
Update on the Phenotypic and Genotypic Spectrum of KIF11-Related Retinopathy


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Clinical Data
2.3. Mutational Screening
2.4. Statistical Analysis
3. Results
3.1. Clinical Features of KIF11-Related Retinopathy
3.2. Ocular Comparison between Two Subgroups
3.3. Genetic Findings
3.4. Extraocular Phenotypic Characteristics
4. Discussion
4.1. Chorioretinal Dysplasia, Not Retinal Folds or Abnormal Vessels, Is the Dominant Ocular Phenotype in KIF11-Associated Retinopathy
4.2. Rare Increase or Straightening of Peripheral Vessels Was Noted in KIF11-Associated Retinopathy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kapitein, L.C.; Peterman, E.J.; Kwok, B.H.; Kim, J.H.; Kapoor, T.M.; Schmidt, C.F. The bipolar mitotic kinesin Eg5 moves on both microtubules that it crosslinks. Nature 2005, 435, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, P.; Simpson, M.A.; Mendola, A.; Vasudevan, P.; Connell, F.C.; van Impel, A.; Moore, A.T.; Loeys, B.L.; Ghalamkarpour, A.; Onoufriadis, A.; et al. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am. J. Hum. Genet. 2012, 90, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzaa, G.M.; Enyedi, L.; Parsons, G.; Collins, S.; Medne, L.; Adams, C.; Ward, T.; Davitt, B.; Bicknese, A.; Zackai, E.; et al. Congenital microcephaly and chorioretinopathy due to de novo heterozygous KIF11 mutations: Five novel mutations and review of the literature. Am. J. Med. Genet. A 2014, 164A, 2879–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robitaille, J.M.; Gillett, R.M.; LeBlanc, M.A.; Gaston, D.; Nightingale, M.; Mackley, M.P.; Parkash, S.; Hathaway, J.; Thomas, A.; Ells, A.; et al. Phenotypic overlap between familial exudative vitreoretinopathy and microcephaly, lymphedema, and chorioretinal dysplasia caused by KIF11 mutations. JAMA Ophthalmol. 2014, 132, 1393–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, C.H.; Huang, S.; Chen, J. Wnt Signaling in vascular eye diseases. Prog. Retin. Eye Res. 2019, 70, 110–133. [Google Scholar] [CrossRef]
- Ranchod, T.M.; Ho, L.Y.; Drenser, K.A.; Capone, A., Jr.; Trese, M.T. Clinical presentation of familial exudative vitreoretinopathy. Ophthalmology 2011, 118, 2070–2075. [Google Scholar] [CrossRef]
- Karjosukarso, D.W.; van Gestel, S.H.C.; Qu, J.; Kouwenhoven, E.N.; Duijkers, L.; Garanto, A.; Zhou, H.; Collin, R.W.J. An FEVR-associated mutation in ZNF408 alters the expression of genes involved in the development of vasculature. Hum. Mol. Genet. 2018, 27, 3519–3527. [Google Scholar] [CrossRef]
- Li, J.K.; Li, Y.; Zhang, X.; Chen, C.L.; Rao, Y.Q.; Fei, P.; Zhang, Q.; Zhao, P.; Li, J. Spectrum of Variants in 389 Chinese Probands With Familial Exudative Vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2018, 59, 5368–5381. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Sun, L.; Hu, A.; Yuan, M.; Yang, Y.; Peng, X.; Ding, X. Mutation Spectrum of the LRP5, NDP, and TSPAN12 Genes in Chinese Patients With Familial Exudative Vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2017, 58, 5949–5957. [Google Scholar] [CrossRef] [Green Version]
- Birtel, J.; Gliem, M.; Mangold, E.; Tebbe, L.; Spier, I.; Muller, P.L.; Holz, F.G.; Neuhaus, C.; Wolfrum, U.; Bolz, H.J.; et al. Novel Insights Into the Phenotypical Spectrum of KIF11-Associated Retinopathy, Including a New Form of Retinal Ciliopathy. Invest. Ophthalmol. Vis. Sci. 2017, 58, 3950–3959. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, C.; Sun, L.; Zhang, A.; Liu, C.; Huang, L.; Ding, X. Symmetry of folds in FEVR: A genotype-phenotype correlation study. Exp. Eye Res. 2019, 186, 107720. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, P.; Allen, R.A.; Straatsma, B.R.; O’Malley, C.C. Paving-Stone Degeneration of the Retina. Arch. Ophthalmol. 1965, 73, 169–182. [Google Scholar] [CrossRef]
- Yuan, M.; Yang, Y.; Yu, S.; Hu, A.; Lu, L.; Ma, J.; Ding, X.; Li, J. Posterior pole retinal abnormalities in mild asymptomatic FEVR. Invest. Ophthalmol. Vis. Sci. 2014, 56, 458–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmour, D.F. Familial exudative vitreoretinopathy and related retinopathies. Eye 2015, 29, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Li, S.; Sun, W.; Xiao, X.; Jia, X.; Liu, M.; Xu, L.; Long, Y.; Zhang, Q. An Ophthalmic Targeted Exome Sequencing Panel as a Powerful Tool to Identify Causative Mutations in Patients Suspected of Hereditary Eye Diseases. Transl. Vis. Sci. Technol. 2019, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Jiang, D.; Xiao, X.; Li, S.; Jia, X.; Sun, W.; Guo, X.; Zhang, Q. Evaluation of 12 myopia-associated genes in Chinese patients with high myopia. Invest. Ophthalmol. Vis. Sci. 2015, 56, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Human Gene Mutation Database. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 17 March 2022).
- Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org (accessed on 17 March 2022).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Sun, L.; Li, S.; Huang, L.; Zhang, T.; Wang, Z.; Yu, B.; Luo, X.; Ding, X. Novel variants in familial exudative vitreoretinopathy patients with KIF11 mutations and the Genotype-Phenotype correlation. Exp. Eye Res. 2020, 199, 108165. [Google Scholar] [CrossRef]
- Li, J.K.; Fei, P.; Li, Y.; Huang, Q.J.; Zhang, Q.; Zhang, X.; Rao, Y.Q.; Li, J.; Zhao, P. Identification of novel KIF11 mutations in patients with familial exudative vitreoretinopathy and a phenotypic analysis. Sci. Rep. 2016, 6, 26564. [Google Scholar] [CrossRef] [Green Version]
- Menon, M.; Mohammadi, S.; Davila-Velderrain, J.; Goods, B.A.; Cadwell, T.D.; Xing, Y.; Stemmer-Rachamimov, A.; Shalek, A.K.; Love, J.C.; Kellis, M.; et al. Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration. Nat. Commun. 2019, 10, 4902. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.E.; Ostergaard, P.; Moore, A.T.; Connell, F.C.; Williams, D.; Quarrell, O.; Brady, A.F.; Spier, I.; Hazan, F.; Moldovan, O.; et al. Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): Review of phenotype associated with KIF11 mutations. Eur. J. Hum. Genet. 2014, 22, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Xiao, X.; Li, S.; Jia, X.; Guo, X.; Zhang, Q. KIF11 mutations are a common cause of autosomal dominant familial exudative vitreoretinopathy. Br. J. Ophthalmol. 2016, 100, 278–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.L.; Parry, N.R.A.; Barton, S.J.; Campbell, C.; Delaney, C.M.; Ellingford, J.M.; Hall, G.; Hardcastle, C.; Morarji, J.; Nichol, E.J.; et al. Panel-Based Clinical Genetic Testing in 85 Children with Inherited Retinal Disease. Ophthalmology 2017, 124, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.W.; Matamoros, A.J. Inhibition of kinesin-5 improves regeneration of injured axons by a novel microtubule-based mechanism. Neural Regen. Res. 2015, 10, 845–849. [Google Scholar] [CrossRef]
- Nishina, S.; Suzuki, Y.; Yokoi, T.; Kobayashi, Y.; Noda, E.; Azuma, N. Clinical features of congenital retinal folds. Am. J. Ophthalmol. 2012, 153, 81–87.e81. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Kramer, J.M.; Merkx, G.; Lugtenberg, D.; Smeets, D.F.; Oortveld, M.A.; Blokland, E.A.; Agrawal, J.; Schenck, A.; van Bokhoven, H.; et al. CDK19 is disrupted in a female patient with bilateral congenital retinal folds, microcephaly and mild mental retardation. Hum. Genet. 2010, 128, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Wheway, G.; Schmidts, M.; Mans, D.A.; Szymanska, K.; Nguyen, T.T.; Racher, H.; Phelps, I.G.; Toedt, G.; Kennedy, J.; Wunderlich, K.A.; et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol. 2015, 17, 1074–1087. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Family ID | Gender | Age at Diagnosis | BCVA (logMAR) OD/OS | Ocular Findings | Other Findings | Source | |
|---|---|---|---|---|---|---|---|
| OD | OS | ||||||
| 24 | M | 1Y | NA | Retinal folds, chorioretinal dysplasia | Chorioretinal dysplasia | None | Previously reported |
| 171 | M | 5M | NA | Retinal folds, chorioretinal dysplasia | End-stage | Microcephaly, Microphthalmia in the left eye | Previously reported |
| 213 | F | 6M | NA | Retinal folds | Retinal folds | Microcephaly, lymphedema | Previously reported |
| 213F | M | 29Y | 0/1 | Retinal folds, chorioretinal dysplasia | Retinal folds, chorioretinal dysplasia | NA | - |
| 227 | F | 6M | NA | Retinal folds | Retinal folds | None | Previously reported |
| 242 | F | 5M | NA | Retinal folds, secondary cataract | Retinal folds | Microcephaly, Microphthalmia in the right eye, lymphedema | Previously reported |
| 242F | M | 25Y | 0/0 | Chorioretinal dysplasia | Chorioretinal dysplasia | NA | Previously reported |
| 242S | M | 8Y | 1.0/0.7 | Chorioretinal dysplasia | Chorioretinal dysplasia | NA | Previously reported |
| 298 | M | 6Y | 0.1/LP | Chorioretinal dysplasia, TEMPVIA, ISPVs | Rhegmatogenous retinal detachment | None | Previously reported |
| 301 | M | 3Y | NA | Retinal folds | Retinal folds | Microcephaly, lymphedema | Previously reported |
| 336 | M | 4M | NA | End-stage | End-stage | Microcephaly, lymphedema | Previously reported |
| 417 | M | 4Y | NA | Retinal folds | Retinal folds | Microcephaly, lymphedema | Previously reported |
| 463 | M | 1Y | NA | Retinal folds, chorioretinal dysplasia | End-stage | Microcephaly, lymphedema | - |
| 463F | M | 37Y | 0.1/0.1 | Chorioretinal dysplasia | Chorioretinal dysplasia | NA | - |
| 481 | M | 2Y | NA | End-stage, secondary cataract | Retinal folds, secondary cataract | Microcephaly, lymphedema | - |
| 492 | M | 3M | NA | Retinal folds, chorioretinal dysplasia | Retinal folds, chorioretinal dysplasia | NA | - |
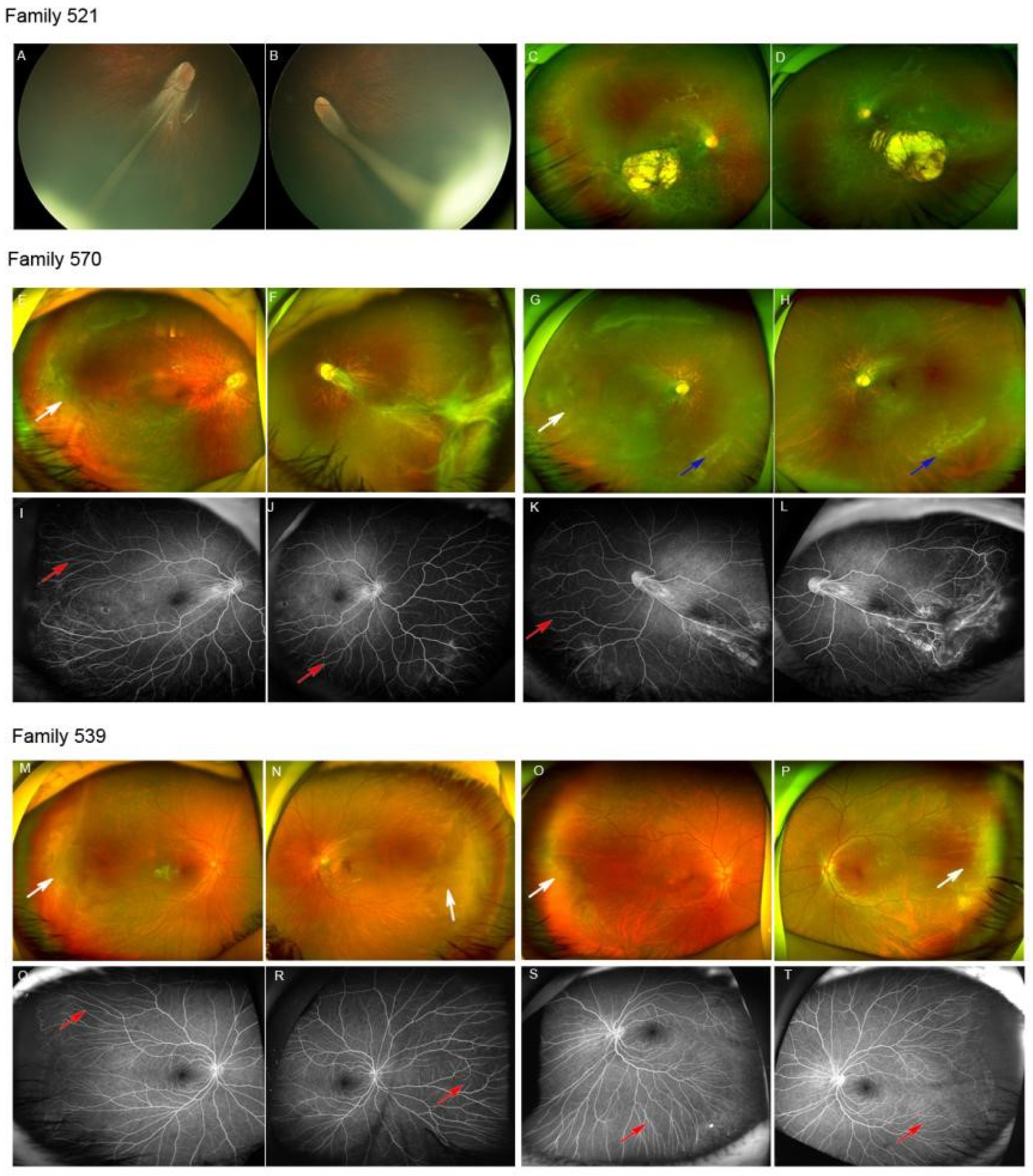
| 521 | F | 5M | NA | Retinal folds | Retinal folds | Microcephaly, lymphedema | - |
| 521F | M | 28Y | NA | Chorioretinal dysplasia, lattice degeneration | Chorioretinal dysplasia, lattice degeneration | NA | - |
| 550 | F | 5M | NA | End-stage | End-stage | Microcephaly, lymphedema | - |
| 577 | F | 4M | NA | Retinal folds | Retinal folds | Microcephaly, lymphedema | - |
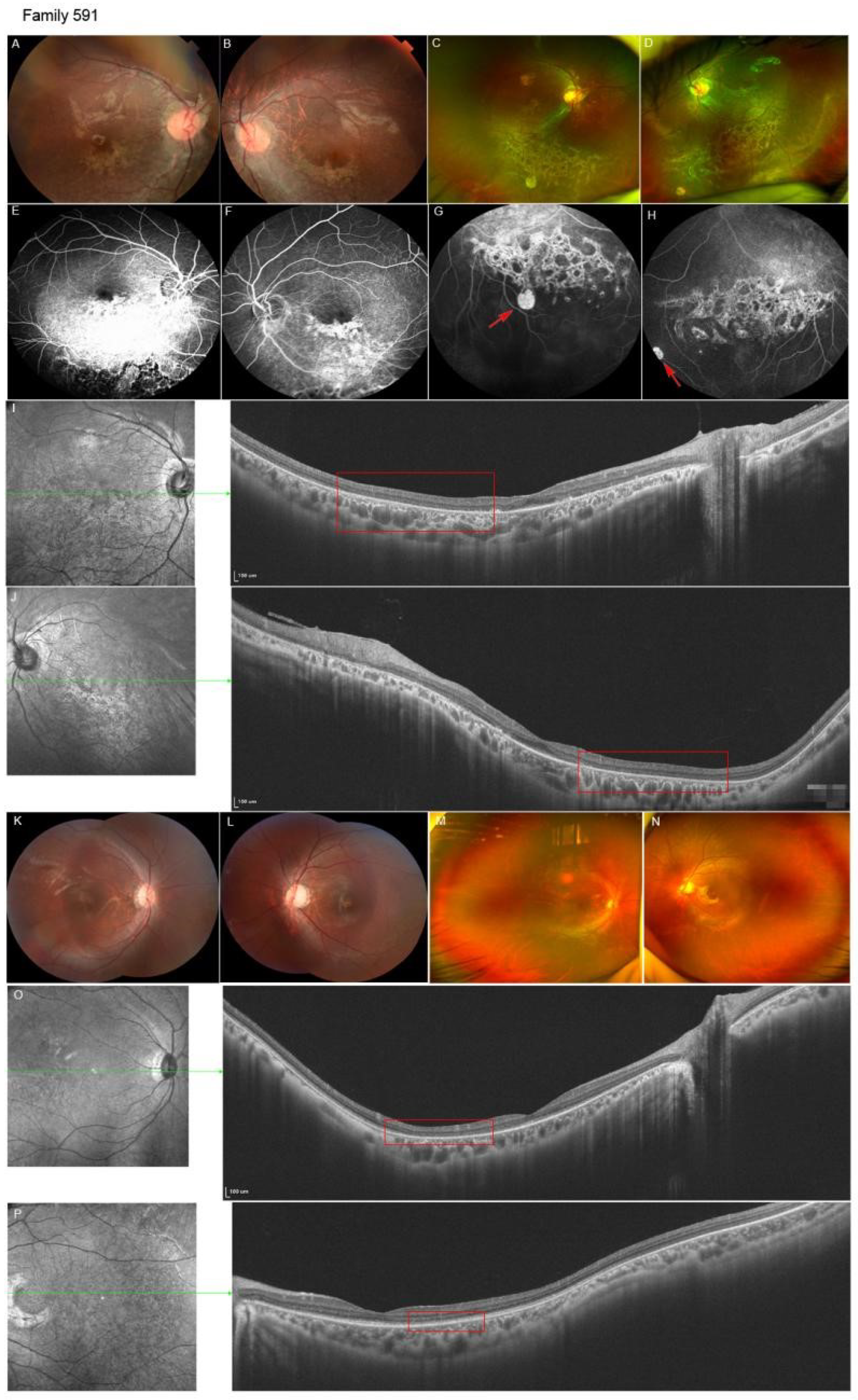
| 591 | M | 8Y | 0.4/0.7 | Chorioretinal dysplasia, ISPVs | Chorioretinal dysplasia, ISPVs | None | - |
| 591B | M | 7Y | 0.8/0.4 | Chorioretinal dysplasia | Chorioretinal dysplasia | NA | - |
| 591F | M | 35Y | NA | Chorioretinal dysplasia, ISPVs | Chorioretinal dysplasia | NA | - |
| 557 | M | 8Y | NA | End-stage | Retinal folds, chorioretinal dysplasia | Microcephaly, lymphedema | - |
| 673 | M | 3Y | NA | End-stage | Retinal folds; chorioretinal dysplasia | Microcephaly, lymphedema | - |
| 673M | F | NA | NA | End-stage | Chorioretinal dysplasia, lattice degeneration | NA | - |
| 680 | M | 3Y | NA | End-stage | End-stage | Microcephaly | - |
| 684 | M | 3Y | NA | Chorioretinal dysplasia | End-stage | Microcephaly | - |
| 688 | M | 2Y | NA | Chorioretinal dysplasia | Retinal folds, chorioretinal dysplasia | Microcephaly, lymphedema | - |
| 564 | M | 5M | NA | End-stage | End-stage | Microcephaly | - |
| 570 | M | 5Y | 0.8/0.8 | Ectopic macula, TEMPVIA, ISPVs, lattice degeneration | Retinal folds, ISPVs, lattice degeneration | None | - |
| 570M | F | 29Y | NA | TEMPVIA, ISPVs, tilted disc, posterior staphyloma, lattice degeneration | Tilted disc, posterior staphyloma, lattice degeneration | NA | - |
| 539 | M | 4Y | NA | TEMPVIA, ISPVs, lattice degeneration | TEMPVIA, ISPVs, lattice degeneration | None | - |
| 539F | M | NA | NA | TEMPVIA, ISPVs, lattice degeneration | TEMPVIA, ISPVs, lattice degeneration | None | - |
| 653 | F | NA | NA | Chorioretinal dysplasia, retinal folds | Chorioretinal dysplasia, retinal folds | Microcephaly, lymphedema | - |
| KIF11 Associated Retinopathy | FEVR Caused by Other Genes | p | |
|---|---|---|---|
| Patients | 35 | 39 | |
| Probands | 25 (71.4%) | 27 (69.2%) | 0.92 |
| Family member | 10 (28.6%) | 12 (30.8%) | |
| Eyes | 70 | 78 | |
| Gender | |||
| Male | 26 (74.2%) | 28 (71.7%) | 0.91 |
| Female | 9 (25.7%) | 11 (28.3%) | |
| Age(years) | 12.40 ± 14.12 (n = 32) | 14.30 ± 14.57 | |
| Probands | 2.42 ± 2.42 (n = 24) | 3.65 ± 3.70 | 0.16 |
| Family member | 24.75 ± 11.32 (n = 8) | 28.25 ± 13.03 | 0.77 |
| Genes | |||
| KIF11 | 35 (100%) | 0 | |
| FZD4 | 0 | 21 (53.8%) | |
| TSPAN12 | 0 | 9 (23.1%) | |
| LRP5 | 0 | 5 (12.8%) | |
| NDP | 0 | 3 (7.7%) | |
| JAG1 | 0 | 1 (2.6%) |
| Phenotypes (Eyes) | KIF11 Associated Retinopathy (n = 70 Eyes) | FEVR Caused by Other Genes (n = 78 Eyes) | p |
|---|---|---|---|
| Chorioretinal dysplasia | 31 (44.2%) | 1 (1.3%) | <0.01 |
| Retinal folds | 24 (34.3%) | 30 (38.5%) | 0.61 |
| Retinal degeneration | 9 (12.9%) | 16 (20.5%) | 0.21 |
| ISPVs | 12 (17.1%) | 39 (50%) | <0.01 |
| End-stage | 14 (20%) | 8 (10.3%) | 0.07 |
| Family Number | cDNA Change | Amino Acid Change | Heredity | Variant Impact | Allele Frequency | Mutation Taster | PROVEN | CADD | Variation Type | Source |
|---|---|---|---|---|---|---|---|---|---|---|
| 463 | c.339delT | p.F113Lfs * 23 | Paternal | Frameshift | NA | DC | NA | NA | P, PVS1 | Novel |
| 481 | c.2905_2909del | K969Rfs * 18 | Paternal | Frameshift | NA | DC | NA | NA | P, PVS1 | Novel |
| 492 | c.1288_1290del | p.430del | De novo | Nonframeshift | NA | DC | D | 22.3 | P, PS2 | Novel |
| 521 | c.2309_2310del | p.K771Ifs * 4 | Paternal | Frameshift | NA | DC | NA | NA | P, PVS1 | Novel |
| 539 | c.3073A>G | p.R1025G | Paternal | Missense | 0.000054 | Polymorphism | D | 23.1 | P, PP5 | Li, J.K. et al. [19] |
| 550 | c.2626C>T | p.Q876 * | De novo | Stop-gain | NA | DC | NA | 36 | P, PVS1 | Novel |
| 577 | c.1382_1383del | p.L461Rfs * 11 | De novo | Frameshift | NA | DC | NA | NA | P, PVS1 | Novel |
| 591 | c.1915dupA | p.I639Nfs * 14 | Paternal | Frameshift | NA | DC | NA | 22.4 | P, PVS1 | Novel |
| 557 | c.1249G>T | p.E417 * | Maternal | Stop-gain | NA | DC | NA | NA | P, PVS1 | Novel |
| 564 | c.116C>G | p.S39 * | De novo | Stop-gain | NA | DC | NA | NA | P, PVS1 | Novel |
| 570 | c.139C>T | p.R47 * | Maternal | Stop-gain | NA | DC | NA | NA | P, PVS1 | Novel |
| 653 | c.134del | p.L45Xfs * 91 | De novo | Frameshift | NA | DC | NA | NA | P, PVS1 | Novel |
| 673 | c.2830C>T | p.R944C | Maternal | Missense | NA | DC | D | 32 | P, PP5 | Ostergaard, P. et al. [2] |
| 680 | c.2266C>T | p.Q756 * | De novo | Stop-gain | NA | DC | NA | 43 | P, PVS1 | Novel |
| 684 | c.2344G>T | p.E782 * | De novo | Stop-gain | NA | DC | NA | 36 | P, PVS1 | Novel |
| 688 | c.1429delA | p.E478Kfs * 61 | De novo | Frameshift | NA | DC | NA | NA | P, PVS1 | Novel |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhang, Z.; Huang, L.; Sun, L.; Li, S.; Zhang, T.; Ding, X. Update on the Phenotypic and Genotypic Spectrum of KIF11-Related Retinopathy. Genes 2022, 13, 713. https://doi.org/10.3390/genes13040713
Wang Y, Zhang Z, Huang L, Sun L, Li S, Zhang T, Ding X. Update on the Phenotypic and Genotypic Spectrum of KIF11-Related Retinopathy. Genes. 2022; 13(4):713. https://doi.org/10.3390/genes13040713
Chicago/Turabian StyleWang, You, Zhaotian Zhang, Li Huang, Limei Sun, Songshan Li, Ting Zhang, and Xiaoyan Ding. 2022. "Update on the Phenotypic and Genotypic Spectrum of KIF11-Related Retinopathy" Genes 13, no. 4: 713. https://doi.org/10.3390/genes13040713
APA StyleWang, Y., Zhang, Z., Huang, L., Sun, L., Li, S., Zhang, T., & Ding, X. (2022). Update on the Phenotypic and Genotypic Spectrum of KIF11-Related Retinopathy. Genes, 13(4), 713. https://doi.org/10.3390/genes13040713