.S.; Wang, Y.; Mullins, D.W.; Fiering, S.; Cheng, C. Systematic pan-cancer analysis reveals immune cell interactions in the tumor microenvironment. Cancer Res. 2017, 77, 1271–1282. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, B.; Peng, Y.; Wu, F.; Li, Q.; Lin, Z.; Xie, S.; Xiao, L.; Lin, X.; Ou, Z.; et al. The prognostic value of TMB and the relationship between TMB and immune infiltration in head and neck squamous cell carcinoma: A gene expression-based study. Oral Oncol. 2020, 110, 104943. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, M.; Liu, Q.; Liu, Y.; Zhu, K.; Chen, L.; Guo, H.; Li, Y.; Shi, B. Identification of gene expression profiles and immune cell infiltration signatures between low and high tumor mutation burden groups in bladder cancer. Int. J. Med. Sci. 2020, 17, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Liu, Z.; Liu, X.; Duan, X.; Huang, Y.; Jin, Z.; Niu, Y.; Zhang, L.; Chen, H. Identification of an Immune-related prognostic signature associated with immune infiltration in melanoma. Front. Genet. 2020, 11, 1002. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhao, W.; Zhou, B.; Su, Z.; Gu, X.; Zhou, Z.; Chen, S. TSNAdb: A database for tumor-specific neoantigens from immunogenomics data analysis. Genom. Proteom. Bioinform. 2018, 16, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.H.; Kao, H.Y.; Lu, Z. PubTator: A web-based text mining tool for assisting biocuration. Nucleic Acids Res. 2013, 41, W518–W522. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Wang, S.; Tong, Y.; Jin, L.; Zong, H.; Zheng, R.; Yang, J.; Zhang, Z.; Ouyang, E.; et al. GIDB: A knowledge database for the automated curation and multidimensional analysis of molecular signatures in gastrointestinal cancer. Database 2019, 2019, baz051. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortimer, T.; Wainwright, E.N.; Patel, H.; Siow, B.M.; Jaunmuktane, Z.; Brandner, S.; Scaffidi, P. Redistribution of EZH2 promotes malignant phenotypes by rewiring developmental programmes. EMBO Rep. 2019, 20, e48155. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Ghazarian, A.A.; Kelly, S.P.; Altekruse, S.F.; Rosenberg, P.S.; McGlynn, K.A. Future of testicular germ cell tumor incidence in the United States: Forecast through 2026. Cancer 2017, 123, 2320–2328. [Google Scholar] [CrossRef] [Green Version]
- Horwich, A.; Shipley, J.; Huddart, R. Testicular germ-cell cancer. Lancet 2006, 367, 754–765. [Google Scholar] [CrossRef]
- Shah, S.; Ward, J.E.; Bao, R.; Hall, C.R.; Brockstein, B.E.; Luke, J.J. Clinical response of a patient to Anti-PD-1 immunotherapy and the immune landscape of testicular germ cell tumors. Cancer Immunol. Res. 2016, 4, 903–909. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbe, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, E.A.; Schweizer, M.T. Durable response to immune checkpoint blockade in a platinum-refractory patient with nonseminomatous germ cell tumor. Clin. Genitourin. Cancer 2017, 15, e855–e857. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the immunotarget registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, V.P.; Luksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Kumar, S.; Cruz, E.; Joshi, S.; Patel, A.; Jahan, R.; Batra, S.K.; Jain, M. Genetic variants of mucins: Unexplored conundrum. Carcinogenesis 2017, 38, 671–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-residing Batf3 dendritic cells are required for effector t cell trafficking and adoptive T cell therapy. Cancer Cell 2017, 31, 711–723.e714. [Google Scholar] [CrossRef] [Green Version]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 2018, 172, 1022–1037.e1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Zhao, B.; Liang, Y.; Ma, B. Identification and validation of a novel 2-LncRNAs signature associated with m6a regulation in colorectal cancer. J. Cancer 2022, 13, 21–33. [Google Scholar] [CrossRef]
- Wu, X.; Yang, T.; Qian, L.; Zhang, D.; Yang, H. Construction of a new tumor immunity-related signature to assess and classify the prognostic risk of colorectal cancer. Int. J. Gen. Med. 2021, 14, 6661–6676. [Google Scholar] [CrossRef] [PubMed]
- Randon, G.; Intini, R.; Cremolini, C.; Elez, E.; Overman, M.J.; Lee, J.; Manca, P.; Bergamo, F.; Pagani, F.; Antista, M.; et al. Tumour mutational burden predicts resistance to EGFR/BRAF blockade in BRAF-mutated microsatellite stable metastatic colorectal cancer. Eur. J. Cancer. 2022, 161, 90–98. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, Y.; Zhang, Z.; Zheng, R.; Huang, D.; Yang, J.; Zong, H.; Tan, F.; Xie, Y.; Huang, H.; et al. ViMIC: A database of human disease-related virus mutations, integration sites and cis-effects. Nucleic Acids Res. 2022, 50, D918–D927. [Google Scholar] [CrossRef] [PubMed]
- Healey Bird, B.; Nally, K.; Ronan, K.; Clarke, G.; Amu, S.; Almeida, A.S.; Flavin, R.; Finn, S. Cancer immunotherapy with immune checkpoint inhibitors-biomarkers of response and toxicity; Current limitations and future promise. Diagnostics 2022, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, M.; Mehnert, J.; Silk, A.W.; Jabbour, S.K.; Ganesan, S.; Popli, P.; Riedlinger, G.; Stephenson, R.; de Meritens, A.B.; Leiser, A.; et al. Real-world application of tumor mutational burden-high (TMB-high) and microsatellite instability (MSI) confirms their utility as immunotherapy biomarkers. ESMO Open 2022, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Coefs | Gene | Coefs | Gene | Coefs | Gene | Coefs |
|---|---|---|---|---|---|---|---|
| FOXD1 | 0.030 | TLE6 | 0.148 | ACCN3 | 0.038 | HOXD13 | 0.025 |
| HOXC6 | 0.027 | CATSPERB | −0.077 | IGF2BP3 | 0.001 | SFTA2 | 0.071 |
| TNNT1 | 0.048 | APC2 | 0.146 | PTGER2 | −0.100 | ATOH1 | −0.037 |
| HOXC4 | 0.017 | ISLR2 | 0.160 | CBLN2 | −0.131 | TMEM61 | −0.116 |
| ANKRD43 | −0.022 | APOLD1 | −0.091 | CD38 | −0.066 |
| Characteristic | Hazard Ratio | p-Value |
|---|---|---|
| Risk Score | ||
| Low Risk Score | 1.00 (reference) | |
| High Risk Score | 3.3807 (0.2958, 5.940) | <0.001 |
| Age | 1.0478 (0.9544, 1.071) | <0.001 |
| Gender | ||
| Female | 1.00 (reference) | |
| Male | 0.9657 (1.0355, 1.540) | 0.884 |
| Tumor Stage | ||
| I | 1.00 (reference) | |
| II | 1.1566 (0.8646, 3.120) | 0.774 |
| III | 1.8502 (0.5405, 4.919) | 0.217 |
| IV | 5.4542 (0.1833, 14.791) | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Tong, Y.; Zong, H.; Xu, X.; Crabbe, M.J.C.; Wang, Y.; Zhang, X. Multi-Level Analysis and Identification of Tumor Mutational Burden Genes across Cancer Types. Genes 2022, 13, 365. https://doi.org/10.3390/genes13020365
Wang S, Tong Y, Zong H, Xu X, Crabbe MJC, Wang Y, Zhang X. Multi-Level Analysis and Identification of Tumor Mutational Burden Genes across Cancer Types. Genes. 2022; 13(2):365. https://doi.org/10.3390/genes13020365
Chicago/Turabian StyleWang, Shuangkuai, Yuantao Tong, Hui Zong, Xuewen Xu, M. James C. Crabbe, Ying Wang, and Xiaoyan Zhang. 2022. "Multi-Level Analysis and Identification of Tumor Mutational Burden Genes across Cancer Types" Genes 13, no. 2: 365. https://doi.org/10.3390/genes13020365
APA StyleWang, S., Tong, Y., Zong, H., Xu, X., Crabbe, M. J. C., Wang, Y., & Zhang, X. (2022). Multi-Level Analysis and Identification of Tumor Mutational Burden Genes across Cancer Types. Genes, 13(2), 365. https://doi.org/10.3390/genes13020365