
Characterization of Accessible Chromatin Regions in Cattle Rumen Epithelial Tissue during Weaning

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Rumen Epithelial Tissue Collection
2.2. ATAC Sequencing
2.3. Data Processing and Mapping
2.4. Peak Calling
2.5. Identification of Differentially Accessible Regions
2.6. Annotation of Differentially Accessible Regions
2.7. Gene Ontology and Pathway Analysis of Differentially Accessible Regions
2.8. Motif Enrichment of Differentially Accessible Regions
2.9. Gene Co-Expression Network Analysis
3. Results
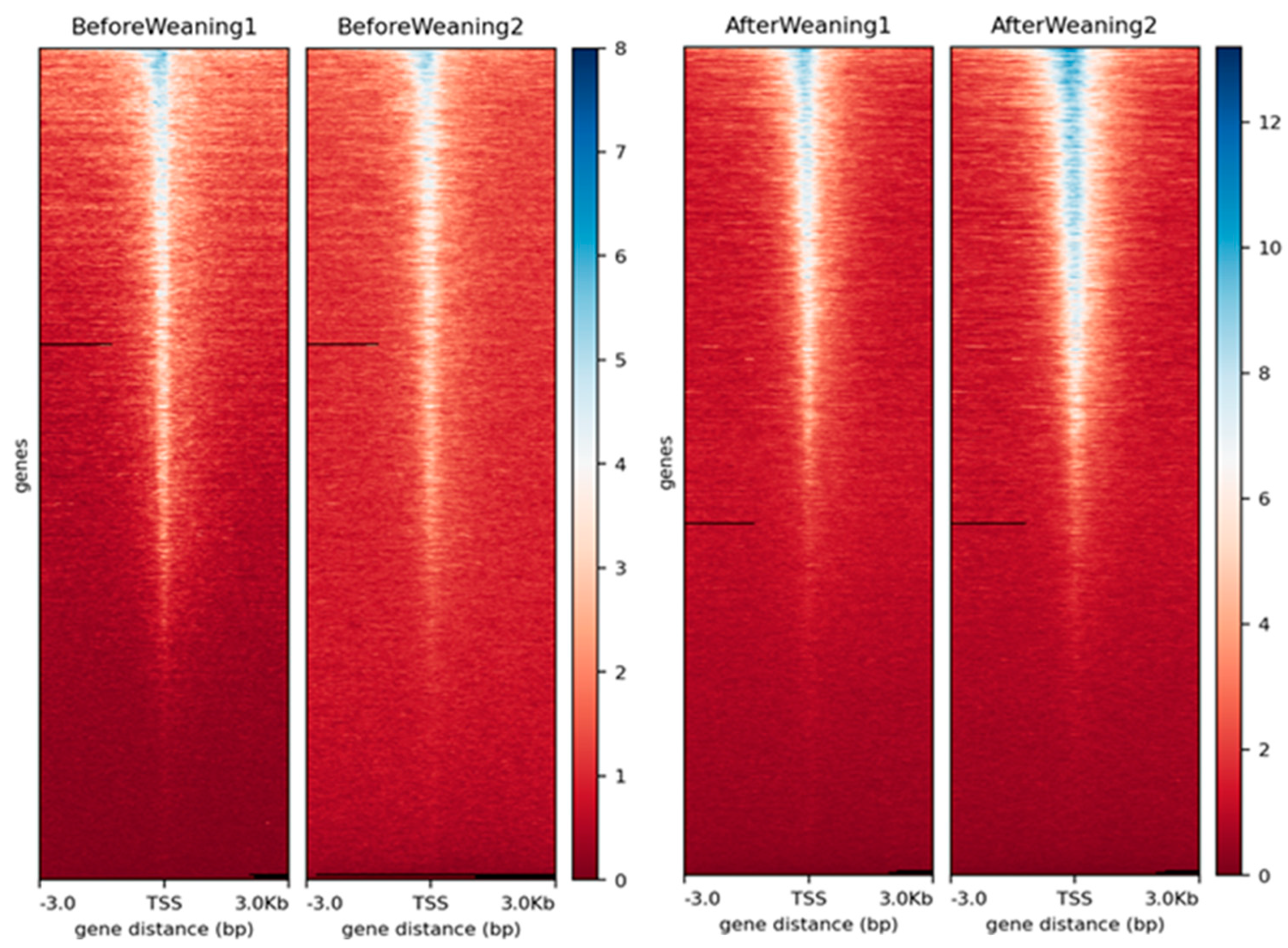
3.1. Data Quality and Peak Calling
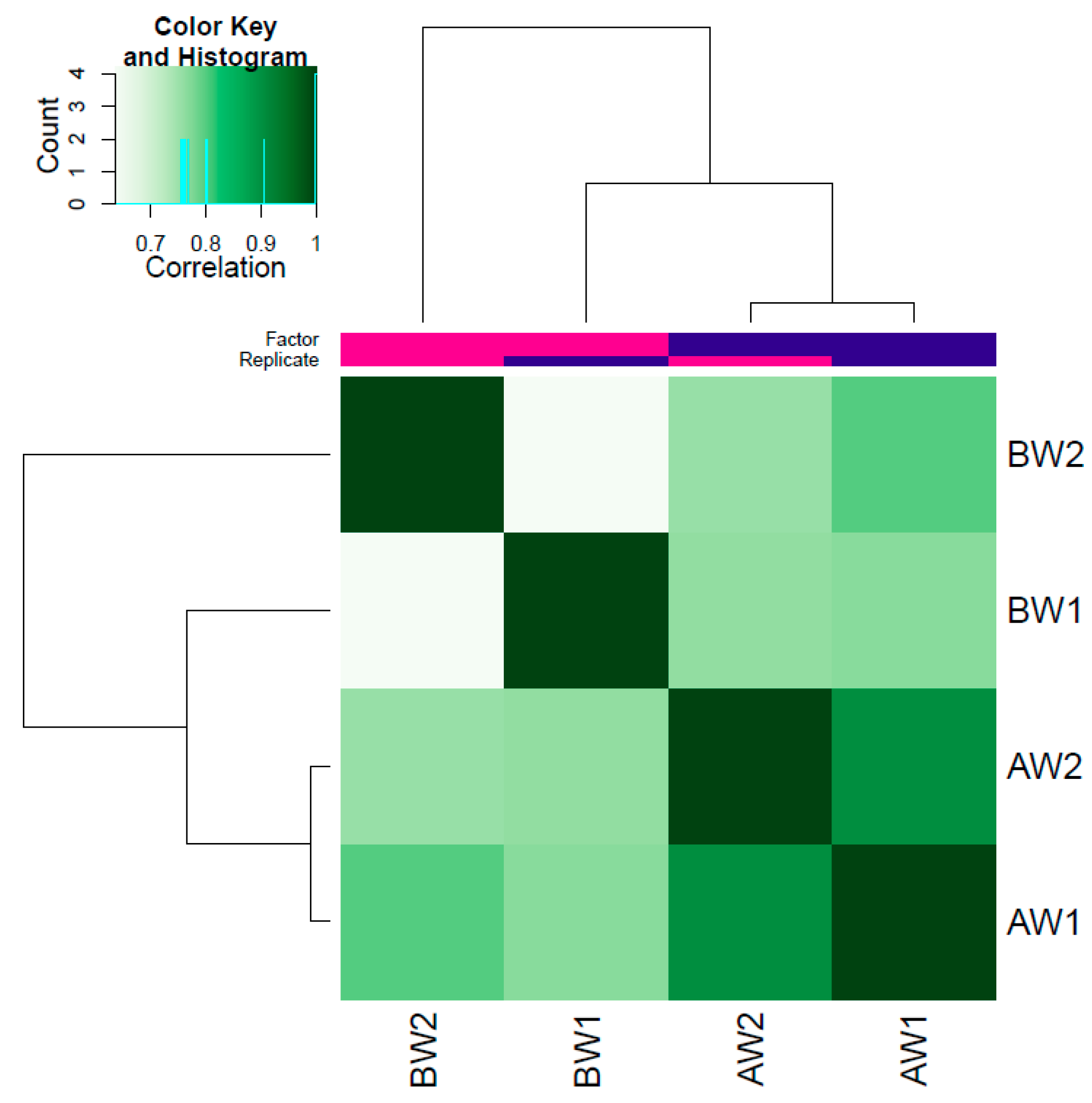
3.2. Differentially Accessible Regions
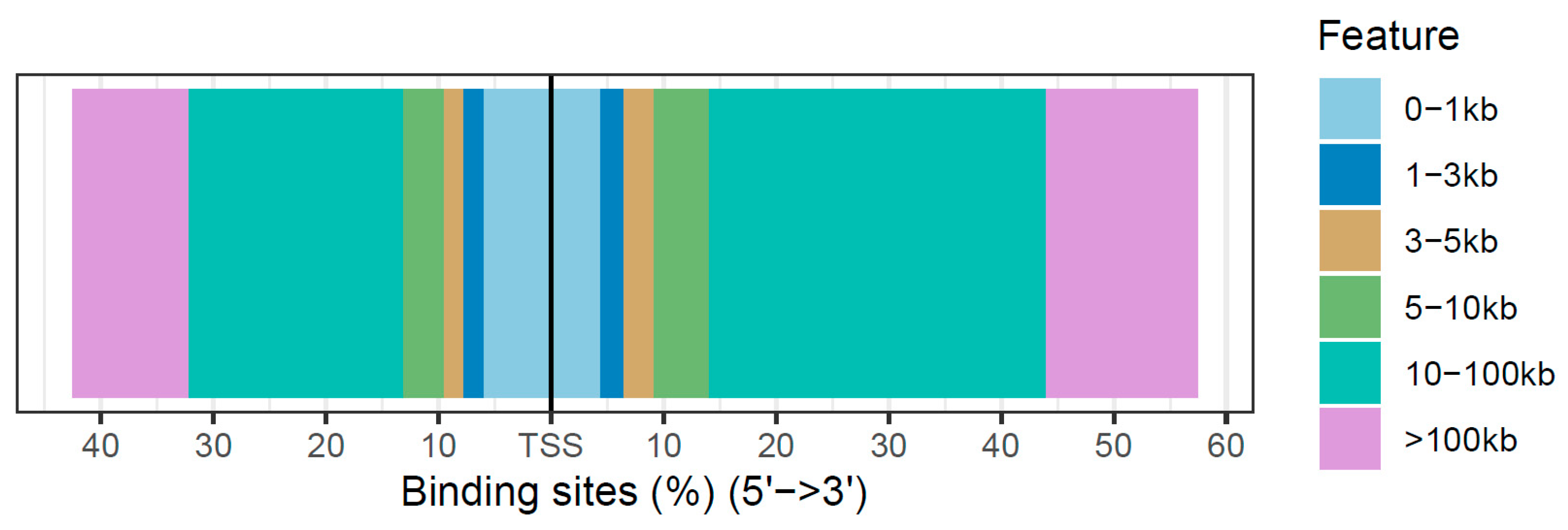
3.3. Annotation of Differentially Accessible Regions
3.4. Functional Annotation of Differentially Accessible Regions
3.5. Pathway Analysis of Differentially Accessible Regions
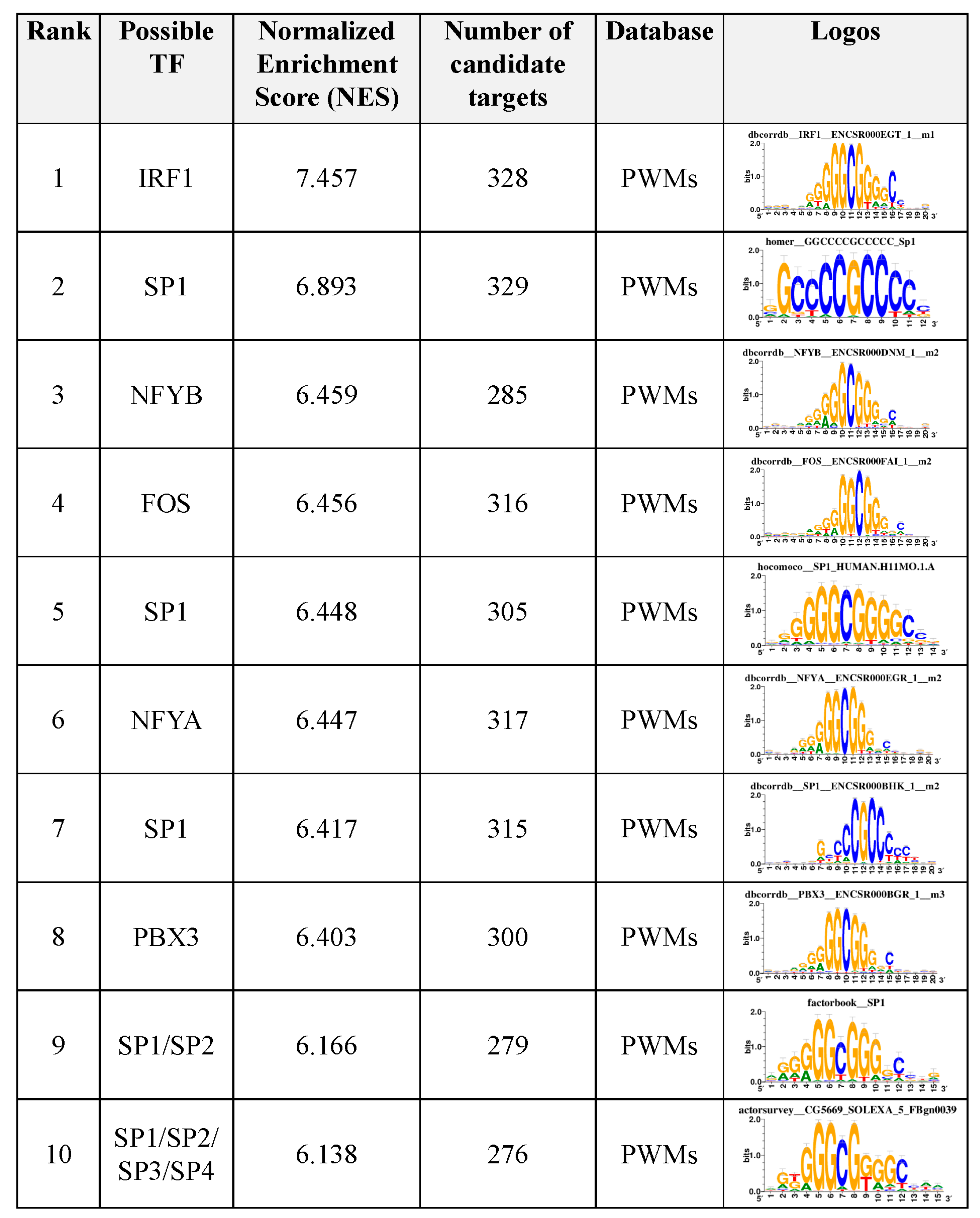
3.6. Motif Enrichment Analysis of Differentially Accessible Regions
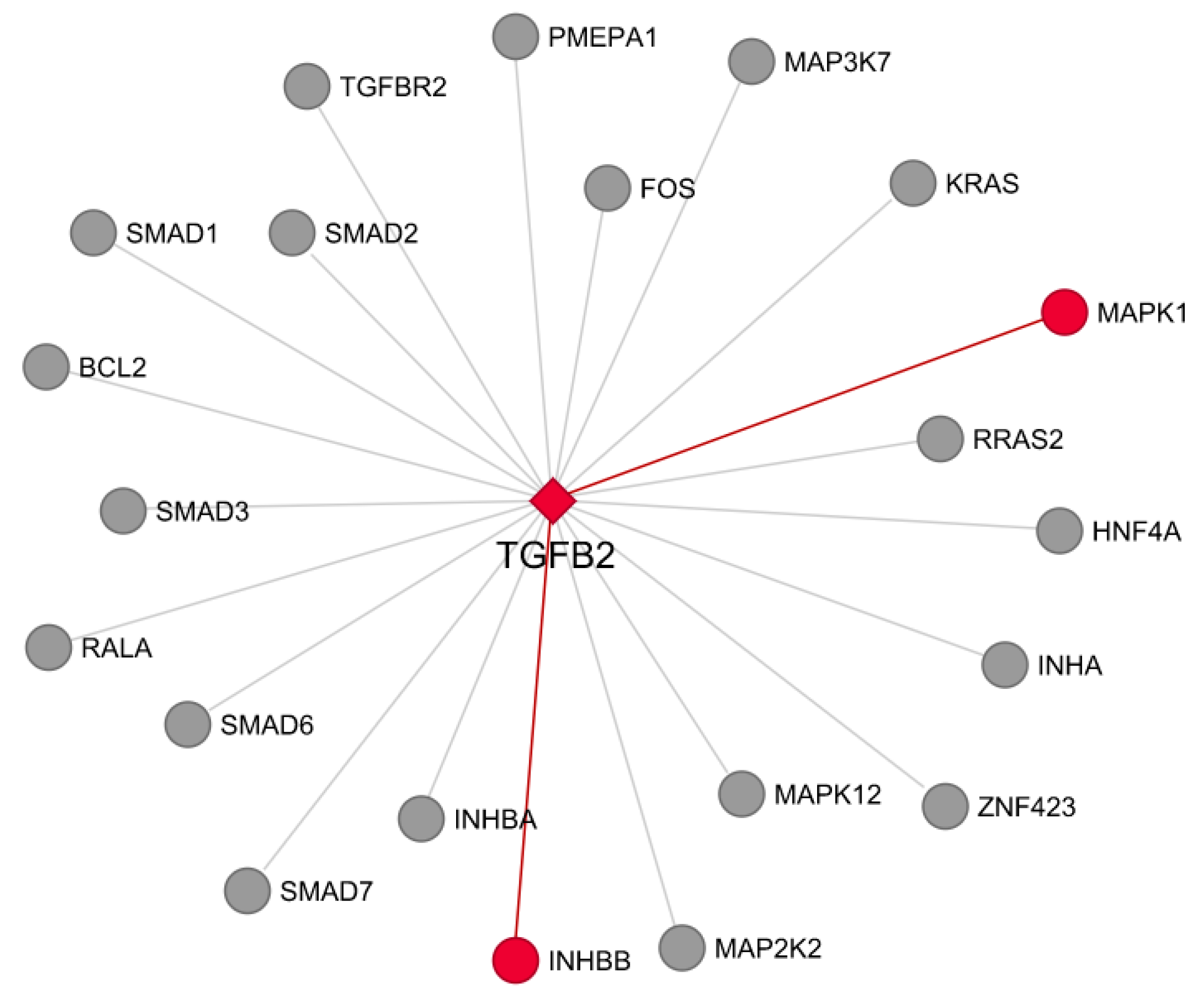
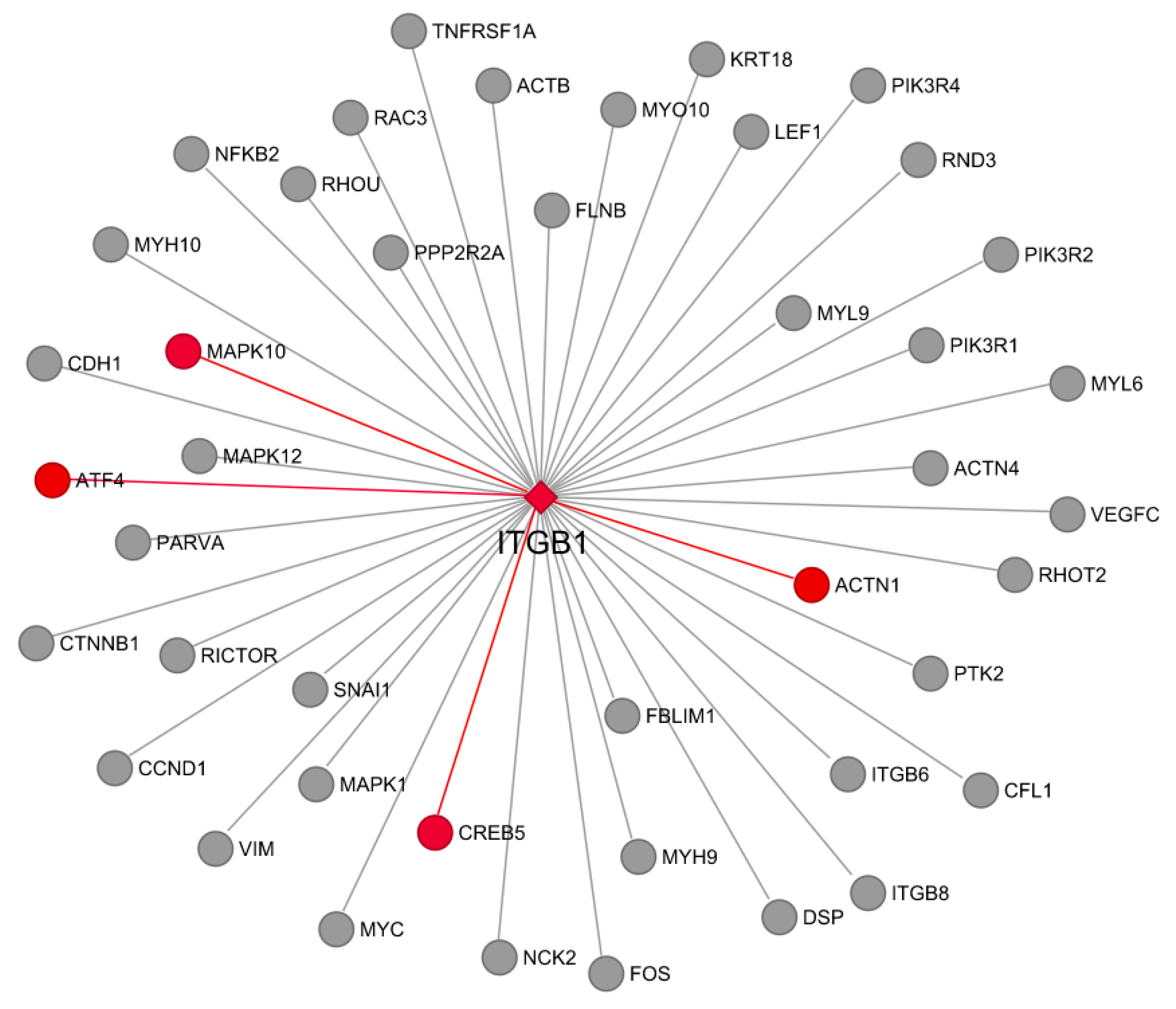
3.7. Co-Expression and Network Visualization of Critical Pathways for Rumen
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newbold, C.J.; Ramos-Morales, E. Review: Ruminal microbiome and microbial metabolome: Effects of diet and ruminant host. Animal 2020, 14, s78–s86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baaske, L.; Gäbel, G.; Dengler, F. Ruminal epithelium: A checkpoint for cattle health. J. Dairy Res. 2020, 87, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Diao, Q.; Zhang, R.; Fu, T. Review of Strategies to Promote Rumen Development in Calves. Animals 2019, 9, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, M.A.; Penner, G.B.; Chaucheyras-Durand, F.; Guan, L.L. Development and physiology of the rumen and the lower gut: Targets for improving gut health. J. Dairy Sci. 2016, 99, 4955–4966. [Google Scholar] [CrossRef] [Green Version]
- Mikuš, T.; Marzel, R.; Mikuš, O. Early weaning: New insights on an ever-persistent problem in the dairy industry. J. Dairy Res. 2020, 87, 88–92. [Google Scholar] [CrossRef]
- Warner, R.G.; Flatt, W.P.; Loosli, J.K. Ruminant Nutrition, Dietary Factors Influencing Development of Ruminant Stomach. J. Agric. Food Chem. 1956, 4, 788–792. [Google Scholar] [CrossRef]
- Connor, E.E.; Baldwin, R.L.; Li, C.-j.; Li, R.W.; Chung, H. Gene expression in bovine rumen epithelium during weaning identifies molecular regulators of rumen development and growth. Funct. Integr. Genom. 2013, 13, 133–142. [Google Scholar] [CrossRef]
- Kato, D.; Suzuki, Y.; Haga, S.; So, K.; Yamauchi, E.; Nakano, M.; Ishizaki, H.; Choi, K.; Katoh, K.; Roh, S.G. Utilization of digital differential display to identify differentially expressed genes related to rumen development. Anim. Sci. J. 2016, 87, 584–590. [Google Scholar] [CrossRef]
- Nishihara, K.; Kato, D.; Suzuki, Y.; Kim, D.; Nakano, M.; Yajima, Y.; Haga, S.; Nakano, M.; Ishizaki, H.; Kawahara-Miki, R.; et al. Comparative transcriptome analysis of rumen papillae in suckling and weaned Japanese Black calves using RNA sequencing. J. Anim. Sci. 2018, 96, 2226–2237. [Google Scholar] [CrossRef]
- Gao, Y.; Fang, L.; Baldwin, R.L., VI; Connor, E.E.; Cole, J.B.; Van Tassell, C.P.; Ma, L.; Li, C.J.; Liu, G.E. Single-cell transcriptomic analyses of dairy cattle ruminal epithelial cells during weaning. Genomics 2021, 113, 2045–2055. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D.; Kim, J.; Taylor, J.F.; Earley, B.; McCabe, M.S.; Lemon, K.; Duffy, C.; McMenamy, M.; Cosby, S.L.; Waters, S.M. ATAC-Seq identifies regions of open chromatin in the bronchial lymph nodes of dairy calves experimentally challenged with bovine respiratory syncytial virus. BMC Genom. 2021, 22, 14. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, P.A.; Naval-Sanchez, M.; Menzies, M.; Nguyen, L.T.; Porto-Neto, L.R.; Fortes, M.R.S.; Reverter, A. Chromatin accessibility and regulatory vocabulary across indicine cattle tissues. Genome Biol. 2021, 22, 273. [Google Scholar] [CrossRef]
- Cao, X.; Cheng, J.; Huang, Y.; Lan, X.; Lei, C.; Chen, H. Comparative Enhancer Map of Cattle Muscle Genome Annotated by ATAC-Seq. Front. Vet. Sci. 2021, 8, 1467. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, S.; Baldwin, R.L., VI; Connor, E.E.; Cole, J.B.; Ma, L.; Fang, L.; Li, C.-j.; Liu, G.E. Functional annotation of regulatory elements in cattle genome reveals the roles of extracellular interaction and dynamic change of chromatin states in rumen development during weaning. BMC Genom. 2022, 114, 110296. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef] [Green Version]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience 2020, 9, giaa021. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Shao, N.Y.; Liu, X.; Maze, I.; Feng, J.; Nestler, E.J. diffReps: Detecting differential chromatin modification sites from ChIP- seq data with biological replicates. PLoS ONE 2013, 8, e65598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodato, N.J.; Rampersaud, A.; Waxman, D.J. Impact of CAR Agonist Ligand TCPOBOP on Mouse Liver Chromatin Accessibility. Toxicol. Sci. 2018, 164, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef] [Green Version]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Herrmann, C.; Van de Sande, B.; Potier, D.; Aerts, S. i-cisTarget: An integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res. 2012, 40, e114. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z. Using VisANT to Analyze Networks. Curr. Protoc. Bioinform. 2014, 45, 8.8.1–8.8.39. [Google Scholar] [CrossRef] [Green Version]
- Vogelmann, R.; Nguyen-Tat, M.D.; Giehl, K.; Adler, G.; Wedlich, D.; Menke, A. TGFβ-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. J. Cell Sci. 2005, 118, 4901–4912. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Radjendirane, V.; Wary, K.K.; Chakrabarty, S. Transforming growth factor β regulates cell-cell adhesion through extracellular matrix remodeling and activation of focal adhesion kinase in human colon carcinoma Moser cells. Oncogene 2004, 23, 5558–5561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, T.; Li, S.; Docheva, D.; Grashoff, C.; Sakai, K.; Kostka, G.; Braun, A.; Pfeifer, A.; Yurchenco, P.D.; Fässler, R. Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 2003, 17, 926–940. [Google Scholar] [CrossRef] [Green Version]
- Hannigan, G.E.; Leung-Hagesteijn, C.; Fitz-Gibbon, L.; Coppolino, M.G.; Radeva, G.; Filmus, J.; Bell, J.C.; Dedhar, S. Regulation of cell adhesion and anchorage-dependent growth by a new β 1-integrin-linked protein kinase. Nature 1996, 379, 91–96. [Google Scholar] [CrossRef]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Halstead, M.M.; Kern, C.; Saelao, P.; Wang, Y.; Chanthavixay, G.; Medrano, J.F.; Van Eenennaam, A.L.; Korf, I.; Tuggle, C.K.; Ernst, C.W.; et al. A comparative analysis of chromatin accessibility in cattle, pig, and mouse tissues. BMC Genom. 2020, 21, 698. [Google Scholar] [CrossRef]
- Landt, S.G.; Marinov, G.K.; Kundaje, A.; Kheradpour, P.; Pauli, F.; Batzoglou, S.; Bernstein, B.E.; Bickel, P.; Brown, J.B.; Cayting, P.; et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012, 22, 1813–1831. [Google Scholar] [CrossRef] [Green Version]
- Levine, M. Transcriptional enhancers in animal development and evolution. Curr. Biol. 2010, 20, R754–R763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.; Cattoglio, C.; Tjian, R. Looping back to leap forward: Transcription enters a new era. Cell 2014, 157, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalili, A.A.; Ahmad, M.R. A Review of Cell Adhesion Studies for Biomedical and Biological Applications. Int. J. Mol. Sci. 2015, 16, 18149–18184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Capelo, A. Dual role for TGF-β1 in apoptosis. Cytokine Growth Factor Rev. 2005, 16, 15–34. [Google Scholar] [CrossRef]
- LeBaron, R.G.; Bezverkov, K.I.; Zimber, M.P.; Pavelec, R.; Skonier, J.; Purchio, A.F. β IG-H3, a novel secretory protein inducible by transforming growth factor-β, is present in normal skin and promotes the adhesion and spreading of dermal fibroblasts in vitro. J. Investig. Dermatol. 1995, 104, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Bond, J.J.; Donaldson, A.J.; Coumans, J.V.F.; Austin, K.; Ebert, D.; Wheeler, D.; Oddy, V.H. Protein profiles of enzymatically isolated rumen epithelium in sheep fed a fibrous diet. J. Anim. Sci. Biotechnol. 2019, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, R.L. The proliferative actions of insulin, insulin-like growth factor-I, epidermal growth factor, butyrate and propionate on ruminal epithelial cells in vitro. Small Rumin. Res. 1999, 32, 261–268. [Google Scholar] [CrossRef]
- Connor, E.E.; Baldwin, R.L., VI; Walker, M.P.; Ellis, S.E.; Li, C.; Kahl, S.; Chung, H.; Li, R.W. Transcriptional regulators transforming growth factor-β1 and estrogen- related receptor-α identified as putative mediators of calf rumen epithelial tissue development and function during weaning. J. Dairy Sci. 2014, 97, 4193–4207. [Google Scholar] [CrossRef]
- Shafaq-Zadah, M.; Gomes-Santos, C.S.; Bardin, S.; Maiuri, P.; Maurin, M.; Iranzo, J.; Gautreau, A.; Lamaze, C.; Caswell, P.; Goud, B.; et al. Persistent cell migration and adhesion rely on retrograde transport of β1 integrin. Nat. Cell Biol. 2016, 18, 54–64. [Google Scholar] [CrossRef]
- Yohe, T.T.; Tucker, H.L.M.; Parsons, C.L.M.; Geiger, A.J.; Akers, R.M.; Daniels, K.M. Short communication: Initial evidence supporting existence of potential rumen epidermal stem and progenitor cells. J. Dairy Sci. 2016, 99, 7654–7660. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.; Zhang, Z.; Wang, Z.; Wang, Y.; Yan, Y.; Li, S.; Tong, H. Platelet endothelial aggregation receptor-1 regulates bovine muscle satellite cell migration and differentiation via integrin β-1 and focal adhesion kinase. Cell Adhes. Migr. 2019, 13, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Garces de Los Fayos Alonso, I.; Liang, H.C.; Turner, S.D.; Lagger, S.; Merkel, O.; Kenner, L. The Role of Activator Protein-1 (AP-1) Family Members in CD30-Positive Lymphomas. Cancers 2018, 10, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczynski, J.; Cook, T.; Urrutia, R. Sp1- and Krüppel-like transcription factors. Genome Biol. 2003, 4, 206. [Google Scholar] [CrossRef] [Green Version]
- Wasylyk, B.; Hahn, S.L.; Giovane, A. The Ets family of transcription factors. Eur. J. Biochem. 1993, 211, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Metges, C.C.; Seyfert, H.-M. Interaction of C/EBP-β and NF-Y factors constrains activity levels of the nutritionally controlled promoter IA expressing the acetyl-CoA carboxylase-α gene in cattle. BMC Mol. Biol. 2012, 13, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savy, V.; Alberio, V.; Canel, N.G.; Ratner, L.D.; Gismondi, M.I.; Ferraris, S.F.; Fernandez-Martin, R.; Knott, J.G.; Bevacqua, R.J.; Salamone, D.F. CRISPR-on for activation of endogenous SMARCA4 and TFAP2C expression in bovine embryos. Reproduction 2020, 159, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Forough, R.; Weylie, B.; Collins, C.; Parker, J.L.; Zhu, J.; Barhoumi, R.; Watson, D.K. Transcription Factor Ets-1 Regulates Fibroblast Growth Factor-1-Mediated Angiogenesis in vivo: Role of Ets-1 in the Regulation of the PI3K/AKT/MMP-1 Pathway. J. Vasc. Res. 2006, 43, 327–337. [Google Scholar] [CrossRef]
- Brey, C.W.; Nelder, M.P.; Hailemariam, T.; Gaugler, R.; Hashmi, S. Kruppel-like family of transcription factors: An emerging new frontier in fat biology. Int. J. Biol. Sci. 2009, 5, 622–636. [Google Scholar] [CrossRef]
- Swamynathan, S.K. Kruppel-like factors: Three fingers in control. Hum. Genom. 2010, 4, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Ghaleb, A.M.; Yang, V.W. Kruppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef]
- Subbalakshmi, A.R.; Sahoo, S.; McMullen, I.; Saxena, A.N.; Venugopal, S.K.; Somarelli, J.A.; Jolly, M.K. KLF4 Induces Mesenchymal-Epithelial Transition (MET) by Suppressing Multiple EMT-Inducing Transcription Factors. Cancers 2021, 13, 5135. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, I.; Imoto, M.; Adjei, P.N.; Gores, G.J.; Subramaniam, M.; Spelsberg, T.C.; Urrutia, R. Overexpression of the TGFβ-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J. Clin. Invest. 1997, 99, 2365–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruberto, A.A.; Grechez-Cassiau, A.; Guerin, S.; Martin, L.; Revel, J.S.; Mehiri, M.; Subramaniam, M.; Delaunay, F.; Teboul, M. KLF10 integrates circadian timing and sugar signaling to coordinate hepatic metabolism. eLife 2021, 10, e65574. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, M.; Harris, S.A.; Oursler, M.J.; Rasmussen, K.; Riggs, B.L.; Spelsberg, T.C. Identification of a novel TGF-β-regulated gene encoding a putative zinc finger protein in human osteoblasts. Nucleic Acids Res. 1995, 23, 4907–4912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, M.; Hawse, J.R.; Rajamannan, N.M.; Ingle, J.N.; Spelsberg, T.C. Functional role of KLF10 in multiple disease processes. Biofactors 2010, 36, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Farre, M.; Kim, J.; Proskuryakova, A.A.; Zhang, Y.; Kulemzina, A.I.; Li, Q.; Zhou, Y.; Xiong, Y.; Johnson, J.L.; Perelman, P.L.; et al. Evolution of gene regulation in ruminants differs between evolutionary breakpoint regions and homologous synteny blocks. Genome Res. 2019, 29, 576–589. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | No. of Reads | No. of Mapped Reads | % of Mapped Reads | No. of MT Reads | % of MT Reads 1 | No. of Duplicate Reads | % of Duplicate Reads 1 | No. of MQ < 10 Reads | % of MQ < 10 Reads 1 | No. of Clean Reads 2 |
|---|---|---|---|---|---|---|---|---|---|---|
| BW1 | 61,550,092 | 59,937,035 | 97.38 | 4,456,644 | 7.44 | 1,537,436 | 2.57 | 17,496,479 | 29.19 | 36,963,828 |
| BW2 | 77,759,870 | 69,274,392 | 89.09 | 4,286,235 | 6.19 | 823,517 | 1.19 | 22,547,946 | 32.55 | 48,310,541 |
| AW1 | 90,778,622 | 88,547,917 | 97.54 | 5,222,533 | 5.90 | 1,261,956 | 1.43 | 26,047,668 | 29.42 | 56,315,258 |
| AW2 | 88,648,740 | 86,493,739 | 97.57 | 6,070,031 | 7.02 | 1,269,929 | 1.47 | 23,577,286 | 27.26 | 55,901,495 |
| Total | 318,737,324 | 304,253,083 | − | 20,035,443 | − | 4,892,838 | − | 89,669,379 | − | 197,491,122 |
| Average | 79,684,331 | 76,063,271 | 95.40 | 5,008,861 | 6.63 | 1,223,210 | 1.66 | 22,417,345 | 29.60 | 49,372,781 |
| Condition | No. of Clean Reads 1 | No. of Clean Reads Used for FRiP 2 | No. of MACS2 Peaks 2 | No. of Assigned Reads in Peaks 2 | FRiP 3 | Average Peak Length | Proportion of Peaks Near TSS (±3 Kb, %) |
|---|---|---|---|---|---|---|---|
| BW1 | 36,963,828 | 36,056,626 | 94,963 | 7,267,896 | 0.20 | 171 | 16.08 |
| BW2 | 48,310,541 | 46,833,278 | 27,640 | 1,483,647 | 0.03 | 210 | 25.33 |
| AW1 | 56,315,258 | 54,744,212 | 65,523 | 4,449,195 | 0.08 | 211 | 18.70 |
| AW2 | 55,901,495 | 54,363,957 | 86,807 | 7,436,305 | 0.13 | 253 | 16.55 |
| Total | 197,491,122 | 191,998,073 | 274,933 | 20,637,043 | − | − | − |
| Average | 49,372,781 | 47,999,518 | 68,733 | 5,159,261 | 0.11 | 211 | 19.17 |
| After Weaning × Before Weaning DARs | No. of DARs | DARs% |
|---|---|---|
| DARs initially identified (p-value < 0.05) | 29,174 | − |
| Significant DARs (FDR < 0.01 and −1 ≤ log2FC ≥ 1) | 3818 | 13.09 |
| Significant DARs that overlapped with peaks | 2907 | 9.96 |
| Unique significant DARs that overlapped with peaks | 2766 | − |
| Induced DARs with log2FC ≥ 1 | 686 | 24.80 |
| Repressed DARs with log2FC ≤ −1 | 2080 | 75.20 |
| Feature | Number | Frequency (%) |
|---|---|---|
| Promoter (<1 kb) | 289 | 10.45 |
| Promoter (1–2 kb) | 51 | 1.84 |
| 5’ UTR | 1 | 0.04 |
| 3’ UTR | 43 | 1.55 |
| Exon | 135 | 4.88 |
| First Intron | 153 | 5.53 |
| Other Intron | 412 | 14.90 |
| Downstream (<1 kb) | 15 | 0.54 |
| Downstream (1–2 kb) | 23 | 0.83 |
| Downstream (2–3 kb) | 22 | 0.80 |
| Distal Intergenic | 1622 | 58.64 |
| Total | 2766 | 100.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boschiero, C.; Gao, Y.; Baldwin, R.L., VI; Ma, L.; Liu, G.E.; Li, C.-J. Characterization of Accessible Chromatin Regions in Cattle Rumen Epithelial Tissue during Weaning. Genes 2022, 13, 535. https://doi.org/10.3390/genes13030535
Boschiero C, Gao Y, Baldwin RL VI, Ma L, Liu GE, Li C-J. Characterization of Accessible Chromatin Regions in Cattle Rumen Epithelial Tissue during Weaning. Genes. 2022; 13(3):535. https://doi.org/10.3390/genes13030535
Chicago/Turabian StyleBoschiero, Clarissa, Yahui Gao, Ransom L. Baldwin, VI, Li Ma, George E. Liu, and Cong-Jun Li. 2022. "Characterization of Accessible Chromatin Regions in Cattle Rumen Epithelial Tissue during Weaning" Genes 13, no. 3: 535. https://doi.org/10.3390/genes13030535
APA StyleBoschiero, C., Gao, Y., Baldwin, R. L., VI, Ma, L., Liu, G. E., & Li, C. -J. (2022). Characterization of Accessible Chromatin Regions in Cattle Rumen Epithelial Tissue during Weaning. Genes, 13(3), 535. https://doi.org/10.3390/genes13030535