
Comparative Chloroplast Genome Analysis of Wax Gourd (Benincasa hispida) with Three Benincaseae Species, Revealing Evolutionary Dynamic Patterns and Phylogenetic Implications

 ,
, 
 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material, DNA Extraction, and Sequencing
2.2. Genome Assembly and Annotations
2.3. Chloroplast Genome Comparison
2.4. Codon Usage and Putative RNA Editing Site
2.5. Repeat Sequences and SSR Analysis
2.6. Comparative Analysis of cp Genomes in Benincaseae
2.7. Phylogenetic Analysis
3. Results
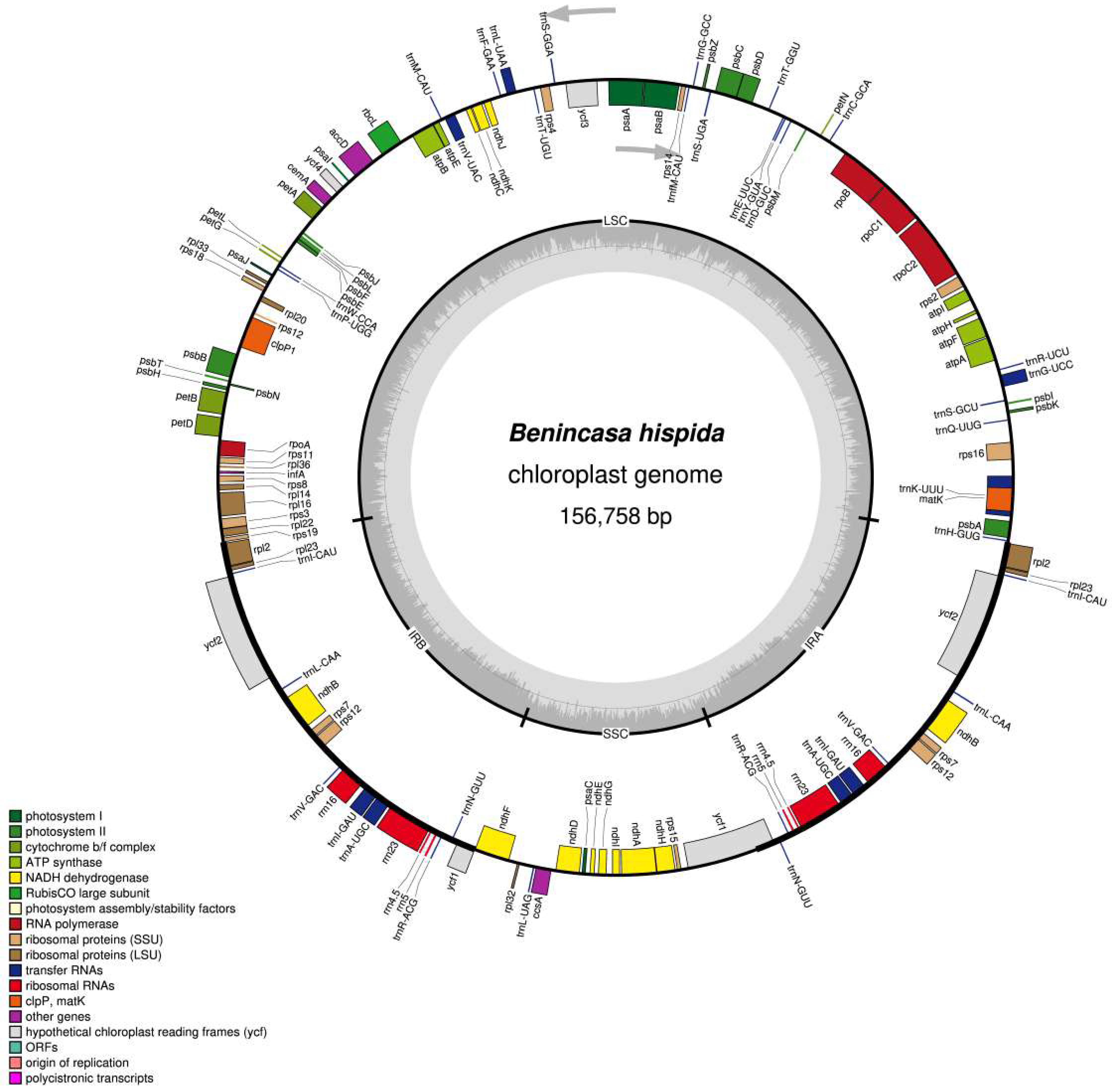
3.1. Chloroplast Genome Assembly, Organization, and Features of Benincasa Hispida
3.2. Codon Usage and Amino Acid Frequencies
3.3. Putative RNA Editing Site within Benincaseae
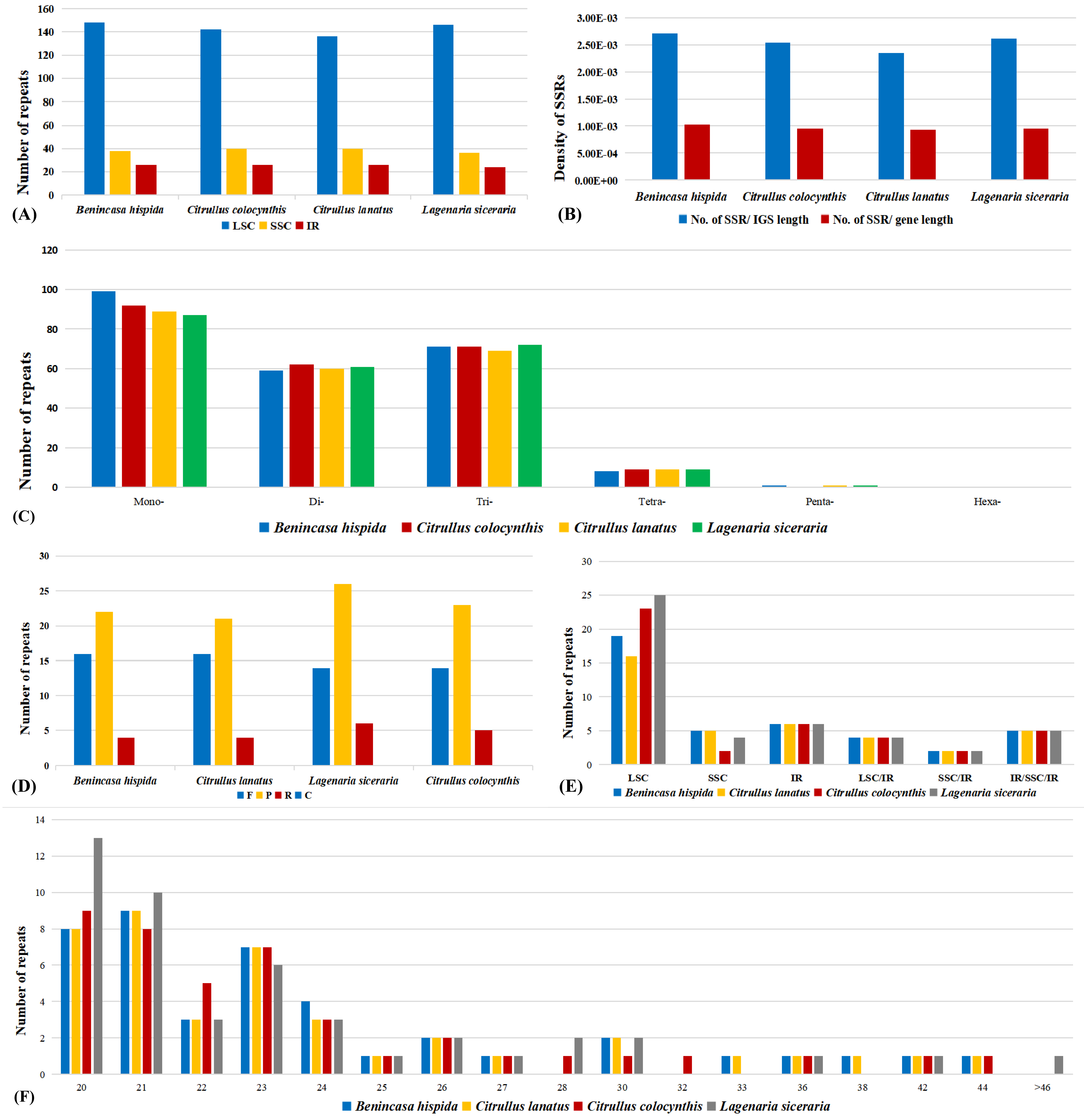
3.4. Repeated Sequence and SSR Analysis
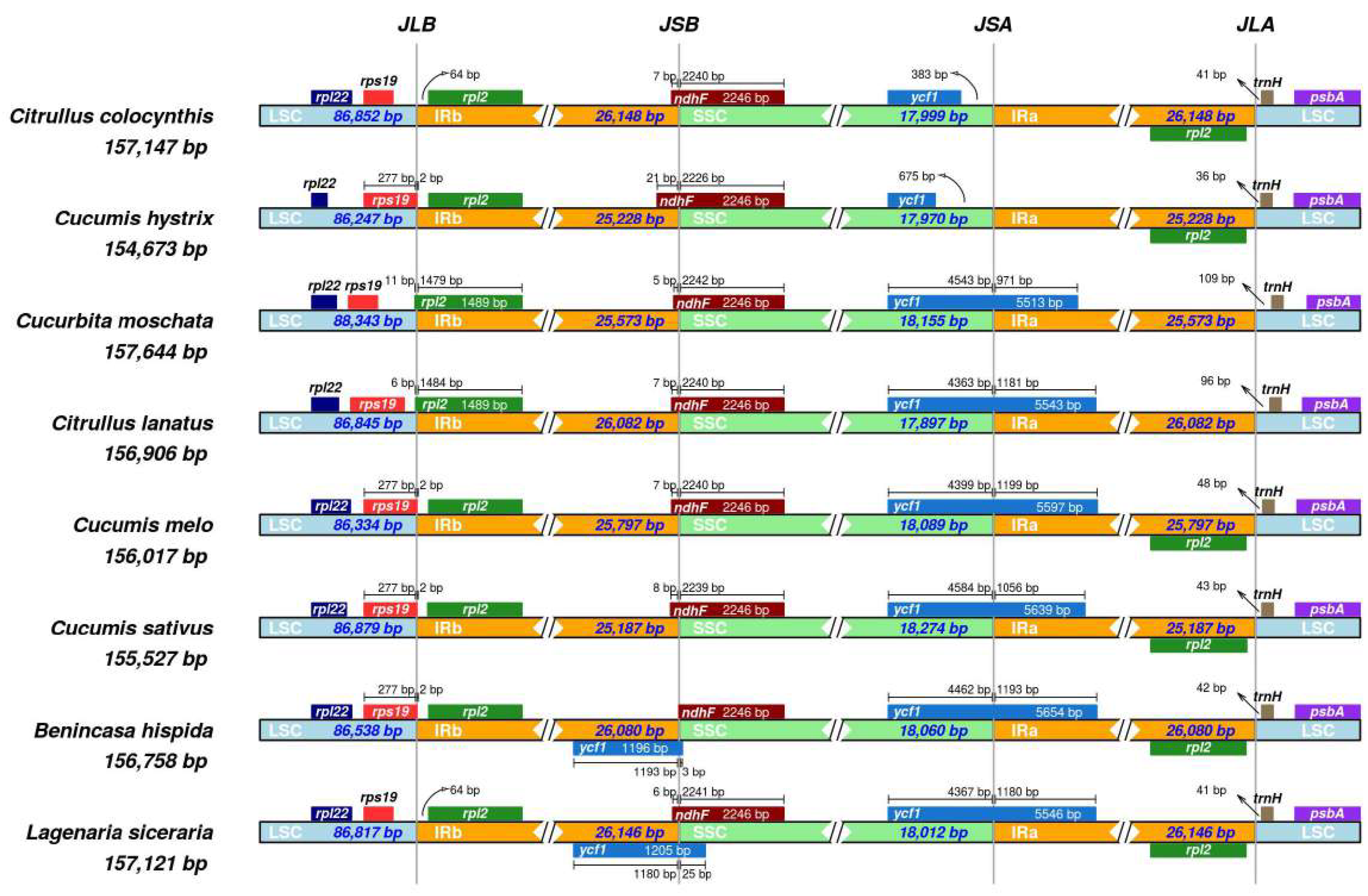
3.5. IR Contraction and Expansion
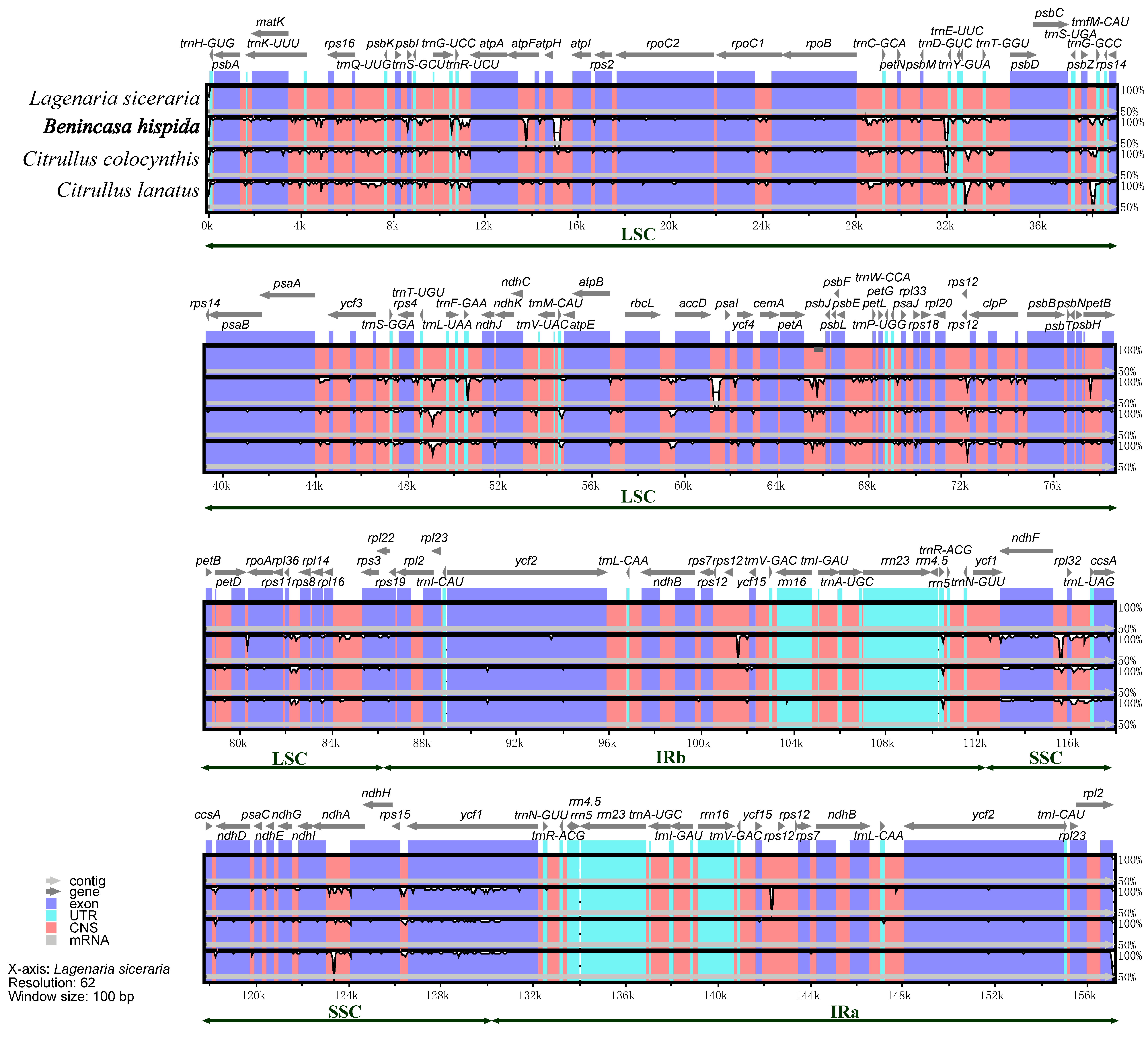
3.6. Divergence Analysis of Chloroplast Genome
3.7. Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Christenhusz, M.J.M.; Byng, J.W. The number of known plants species in the world and its annual increase. Phytotaxa 2016, 261, 201–217. [Google Scholar] [CrossRef] [Green Version]
- Ward, B.L.; Anderson, R.S.; Bendich, A.J. The mitochondrial genome is large and variable in a family of plants (Cucurbitaceae). Cell 1981, 25, 793–803. [Google Scholar] [CrossRef]
- Nandecha, C.; Nahata, A.; Dixit, V.K. Effect of Benincasa hispida fruits on testosterone-induced prostatic hypertrophy in albino rats. Curr. Ther. Res. 2010, 71, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Kocyan, A.; Zhang, L.-B.; Schaefer, H.; Renner, S.S. A multi-locus chloroplast phylogeny for the Cucurbitaceae and its implications for character evolution and classification. Mol. Phylogenet. Evol. 2007, 44, 553–577. [Google Scholar] [CrossRef]
- Renner, S.S.; Schaefer, H. Phylogeny and Evolution of the Cucurbitaceae. In Genetics and Genomics of Cucurbitaceae. Plant Genetics and Genomics: Crops and Models; Grumet, R., Katzir, N., Garcia-Mas, J., Eds.; Springer: Cham, The Netherlands, 2016; Volume 20, pp. 155–172. [Google Scholar] [CrossRef]
- Guo, J.; Xu, W.; Hu, Y.; Huang, J.; Zhao, Y.; Zhang, L.; Huang, C.-H.; Ma, H. Phylotranscriptomics in Cucurbitaceae reveal multiple whole-genome duplications and key morphological and molecular innovations. Mol. Plant 2020, 13, 1117–1133. [Google Scholar] [CrossRef]
- Steward, F.C. Some Economic Plants: Tropical Crops: Dicotyledons. J. W. Purseglove. Wiley, New York, 1968; 2 vols., xx + 719 pp., illus. $8.50 each. Science 1969, 163, 1050–1051. [Google Scholar] [CrossRef]
- Thomas, T.D.; Sreejesh, K.R. Callus induction and plant regeneration from cotyledonary explants of ash gourd (Benincasa hispida L.). Sci. Hortic. 2004, 100, 359–367. [Google Scholar] [CrossRef]
- Naik, R.; Buha, M.; Acharya, R.; Borkar, S.D. Role of vegetables (Shaka Dravyas) in prevention and management of gastro—Intestinal tract diseases: A critical review. J. Res. Tradit. Med. 2016, 2, 103–112. [Google Scholar] [CrossRef]
- Al-Snafi, A.E. The Pharmacological Importance of Benincasa hispida. A review. Int. J. Pharma Sci. Res. 2013, 4, 975–9492. Available online: https://www.researchgate.net/publication/313676687_The_Pharmacological_Importance_of_Benincasa_hispida_A_review (accessed on 13 December 2021).
- Rachchh, M.A.; Jain, S.M. Gastroprotective effect of Benincasa hispida fruit extract. Indian J. Pharmacol. 2008, 40, 271–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhingra, D.; Joshi, P. Antidepressant-like activity of Benincasa hispida fruits in mice: Possible involvement of monoaminergic and GABAergic systems. J. Pharmacol. Pharmacother. 2012, 3, 60–62. [Google Scholar] [CrossRef] [Green Version]
- Jayasree, T.; Kishore, K.K.; Vinay, M.; Vasavi, P.; Dixit, R. Evaluation of the diuretic effect of the chloroform extract of the Benincasa hispida rind (pericarp) extract in guinea-pigs. J. Clin. Diagn. Res. 2011, 5, 578–582. [Google Scholar]
- Lee, K.-H.; Choi, H.-R.; Kim, C.-H. Anti-angiogenic effect of the seed extract of Benincasa hispida Cogniaux. J. Ethnopharmacol. 2005, 97, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Qadrie, Z.L.; Hawisa, N.T.; Khan, M.W.A.; Samuel, M.; Anandan, R. Antinociceptive and anti-pyretic activity of Benincasa hispida (thunb.) cogn. In Wistar albino rats. Pak. J. Pharm. Sci. 2009, 22, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, D.; Lavarasan, R.J.; Babu, S.C.; Refai, M.; Ansari, L. Antimicrobial studies on methanol extract of Benincasa hispida cogn., fruit. Anc. Sci. Life 2003, 22, 98–100. [Google Scholar] [PubMed]
- Bimakr, M.; Rahman, R.A.; Taip, F.S.; Adzahan, N.M.; Sarker, M.Z.I.; Ganjloo, A. Optimization of ultrasound-assisted extraction of crude oil from winter melon (Benincasa hispida) seed using response surface methodology and evaluation of its antioxidant activity, total phenolic content and fatty acid composition. Molecules 2012, 17, 11748–11762. [Google Scholar] [CrossRef] [PubMed]
- Grover, J.K.; Adiga, G.; Vats, V.; Rathi, S.S. Extracts of Benincasa hispida prevent development of experimental ulcers. J. Ethnopharmacol. 2001, 78, 159–164. [Google Scholar] [CrossRef]
- Palmer, J.D. Plastid chromosomes: Structure and evolution. Mol. Biol. Plast. 1991, 7, 5–53. [Google Scholar] [CrossRef]
- Ahmed, I.; Biggs, P.J.; Matthews, P.J.; Collins, L.J.; Hendy, M.D.; Lockhart, P.J. Mutational dynamics of aroid chloroplast genomes. Genome Biol. Evol. 2012, 4, 1316–1323. [Google Scholar] [CrossRef] [Green Version]
- Sloan, D.B.; Triant, D.A.; Forrester, N.J.; Bergner, L.M.; Wu, M.; Taylor, D.R. A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol. Phylogenetics Evol. 2013, 72, 82–89. [Google Scholar] [CrossRef]
- Ahmed, I. Chloroplast genome sequencing: Some reflections. J. Next Gener. Seq. Appl. 2015, 2, 2469–9853. [Google Scholar] [CrossRef] [Green Version]
- Lössl, A.G.; Waheed, M.T. Chloroplast-derived vaccines against human diseases: Achievements, challenges and scopes. Plant Biotechnol. J. 2011, 9, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Xu, Y.; Wang, J.; Liu, W.; Zhou, Q.; Luo, S.; Huang, W.; He, X.; Li, Q.; Peng, Q.; et al. The wax gourd genomes offer insights into the genetic diversity and ancestral cucurbit karyotype. Nat. Commun. 2019, 10, 5158. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Wang, Y.; Wang, S.; Liu, Y.; Yuan, Q.; Sun, J.; Guo, L. Chloroplast genome variation and phylogenetic relationships of Atractylodes species. BMC Genom. 2021, 22, 103. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. In Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010; Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 13 December 2021).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Guo, S.; Xu, J.; He, L.; Carlson, J.E.; Hou, X. Phylogenetic analysis based on chloroplast genome uncover evolutionary relationship of all the nine species and six cultivars of tree peony. Ind. Crops Prod. 2020, 153, 112567. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Muraguri, S.; Xu, W.; Chapman, M.; Muchugi, A.; Oluwaniyi, A.; Oyebanji, O.; Liu, A. Intraspecific variation within Castor bean (Ricinus communis L.) based on chloroplast genomes. Ind. Crop. Prod. 2020, 155, 112779. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 0955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Lehwark, P.; Greiner, S. GB2sequin—A file converter preparing custom GenBank files for database submission. Genomics 2019, 111, 759–761. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: Bioinformatics Software for Sequence Data Analysis. 2012. Available online: https://www.geneious.com/ (accessed on 13 December 2021).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. Irscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R.; Goldman, N.; Pedersen, A.-M.K. Codon-Substitution Models for Heterogeneous Selection Pressure at Amino Acid Sites. Genetics 2000, 155, 431–449. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Wen, D.; Yu, Y.; Meudt, H.M.; Nakhleh, L. Bayesian inference of phylogenetic networks from bi-allelic genetic markers. PLOS Comput. Biol. 2018, 14, e1005932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poczai, P.; Hyvönen, J. The complete chloroplast genome sequence of the CAM epiphyte Spanish moss (Tillandsia usneoides, Bromeliaceae) and its comparative analysis. PloS ONE 2017, 12, e0187199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.-B.; Zhou, X.-Y.; Li, J.-W. Development of novel chloroplast microsatellite markers for Cucumis from sequence database. Biol. Plant. 2009, 53, 793–796. [Google Scholar] [CrossRef]
- Bhowmick, B.K.; Jha, S. Differential heterochromatin distribution, flow cytometric genome size and meiotic behavior of chromosomes in three Cucurbitaceae species. Sci. Hortic. 2015, 193, 322–329. [Google Scholar] [CrossRef]
- Bausher, M.G.; Singh, N.D.; Lee, S.-B.; Jansen, R.K.; Daniell, H. The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var ‘Ridge Pineapple’: Organization and phylogenetic relationships to other angiosperms. BMC Plant Biol. 2006, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehmood, F.; Abdullah, S.I.; Ahmed, I.; Waheed, M.T.; Mirza, B. Characterization of Withania somnifera chloroplast genome and its comparison with other selected species of Solanaceae. Genomics 2020, 112, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Guo, L.; Zhao, W.; Xu, J.; Li, Y.; Zhang, X.; Shen, X.; Wu, M.; Hou, X. Complete chloroplast genome sequence and phylogenetic analysis of Paeonia ostii. Molecules 2018, 23, 246. [Google Scholar] [CrossRef] [Green Version]
- Daniell, H.; Jin, S.; Zhu, X.; Gitzendanner, M.A.; Soltis, D.E.; Soltis, P.S. Green giant—A tiny chloroplast genome with mighty power to produce high-value proteins: History and phylogeny. Plant Biotechnol. J. 2021, 19, 430–447. [Google Scholar] [CrossRef]
- Yang, X.; Luo, X.; Cai, X. Analysis of codon usage pattern in Taenia saginata based on a transcriptome dataset. Parasites Vectors 2014, 7, 527. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, K. Codon evolution in double-stranded organelle DNA: Strong regulation of homonucleotides and their analog alternations. Nat. Sci. 2010, 2, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, C.; Guo, X.; Liu, Q.; Wang, K. Complete chloroplast genome of Camellia japonica genome structures, comparative and phylogenetic analysis. PloS ONE 2019, 14, e0216645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saina, J.K.; Li, Z.-Z.; Gichira, A.W.; Liao, Y.-Y. The complete chloroplast genome sequence of tree of heaven (Ailanthus altissima (Mill.) (Sapindales: Simaroubaceae), an important pantropical tree. Int. J. Mol. Sci. 2018, 19, 929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yu, H.; Wang, J.; Lei, W.; Gao, J.; Qiu, X.; Wang, J. The complete chloroplast genome sequences of the medicinal plant Forsythia suspensa (Oleaceae). Int. J. Mol. Sci. 2017, 18, 2288. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, I.; Matthews, P.J.; Biggs, P.; Naeem, M.; McLenachan, P.A.; Lockhart, P.J. Identification of chloroplast genome loci suitable for high-resolution phylogeographic studies of Colocasia esculenta (L.) Schott (Araceae) and closely related taxa. Mol. Ecol. Resour. 2013, 13, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Cavender-Bares, J.; González-Rodríguez, A.; Eaton, D.A.R.; Hipp, A.A.L.; Beulke, A.; Manos, P.S. Phylogeny and biogeography of the American live oaks (Quercus subsection Virentes): A genomic and population genetics approach. Mol. Ecol. 2015, 24, 3668–3687. [Google Scholar] [CrossRef]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme reconfiguration of plastid genomes in the angiosperm family geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 2010, 28, 583–600. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Zhang, Y.; Xu, J.; Li, W.; Li, M. Characterization of the complete chloroplast genome sequence of Dalbergia species and its phylogenetic implications. Sci. Rep. 2019, 9, 20401. [Google Scholar] [CrossRef]
- Jeon, J.-H.; Kim, S.-C. Comparative analysis of the complete chloroplast genome sequences of three closely related east-Asian wild roses (Rosa sect. Synstylae; Rosaceae). Genes 2019, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullah, S.I.; Mehmood, F.; Ali, Z.; Malik, M.S.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Comparative analyses of chloroplast genomes among three Firmiana species: Identification of mutational hotspots and phylogenetic relationship with other species of Malvaceae. Plant Gene 2019, 19, 100199. [Google Scholar] [CrossRef]
- Shen, X.; Wu, M.; Liao, B.; Liu, Z.; Bai, R.; Xiao, S.; Li, X.; Zhang, B.; Xu, J.; Chen, S. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 2017, 22, 1330. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [Green Version]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; He, P.; Li, P.; Lee, J.; Soltis, D.E.; Fu, C. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genom. 2018, 19, 235. [Google Scholar] [CrossRef]
- Zhu, B.; Qian, F.; Hou, Y.; Yang, W.; Cai, M.; Wu, X. Complete chloroplast genome features and phylogenetic analysis of Eruca sativa (Brassicaceae). PloS ONE 2021, 16, e0248556. [Google Scholar] [CrossRef]
- Menezes, A.P.A.; Resende-Moreira, L.C.; Buzatti, R.S.O.; Nazareno, A.G.; Carlsen, M.; Lobo, F.P.; Kalapothakis, E.; Lovato, M.B. Chloroplast genomes of Byrsonima species (Malpighiaceae): Comparative analysis and screening of high divergence sequences. Sci. Rep. 2018, 8, 2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahzadi, I.; Abdullah, M.F.; Ali, Z.; Ahmed, I.; Mirza, B. Chloroplast genome sequences of Artemisia maritima and Artemisia absinthium: Comparative analyses, mutational hotspots in genus Artemisia and phylogeny in family Asteraceae. Genomics 2020, 112, 1454–1463. [Google Scholar] [CrossRef]
- Nazareno, A.G.; Carlsen, M.; Lohmann, L.G. Complete chloroplast genome of Tanaecium tetragonolobum: The first ignoniaceae plastome. PloS ONE 2015, 10, e0129930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.K.; Saski, C.; Lee, S.-B.; Hansen, A.K.; Daniell, H. Complete plastid genome sequences of three rosids (Castanea, Prunus, Theobroma): Evidence for at least two independent transfers of rpl22 to the nucleus. Mol. Biol. Evol. 2011, 28, 835–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, Y.; Zang, M.; Li, M.; Fang, Y. Complete chloroplast genome sequence and phylogenetic analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. The chloroplast genome sequence of bittersweet (Solanum dulcamara): Plastid genome structure evolution in Solanaceae. PloS ONE 2018, 13, e0196069. [Google Scholar] [CrossRef]
- Du, Y.-P.; Bi, Y.; Yang, F.-P.; Zhang, M.-F.; Chen, X.-Q.; Xue, J.; Zhang, X.-H. Complete chloroplast genome sequences of Lilium: Insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 2017, 7, 5751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, Y.; Gao, L.; Liu, B.; Yang, Y.; Kong, S.; Sun, Y.; Yang, Y.; Wu, X. Complete chloroplast genome sequences of four Allium species: Comparative and phylogenetic analyses. Sci. Rep. 2019, 9, 12250. [Google Scholar] [CrossRef] [Green Version]
- Xiao-Ming, Z.; Junrui, W.; Li, F.; Sha, L.; Hongbo, P.; Lan, Q.; Jing, L.; Yan, S.; Weihua, Q.; Lifang, Z.; et al. Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci. Rep. 2017, 7, 1555. [Google Scholar] [CrossRef]
- Kode, V.; Mudd, E.A.; Iamtham, S.; Day, A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005, 44, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Nie, L.; Sun, W.; Xu, Z.; Wang, Y.; Yu, J.; Song, J.; Yao, H. Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family Zingiberaceae based on the complete chloroplast genome. Plants 2019, 8, 283. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Zhang, L.; Qi, J.; Zhang, L. Complete chloroplast genome sequence of Hibiscus cannabinus and comparative analysis of the Malvaceae family. Front. Genet. 2020, 11, 277. [Google Scholar] [CrossRef] [PubMed]
- Levi, A.; Harris, K.R.; Wechter, W.P.; Kousik, C.S.; Thies, J.A. DNA markers and pollen morphology reveal that Praecitrullus fistulosus is more closely related to Benincasa hispida than to Citrullus spp. Genet. Resour. Crop Evol. 2010, 57, 1191–1205. [Google Scholar] [CrossRef]
- Rodríguez-Moreno, L.; González, V.M.; Benjak, A.; Martí, M.C.; Puigdomènech, P.; Aranda, M.A.; Garcia-Mas, J. Determination of the melon chloroplast and mitochondrial genome sequences reveals that the largest reported mitochondrial genome in plants contains a significant amount of DNA having a nuclear origin. BMC Genom. 2011, 12, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneidak, S.; Khalik, K.A. Seed coat diversity in some tribes of Cucurbitaceae: Implications for taxonomy and species identification. Acta Bot. Bras. 2015, 29, 129–142. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Tembrock, L.R.; Zheng, S.; Wu, Z. The complete chloroplast genome of Catha edulis: A comparative analysis of genome features with related species. Int. J. Mol. Sci. 2018, 19, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logacheva, M.D.; Penin, A.; Samigullin, T.H.; Vallejo-Roman, C.M.; Antonov, A.S. Phylogeny of Flowering Plants by the Chloroplast Genome Sequences: In Search of a “Lucky Gene”. Biochemistry 2007, 72, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Song, W.C.; Ji, C.X.; Chen, Z.M.; Cai, H.H.; Wu, X.M.; Shi, C.; Wang, S. Comparative analysis the complete chloroplast genomes of nine Musa Species: Genomic features, comparative analysis, and phylogenetic implications. Front Plant Sci. 2022, 13, 62. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Benincasa hispida | |
|---|---|---|
| Size (base pair, bp) | 156,758 | |
| LSC length (bp) | 86,538 | |
| SSC length (bp) | 18,060 | |
| IR length (bp) | 26,080 | |
| Number of genes | 131 | |
| Number of protein-coding genes | 86 | |
| Number of tRNA genes | 37 | |
| Number of rRNA genes | 8 | |
| Duplicate genes | 18 | |
| GC content | Total (%) | 37.2 |
| LSC (%) | 35 | |
| SSC (%) | 31.7 | |
| IR (%) | 42.9 | |
| CDS (%) | 37.9 | |
| rRNA (%) | 55.2 | |
| tRNA (%) | 53.2 | |
| ALL gene % | 39.4 | |
| Protein-coding part (CDS) (% bp) | 51.1 | |
| All genes (% bp) | 71.6 | |
| Non-coding region (% bp) | 28.4 | |
| Category of Genes | Group of Genes | Gene Name |
|---|---|---|
| Photosynthesis-related genes | Large subunit of rubisco | rbcL |
| Photosystem I | psaA, psaB, psaC, psaI, psaJ | |
| Assembly/srability of photosystem I | ycf3 **, ycf4 | |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | |
| Cytochrome b6/f complex | petA, petB *, petD *, petG, petL, petN | |
| Cytochrome c synthesis | ccsA | |
| NADH dehydrogenase | ndhA *, ndhB *, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Transcription and translation related genes | RNA polymerase subunits/transcription | rpoA, rpoB, rpoC1 *, rpoC2 |
| Small subunit of ribosomal proteins | rps11, rps12 * (*2), rps14, rps15, rps16 *, rps18, rps19, rps2, rps3, rps4, rps7 (*2), rps8 | |
| Large subunit of ribosomal proteins | rpl14, rpl16 *, rpl2 * (*2), rpl20, rpl22, rpl23 (*2), rpl32, rpl33, rpl36 | |
| Translation initiation factor | infA | |
| RNA genes | Ribosomal RNA | rrn16 (*2), rrn23 (*2), rrn4.5 (*2), rrn5 (*2) |
| transfer RNA | trnA-UGC * (*2), trnR-ACG (*2), trnR-UCU, trnN-GUU (*2), trnD-GUC, trnC-GCA, trnQ-UUG, trnE-UUC, trnG-GCC, trnG-UCC *, trnH-GUG, trnI-CAU (*2), trnI-GAU * (*2), trnL-CAA (*2), trnL-UAA *, trnL-UAG, trnK-UUU *, trnfM-CAU, trnM-CAU, trnF-GAA, trnP-UGG, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnW-CCA, trnY-GUA, trnV-GAC (*2), trnV-UAC * | |
| Other genes | RNA processing | matK |
| Carbon metabolism | cemA | |
| Fatty acid synthesis | accD | |
| Proteolysis | clpP1 ** | |
| Component of TIC complex | ycf1 (*2) | |
| Hypothetical proteins | ycf2 (*2) |
| Gene Name | Models | np | ln L | Likelihood RatioTest p-Value | Positively Selected Sites | |
|---|---|---|---|---|---|---|
| AA-Site | Score | |||||
| accD | M8 (beta) | 10 | −2173.400149 | 0.007931755 | 159 W | 0.984 * |
| M7 (beta & ω > 1) | 8 | −2178.23703 | ||||
| clpP | M8 (beta) | 10 | −926.578492 | 0.070969008 | ||
| M7 (beta & ω > 1) | 8 | −929.224004 | ||||
| rps4 | M8 (beta) | 10 | −877.349259 | 0.030217199 | 158 Q | 0.971 * |
| M7 (beta & ω > 1) | 8 | −880.848603 | ||||
| ycf1 | M8 (beta) | 10 | −6139.981658 | 0.156641895 | ||
| M7 (beta & ω > 1) | 8 | −6141.835451 | ||||
| ycf2 | M8 (beta) | 10 | −9230.970637 | 0.063743376 | ||
| M7 (beta & ω > 1) | 8 | −9233.723527 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, W.; Chen, Z.; He, L.; Feng, Q.; Zhang, H.; Du, G.; Shi, C.; Wang, S. Comparative Chloroplast Genome Analysis of Wax Gourd (Benincasa hispida) with Three Benincaseae Species, Revealing Evolutionary Dynamic Patterns and Phylogenetic Implications. Genes 2022, 13, 461. https://doi.org/10.3390/genes13030461
Song W, Chen Z, He L, Feng Q, Zhang H, Du G, Shi C, Wang S. Comparative Chloroplast Genome Analysis of Wax Gourd (Benincasa hispida) with Three Benincaseae Species, Revealing Evolutionary Dynamic Patterns and Phylogenetic Implications. Genes. 2022; 13(3):461. https://doi.org/10.3390/genes13030461
Chicago/Turabian StyleSong, Weicai, Zimeng Chen, Li He, Qi Feng, Hongrui Zhang, Guilin Du, Chao Shi, and Shuo Wang. 2022. "Comparative Chloroplast Genome Analysis of Wax Gourd (Benincasa hispida) with Three Benincaseae Species, Revealing Evolutionary Dynamic Patterns and Phylogenetic Implications" Genes 13, no. 3: 461. https://doi.org/10.3390/genes13030461
APA StyleSong, W., Chen, Z., He, L., Feng, Q., Zhang, H., Du, G., Shi, C., & Wang, S. (2022). Comparative Chloroplast Genome Analysis of Wax Gourd (Benincasa hispida) with Three Benincaseae Species, Revealing Evolutionary Dynamic Patterns and Phylogenetic Implications. Genes, 13(3), 461. https://doi.org/10.3390/genes13030461