
The Modified Shields Classification and 12 Families with Defined DSPP Mutations

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Enrollment of Human Subjects
2.2. Genomic DNA Extraction
2.3. Identifying DSPP Disease-Causing Mutation in 12 Families
2.4. Whole-Exome Sequencing and Bioinformatics Analysis
2.5. Segregation Analyses using Sanger Sequencing
2.6. Single Molecule Real-Time (SMRT) DNA Sequencing
3. Results
3.1. DSPP Mutations Causing Inherited Dental Defects
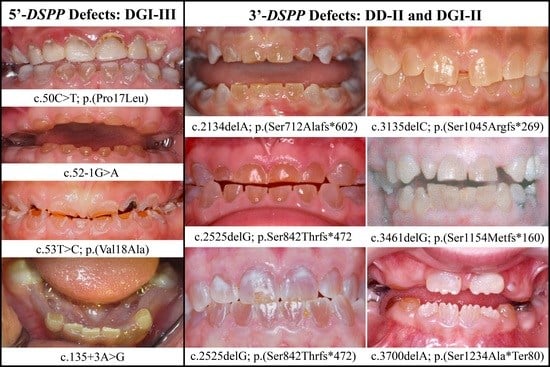
3.2. Four Families with DGI-III: 5′ DSPP Mutations
3.3. Eight Families with 3′ DSPP Mutations Causing DD-II or DGI-II
4. Discussion
4.1. A Diagnosis of DGI-III
4.2. A Diagnosis of DGI-II
4.3. Genetic Algorithm for Applying Shields Classification for Diagnosis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldberg, M.; Kulkarni, A.B.; Young, M.; Boskey, A. Dentin: Structure, composition and mineralization. Front Biosci. (Elite Ed.) 2011, 3, 711–735. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Chai, Y. Regulatory mechanisms of jaw bone and tooth development. Curr. Top. Dev. Biol. 2019, 133, 91–118. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Hu, Y.; Zhang, H.; Xu, Q.; Smith, C.E.; Zhang, C.; Kim, J.-W.; Wang, S.-K.; Saunders, T.L.; Lu, Y.; et al. Mouse Dspp frameshift model of human dentinogenesis imperfecta. Sci. Rep. 2021, 11, 20653. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.M.; Nalbandian, J. Development of Dentine and Pulp. In Teeth; Springer: Berlin/Heidelberg, Germany, 1989; pp. 73–171. [Google Scholar] [CrossRef]
- Simmer, J.P.; Richardson, A.S.; Hu, Y.-Y.; Smith, C.E.; Hu, J.C.-C. A post-classical theory of enamel biomineralization… and why we need one. Int. J. Oral Sci. 2012, 4, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.E.; Hu, Y.; Hu, J.C.; Simmer, J.P. Ultrastructure of early amelogenesis in wild-type, Amelx−/−, and Enam−/− mice: Enamel ribbon initiation on dentin mineral and ribbon orientation by ameloblasts. Mol. Genet. Genomic Med. 2016, 4, 662–683. [Google Scholar] [CrossRef] [PubMed]
- Simmer, J.P.; Hu, J.C.-C.; Hu, Y.; Zhang, S.; Liang, T.; Wang, S.-K.; Kim, J.-W.; Yamakoshi, Y.; Chun, Y.-H.; Bartlett, J.D.; et al. A Genetic Model for the Secretory Stage of Dental Enamel Formation. J Struct. Biol. 2021, 213, 107805. [Google Scholar] [CrossRef]
- Munhoz, C.O.G.; Leblond, C.P. Deposition of calcium phosphate into dentin and enamel as shown by radioautography of sections of incisor teeth following injection of45Ca into rats. Calcif. Tissue Res. 1974, 15, 221–235. [Google Scholar] [CrossRef]
- Habelitz, S.; Balooch, M.; Marshall, S.J.; Balooch, G.; Marshall, G.W., Jr. In situ atomic force microscopy of partially demineralized human dentin collagen fibrils. J. Struct. Biol. 2002, 138, 227–236. [Google Scholar] [CrossRef]
- Takuma, S.; Eda, S. Structure and Development of the Peritubular Matrix in Dentin. J. Dent. Res. 1966, 45, 683–692. [Google Scholar] [CrossRef]
- Li, C.; Jing, Y.; Wang, K.; Ren, Y.; Liu, X.; Wang, X.; Wang, Z.; Zhao, H.; Feng, J. Dentinal mineralization is not limited in the mineralization front but occurs along with the entire odontoblast process. Int. J. Biol. Sci. 2018, 14, 693–704. [Google Scholar] [CrossRef]
- Linde, A. Dentin matrix proteins: Composition and possible functions in calcification. Anat. Rec. 1989, 224, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Linde, A.; Bhown, M.; Butler, W. Noncollagenous proteins of dentin. A re-examination of proteins from rat incisor dentin utilizing techniques to avoid artifacts. J. Biol. Chem. 1980, 255, 5931–5942. [Google Scholar] [CrossRef]
- MacDougall, M.; Simmons, D.; Luan, X.; Nydegger, J.; Feng, J.; Gu, T.T. Dentin Phosphoprotein and Dentin Sialoprotein Are Cleavage Products Expressed from a Single Transcript Coded by a Gene on Human Chromosome 4. J. Biol. Chem. 1997, 272, 835–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, S.; Simmer, J.P.; Hu, J.C.-C.; Richardson, A.S.; Yamakoshi, F.; Yamakoshi, Y. Astacin proteases cleave dentin sialophosphoprotein (Dspp) to generate dentin phosphoprotein (Dpp). J. Bone Miner. Res. 2010, 26, 220–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D′Souza, R.N.; Bronckers, A.L.; Happonen, R.P.; Doga, D.A.; Farach-Carson, M.; Butler, W.T. Developmental expression of a 53 KD dentin sialoprotein in rat tooth organs. J. Histochem. Cytochem. 1992, 40, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, H.; Hou, H.; Veis, A.; Butler, W. Cloning and sequence determination of rat dentin sialoprotein, a novel dentin protein. J. Biol. Chem. 1994, 269, 3698–3702. [Google Scholar] [CrossRef]
- Yamakoshi, Y.; Hu, J.C.-C.; Fukae, M.; Iwata, T.; Kim, J.-W.; Zhang, H.; Simmer, J.P. Porcine Dentin Sialoprotein Is a Proteoglycan with Glycosaminoglycan Chains Containing Chondroitin 6-Sulfate. J. Biol. Chem. 2005, 280, 1552–1560. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Sun, Y.; Prasad, M.; Wang, X.; Yamoah, A.; Li, Y.; Feng, J.; Qin, C. Glycosaminoglycan Chain of Dentin Sialoprotein Proteoglycan. J. Dent. Res. 2010, 89, 808–812. [Google Scholar] [CrossRef]
- Yamakoshi, Y.; Nagano, T.; Hu, J.C.; Yamakoshi, F.; Simmer, J.P. Porcine dentin sialoprotein glycosylation and glycosaminoglycan attachments. BMC Biochem. 2011, 12, 6. [Google Scholar] [CrossRef] [Green Version]
- Veis, A.; Perry, A. The Phosphoprotein of the Dentin Matrix. Biochemistry 1967, 6, 2409–2416. [Google Scholar] [CrossRef]
- Yamakoshi, Y.; Simmer, J.P. Structural features, processing mechanism and gene splice variants of dentin sialophosphoprotein. Jpn. Dent. Sci. Rev. 2018, 54, 183–196. [Google Scholar] [CrossRef]
- Jonsson, M.; Fredriksson, S.; Jontell, M.; Linde, A. Isoelectric focusing of the phosphoprotein of rat-incisor dentin in ampholine and acid pH gradients: Evidence for carrier ampholyte—protein complexes. J. Chromatogr. A 1978, 157, 235–242. [Google Scholar] [CrossRef]
- Yamakoshi, Y.; Lu, Y.; Hu, J.C.; Kim, J.W.; Iwata, T.; Kobayashi, K.; Nagano, T.; Yamakoshi, F.; Hu, Y.; Fukae, M.; et al. Porcine dentin sialophosphoprotein: Length polymorphisms, glycosylation, phosphorylation, and stability. J. Biol. Chem. 2008, 283, 14835–14844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, D.A.; Suzanne Hart, P.; Hart, T.C.; Hartsfield, J.K.; Wilson, A.; Wright, J.T.; Fisher, L.W. A comprehensive analysis of normal variation and disease-causing mutations in the human DSPP gene. Hum. Mutat. 2008, 29, 1392–1404. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Kawasaki, K.; Lee, M.; Reid, B.M.; Nunez, S.M.; Choi, M.; Seymen, F.; Koruyucu, M.; Kasimoglu, Y.; Estrella-Yuson, N.; et al. The dentin phosphoprotein repeat region and inherited defects of dentin. Mol. Genet. Genom. Med. 2015, 4, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.E.; Hauge, M. Osteogenesis imperfecta: A genetic, radiological, and epidemiological study. Clin. Genet. 2008, 36, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, K.; Åström, E.; Rubin, C.J.; Grigelioniene, G.; Malmgren, B.; Ljunggren, Ö.; Kindmark, A. Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur. J. Hum. Genet. 2015, 23, 1042–1050. [Google Scholar] [CrossRef] [Green Version]
- Marçal, F.F.; Ribeiro, E.M.; Costa, F.W.G.; Fonteles, C.S.R.; Teles, G.S.; Silva, P.G.D.B.; Junior, C.M.C.; Ribeiro, T.R. Dental alterations on panoramic radiographs of patients with osteogenesis imperfecta in relation to clinical diagnosis, severity, and bisphosphonate regimen aspects: A STROBE-compliant case-control study. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2019, 128, 621–630. [Google Scholar] [CrossRef]
- Andersson, K.; Dahllöf, G.; Lindahl, K.; Kindmark, A.; Grigelioniene, G.; Åström, E.; Malmgren, B. Mutations in COL1A1 and COL1A2 and dental aberrations in children and adolescents with osteogenesis imperfecta—A retrospective cohort study. PLoS ONE 2017, 12, e0176466. [Google Scholar] [CrossRef]
- Pallos, D.; Hart, P.S.; Cortelli, J.R.; Vian, S.; Wright, J.T.; Korkko, J.; Brunoni, D.; Hart, T.C. Novel COL1A1 mutation (G559C) correction of G599C] associated with mild osteogenesis imperfecta and dentinogenesis imperfecta. Arch. Oral Biol. 2001, 46, 459–470. [Google Scholar] [CrossRef]
- Wang, S.-K.; Chan, H.-C.; Makovey, I.; Simmer, J.P.; Hu, J.C.-C. Novel PAX9 and COL1A2 Missense Mutations Causing Tooth Agenesis and OI/DGI without Skeletal Abnormalities. PLoS ONE 2012, 7, e51533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, A.C.; Marini, J. Evaluation of oral problems in an osteogenesis imperfecta population. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 1999, 87, 189–196. [Google Scholar] [CrossRef]
- Hart, P.S.; Hart, T.C. Disorders of Human Dentin. Cells Tissues Organs 2007, 186, 70–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-W.; Simmer, J.P. Hereditary Dentin Defects. J. Dent. Res. 2007, 86, 392–399. [Google Scholar] [CrossRef]
- Kang, H.; AC, S.A.; Marini, J.C. Osteogenesis imperfecta: New genes reveal novel mechanisms in bone dysplasia. Transl. Res. 2017, 181, 27–48. [Google Scholar] [CrossRef]
- Witkop, C. Hereditary defects in enamel and dentin. Hum. Hered. 1957, 7, 236–239. [Google Scholar] [CrossRef]
- Malmgren, B.; Norgren, S. Dental aberrations in children and adolescents with osteogenesis imperfecta. Acta Odontol. Scand. 2002, 60, 65–71. [Google Scholar] [CrossRef]
- Bixler, D. Heritable disorders affecting dentin. In Oral Facial Genetics; Stewart, R.E., Prescott, G.H., Eds.; C.V. Mosby Co.: St. Louis, MO, USA, 1976; pp. 227–261. [Google Scholar]
- Witkop, C. A study of tri-racial isolates in eastern United States. Hum. Hered. 1956, 6, 410–412. [Google Scholar] [CrossRef]
- Becks, H. Histologic Study of Tooth Structure in Osteogenesis Imperfecta. Dent. Cosm. 1931, 73, 437. [Google Scholar]
- Roberts, E.; Schour, I. Hereditary opalescent dentine (Dentinogenesis imperfecta). Am. J. Orthod. Oral Surg. 1939, 25, 267–276. [Google Scholar] [CrossRef]
- Hodge, H.C.; Finn, S.B. Hereditary Opalescent Dentin a Dominant Hereditary Tooth Anomaly in Man. J. Hered. 1938, 29, 359–364. [Google Scholar] [CrossRef]
- Hursey, R.J.; Witkop, C.J.; Miklashek, D.; Sackett, L.M. Dentinogenesis imperfecta in a racial isolate with multiple hereditary defects. Oral Surgery, Oral Med. Oral Pathol. 1956, 9, 641–658. [Google Scholar] [CrossRef]
- Witkop, C.J.; Dyson, H.R.; Sackett, L.M. A study of hereditary defects occurring in a racial isolate residing in Southern Maryland; special report. Clin. Proc.-Child. Hosp. Dist. Columbia 1956, 12, 29–33. [Google Scholar] [PubMed]
- Witkop, C.J.; MacLean, C.J.; Schmidt, P.J.; Henry, J.L. Medical and dental findings in the Brandywine isolate. Ala. J. Med. Sci. 1966, 3, 382–403. [Google Scholar] [PubMed]
- Levin, L.; Leaf, S.H.; Jelmini, R.J.; Rose, J.J.; Rosenbaum, K.N. Dentinogenesis imperfecta in the Brandywine isolate (DI type III): Clinical, radiologic, and scanning electron microscopic studies of the dentition. Oral Surg. Oral Med. Oral Pathol. 1983, 56, 267–274. [Google Scholar] [CrossRef]
- Richardson, A.S.; Fantin, T.D. Anomalous dysplasia of dentine: Report of case. J. Can. Dent. Assoc. 1970, 36. [Google Scholar]
- Rao, S.R.; Witkop, C.J., Jr.; Yamane, G.M. Pulpal dysplasia. Oral Surg. Oral Med. Oral Pathol. 1970, 30, 682–689. [Google Scholar] [CrossRef]
- Shields, E.; Bixler, D.; El-Kafrawy, A. A proposed classification for heritable human dentine defects with a description of a new entity. Arch. Oral Biol. 1973, 18, 543-IN7. [Google Scholar] [CrossRef]
- Basel, D.; Steiner, R.D. Osteogenesis imperfecta: Recent findings shed new light on this once well-understood condition. Genet Med. 2009, 11, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. Part. A 2019, 179, 2393–2419. [Google Scholar] [CrossRef]
- Witkop, C.J., Jr. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: Problems in classification. J. Oral Pathol. Med. 1988, 17, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Yu, C.; Chou, X.; Yuan, W.; Wang, Y.; Bu, L.; Fu, G.; Qian, M.; Yang, J.; Shi, Y.; et al. Dentinogenesis imperfecta 1 with or without progressive hearing loss is associated with distinct mutations in DSPP. Nat. Genet. 2001, 27, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, J.; Li, C.; Gao, S.; Qiu, C.; Liu, P.; Wu, G.; Qiang, B.; Lo, W.H.; Shen, Y. DSPP mutation in dentinogenesis imperfecta Shields type II. Nat. Genet. 2001, 27, 151–152. [Google Scholar] [CrossRef] [PubMed]
- Rajpar, M.H.; Koch, M.J.; Davies, R.M.; Mellody, K.T.; Kielty, C.M.; Dixon, M.J. Mutation of the signal peptide region of the bicistronic gene DSPP affects translocation to the endoplasmic reticulum and results in defective dentine biomineralization. Hum. Mol. Genet. 2002, 11, 2559–2565. [Google Scholar] [CrossRef] [Green Version]
- Malmgren, B.; Lindskog, S.; Elgadi, A.; Norgren, S. Clinical, histopathologic, and genetic investigation in two large families with dentinogenesis imperfecta type II. Hum. Genet. 2004, 114, 491–498. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.; Liu, J.; Zhao, Z.; Qu, E.; Wang, X.; Chang, W.; Xu, C.; Wang, Q.K.; Liu, M. A novel DSPP mutation is associated with type II dentinogenesis imperfecta in a Chinese family. BMC Med. Genet. 2007, 8, 52. [Google Scholar] [CrossRef] [Green Version]
- Qu, E.-J.; Zhang, H.-B.; Chen, L.-Y.; Gu, L.-B. Mutation analysis of a Chinese family with genetic dentinogenesis imperfecta. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2009, 26. [Google Scholar] [CrossRef]
- Taleb, K.; Lauridsen, E.; Daugaard-Jensen, J.; Nieminen, P.; Kreiborg, S. Dentinogenesis imperfecta type II- genotype and phenotype analyses in three Danish families. Mol. Genet. Genom. Med. 2018, 6, 339–349. [Google Scholar] [CrossRef]
- Li, D.; Du, X.; Zhang, R.; Shen, B.; Huang, Y.; Valenzuela, R.K.; Wang, B.; Zhao, H.; Liu, Z.; Li, J.; et al. Mutation identification of the DSPP in a Chinese family with DGI-II and an up-to-date bioinformatic analysis. Genomics 2012, 99, 220–226. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Lee, K.E.; Song, S.J.; Hyun, H.K.; Lee, S.H.; Kim, J.W. A DSPP mutation causing dentinogenesis imperfecta and characterization of the mutational effect. Biomed. Res. Int. 2013, 2013, 948181. [Google Scholar]
- Hu, A.; Li, X.; Chen, D.; Lu, T.; Huang, J.; Xu, X.; Chen, D.; Xiong, F. Analysis of DSPP gene mutation in a Chinese pedigree affected with hereditary dentinogenesis imperfecta. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2018, 35, 511–514. [Google Scholar] [PubMed]
- Wang, H.; Hou, Y.; Cui, Y.; Huang, Y.; Shi, Y.; Xia, X.; Lu, H.; Wang, Y.; Li, X. A novel splice site mutation in the dentin sialophosphoprotein gene in a Chinese family with dentinogenesis imperfecta type II. Mutat. Res. Mol. Mech. Mutagen. 2009, 662, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-K.; Hu, J.C.-C.; Lee, K.-E.; Simmer, J.P.; Kim, J.-W. A Dentin Sialophosphoprotein Mutation That Partially Disrupts a Splice Acceptor Site Causes Type II Dentin Dysplasia. J. Endod. 2008, 34, 1470–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-W.; Nam, S.-H.; Jang, K.-T.; Lee, S.-H.; Kim, C.-C.; Hahn, S.-H.; Hu, J.C.-C.; Simmer, J.P. A novel splice acceptor mutation in the DSPP gene causing dentinogenesis imperfecta type II. Qual. Life Res. 2004, 115, 248–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holappa, H.; Nieminen, P.; Tolva, V.; Lukinmaa, P.-L.; Alaluusua, S. Splicing site mutations in dentin sialophosphoprotein causing dentinogenesis imperfecta type II. Eur. J. Oral Sci. 2006, 114, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, Y.; Liu, H.; Yang, J.; Zhang, F.; Feng, H. Phenotype and genotype analyses in seven families with dentinogenesis imperfecta or dentin dysplasia. Oral Dis. 2016, 23, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, Y.; Gao, J.; Li, S.; Zhao, X.; Zhang, X. Identification of a novel mutation of DSPP gene in a Chinese family affected with dentinogenesis imperfecta shields type II. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2016, 33, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Hu, J.C.-C.; Lee, J.-I.; Moon, S.-K.; Kim, Y.-J.; Jang, K.-T.; Lee, S.-H.; Kim, C.-C.; Hahn, S.-H.; Simmer, J.P. Mutational hot spot in the DSPP gene causing dentinogenesis imperfecta type II. Qual. Life Res. 2004, 116, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Wang, C.; Peng, B.; Ye, X.; Zhao, G.; Fan, M.; Fu, Q.; Bian, Z. Phenotypes and genotypes in 2 DGI families with different DSPP mutations. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2006, 102, 360–374. [Google Scholar] [CrossRef]
- Kida, M.; Tsutsumi, T.; Shindoh, M.; Ikeda, H.; Ariga, T. De novo mutation in the DSPP gene associated with dentinogenesis imperfecta type II in a Japanese family. J. Oral Sci. 2009, 117, 691–694. [Google Scholar] [CrossRef]
- Lee, S.-K.; Lee, K.-E.; Jeon, D.; Lee, G.; Lee, H.; Shin, C.-U.; Jung, Y.-J.; Hahn, S.-H.; Kim, J.-W. A Novel Mutation in the DSPP Gene Associated with Dentinogenesis Imperfecta Type II. J. Dent. Res. 2009, 88, 51–55. [Google Scholar] [CrossRef]
- Lee, S.-K.; Lee, K.-E.; Hwang, Y.-H.; Kida, M.; Tsutsumi, T.; Ariga, T.; Park, J.-C.; Kim, J.-W. Identification of the DSPP mutation in a new kindred and phenotype-genotype correlation. Oral Dis. 2010, 17, 314–319. [Google Scholar] [CrossRef]
- Du, Q.; Cao, L.; Liu, Y.; Pang, C.; Wu, S.; Zheng, L.; Jiang, W.; Na, X.; Yu, J.; Wang, S.; et al. Phenotype and molecular characterizations of a family with dentinogenesis imperfecta shields type II with a novel DSPP mutation. Ann. Transl. Med. 2021, 9, 1672. [Google Scholar] [CrossRef]
- Wang, S.-K.; Chan, H.-C.; Rajderkar, S.; Milkovich, R.N.; Uston, K.A.; Kim, J.-W.; Simmer, J.P.; Hu, J.C.-C. Enamel malformations associated with a defined dentin sialophosphoprotein mutation in two families. Eur. J. Oral Sci. 2011, 119, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, J.; Ma, Y.; Du, W.; Zhao, S.; Zhang, Z.; Zhang, X.; Liu, Y.; Xiao, H.; Wang, H.; et al. A Novel Splicing Mutation Alters DSPP Transcription and Leads to Dentinogenesis Imperfecta Type II. PLoS ONE 2011, 6, e27982. [Google Scholar] [CrossRef]
- Bai, H.; Agula, H.; Wu, Q.; Zhou, W.; Sun, Y.; Qi, Y.; Latu, S.; Chen, Y.; Mutu, J.; Qiu, C. A novel DSPP mutation causes dentinogenesis imperfecta type II in a large Mongolian family. BMC Med. Genet. 2010, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Marschall, Z.; Mok, S.; Phillips, M.D.; McKnight, D.A.; Fisher, L.W. Rough endoplasmic reticulum trafficking errors by different classes of mutant dentin sialophosphoprotein (DSPP) cause dominant negative effects in both dentinogenesis imperfecta and dentin dysplasia by entrapping normal DSPP. J. Bone Miner. Res. 2012, 27, 1309–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, D.; Simmer, J.; Hart, P.; Hart, T.; Fisher, L. Overlapping DSPP Mutations Cause Dentin Dysplasia and Dentinogenesis Imperfecta. J. Dent. Res. 2008, 87, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, P.; Papagiannoulis-Lascarides, L.; Waltimo-Sirén, J.; Ollila, P.; Karjalainen, S.; Arte, S.; Veerkamp, J.; Walton, V.T.; Küstner, E.C.; Siltanen, T.; et al. Frameshift mutations in dentin phosphoprotein and dependence of dentin disease phenotype on mutation location. J. Bone Miner. Res. 2010, 26, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Porntaveetus, T.; Osathanon, T.; Nowwarote, N.; Pavasant, P.; Srichomthong, C.; Suphapeetiporn, K.; Shotelersuk, V. Dental properties, ultrastructure, and pulp cells associated with a novel DSPP mutation. Oral Dis. 2018, 24, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, C.N.; Fan, M.W.; Su, B.; Bian, Z. Dentin phosphoprotein frameshift mutations in hereditary dentin disorders and their variation patterns in normal human population. J. Med. Genet. 2008, 45, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Hong, J.; Seymen, F.; Kim, Y.J.; Kang, J.; Koruyucu, M.; Tuloglu, N.; Bayrak, S.; Song, J.; Shin, T.J.; et al. Novel frameshift mutations in DSPP cause dentin dysplasia type II. Oral Dis. 2019, 25, 2044–2046. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-E.; Kang, H.-Y.; Lee, S.-K.; Yoo, S.-H.; Lee, J.-C.; Hwang, Y.-H.; Nam, K.; Kim, J.-S.; Park, J.-C. Novel dentin phosphoprotein frameshift mutations in dentinogenesis imperfecta type II. Clin. Genet. 2011, 79, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Bloch-Zupan, A.; Huckert, M.; Stoetzel, C.; Meyer, J.; Geoffroy, V.; Razafindrakoto, R.W.; Ralison, S.N.; Randrianaivo, J.C.; Ralison, G.; Andriamasinoro, R.O.; et al. Detection of a Novel DSPP Mutation by NGS in a Population Isolate in Madagascar. Front. Physiol. 2016, 7, 70. [Google Scholar] [PubMed] [Green Version]
- Dong, J.; Gu, T.; Jeffords, L.; MacDougall, M. Dentin phosphoprotein compound mutation in dentin sialophosphoprotein causes dentinogenesis imperfecta type III. Am. J. Med. Genet. Part. A 2004, 132, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Zhang, H.; Xu, Q.; Wang, S.; Qin, C.; Lu, Y. Mutant Dentin Sialophosphoprotein Causes Dentinogenesis Imperfecta. J. Dent. Res. 2019, 98, 912–919. [Google Scholar] [CrossRef]
- Liang, T.; Xu, Q.; Zhang, H.; Wang, S.; Diekwisch, T.G.H.; Qin, C.; Lu, Y. Enamel Defects Associated With Dentin Sialophosphoprotein Mutation in Mice. Front. Physiol. 2021, 12, 724098. [Google Scholar] [CrossRef]
- Suzuki, S.; Nakata, M.; Eto, K. Clinical and histologic observations of opalescent dentin associated with enamel defects. Oral Surg. Oral Med. Oral Pathol. 1977, 44, 767–774. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Seymen, F.; Lin, B.P.-J.; Kızıltan, B.; Gencay, K.; Simmer, J.P.; Hu, J.-C. ENAM Mutations in Autosomal-dominant Amelogenesis Imperfecta. J. Dent. Res. 2005, 84, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreenath, T.; Thyagarajan, T.; Hall, B.; Longenecker, G.; D′Souza, R.; Hong, S.; Wright, J.T.; MacDougall, M.; Sauk, J.; Kulkarni, A.B. Dentin Sialophosphoprotein Knockout Mouse Teeth Display Widened Predentin Zone and Develop Defective Dentin Mineralization Similar to Human Dentinogenesis Imperfecta Type III. J. Biol. Chem. 2003, 278, 24874–24880. [Google Scholar] [CrossRef] [Green Version]
- Marinakis, N.M.; Svingou, M.; Veltra, D.; Kekou, K.; Sofocleous, C.; Tilemis, F.; Kosma, K.; Tsoutsou, E.; Fryssira, H.; Traeger-Synodinos, J. Phenotype-driven variant filtration strategy in exome sequencing toward a high diagnostic yield and identification of 85 novel variants in 400 patients with rare Mendelian disorders. Am. J. Med. Genet. Part. A 2021, 185, 2561–2571. [Google Scholar] [CrossRef]
- De La Dure-Molla, M.; Fournier, B.; Berdal, A. Isolated dentinogenesis imperfecta and dentin dysplasia: Revision of the classification. Eur. J. Hum. Genet. 2014, 23, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Begue-Kirn, C.; Krebsbach, P.H.; Bartlett, J.D.; Butler, W.T. Dentin sialoprotein, dentin phosphoprotein, enamelysin and ameloblastin: Tooth-specific molecules that are distinctively expressed during murine dental differentiation. Eur. J. Oral Sci. 1998, 106, 963–970. [Google Scholar] [CrossRef]
- Smith, C.E.L.; Poulter, J.A.; Antanaviciute, A.; Kirkham, J.; Brookes, S.J.; Inglehearn, C.F.; Mighell, A.J. Amelogenesis Imperfecta; Genes, Proteins, and Pathways. Front. Physiol. 2017, 8, 435. [Google Scholar] [CrossRef] [Green Version]
- Bencharit, S.; Border, M.B.; Mack, C.R.; Byrd, W.C.; Wright, J.T. Full-Mouth Rehabilitation for a Patient with Dentinogenesis Imperfecta: A Clinical Report. J. Oral Implant. 2014, 40, 593–600. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Location | Gene (NG_011595.1) | cDNA (NM_014208.3) | Protein (NP_055023.2) | References |
|---|---|---|---|---|---|
| 1 | Exon 2 | g.7396T>G | c.16T>G | p.(Tyr6Asp) | [56] |
| 2 | Exon 2 | g.7424C>T | c.44C>T | p.(Ala15Val) | [57] |
| 3 | Exon 2 | g.7429C>A | c.49C>A | p.(Pro17Thr) | [54] |
| 4 | Exon 2 | g.7429C>T | c.49C>T | p.(Pro17Ser) | [25,34,58,59,60] |
| 5 | Exon 2 | g.7430C>T | c.50C>T | p.(Pro17Leu) | Family 1, [61,62,63] |
| 6 | Intron 2 | g.8552_8574del23 | c.52-25_52-2del23 | p.(?) | [64] |
| 7 | Intron 2 | g.8571T>G | c.52-6T>G | p.(?) | [65] |
| 8 | Intron 2 | g.8574C>G | c.52-3C>G | p.(?) | [66] |
| 9 | Intron 2 | g.8574C>A | c.52-3C>A | p.(?) | [67] |
| 10 | Intron 2 | g.8575A>G | c.52-2A>G | p.(?) | [60,68] |
| 11 | Intron 2 | g.8576G>A | c.52-1G>A | p.(?) | Family 2, [69] |
| 12 | Exon 3 | g.8577G>T | c.52G>T | p.(Val18Phe) | [54,67,68,70,71] |
| 13 | Exon 3 | g.8578T>A | c.53T>A | p.(Val18Asp) | [72,73,74] |
| 14 | Exon 3 | g.8578T>G | c.53T>G | p.(Val18Gly) | [75] |
| 15 | Exon 3 | g.8578T>C | c.53T>C | p.(Val18Ala) | Family 3 |
| 16 | Exon 3 | g.8658C>T | c.133C>T | p.(Gln45*) | [55,71] |
| 17 | Exon 3 | g.8660C>T | c.135G>T | p.(Gln45His) | [60] |
| 18 | Intron 3 | g.8661G>A | c.135+1G>A | p.(?) | [54,76] |
| 19 | Intron 3 | g.8661G>T | c.135+1G>T | p.(?) | [25] |
| 20 | Intron 3 | g.8662T>C | c.135+2T>C | p.(?) | [77] |
| 21 | Intron 3 | g.8663A>G | c.135+3A>G | p.(?) | Family 4 [78] |
| # | Gene (NG_011595.1) | cDNA (NM_014208.3) | Protein (NP_055023.2) | References |
|---|---|---|---|---|
| 1 | g.10820delT | c.1686delT | p.(Asp562Glufs*752) | [81] |
| 2 | g.10964delC | c.1830delC | p.(Ser610Argfs*704) | [81] |
| 3 | g.11004_11007delTCAG | c.1870_1873delTCAG | p.(Ser624Thrfs*689) | [25] |
| 4 | g.11008_11011delACAG | c.1874_1877delACAG | p.(Asp625Alafs*688) | [68] |
| 5 | g.11049_11052delAAGT | c.1915_1918delAAGT | p.(Lys639Glnfs*674) | Family 5 [82] |
| 6 | g.11052_11055delTCAG | c.1918_1921delTCAG | p.(Ser640Thrfs*673) | Fam 6 [25,81] |
| 7 | g.11056_11059delACAG | c.1922_1925delACAG | p.(Asp641Alafs*672) | [81] |
| 8 | g.11174delC | c.2040delC | p.(Ser680Argfs*634) | [83] |
| 9 | g.11197delA | c.2063delA | p.(Asp688Valfs*626) | [81] |
| 10 | g.11268delA | c.2134delA | p.(Ser712Alafs*602) | Family 7 [84] |
| 11 | g.11406delA | c.2272delA | p.(Ser758Alafs*556) | [25] |
| 12 | g.11483delT | c.2349delT | p.(Ser783Argfs*531) | [81] |
| 13 | g.11659delG | c.2525delG | p.(Ser842Thrfs*472) | Fam 8&9 [25] |
| 14 | g.11727delA | c.2593delA | p.(Ser865Valfs*449) | [83] |
| 15 | g.11800delG | c.2666delG | p.(Ser889Thrfs*425) | [81] |
| 16 | g.11818delG | c.2684delG | p.(Ser895Metfs*419) | [68,83] |
| 17 | g.11822delT | c.2688delT | p.(Asp896Glufs*418) | [85] |
| 18 | g.12269delC | c.3135delC | p.(Ser1045Argfs*269) | Fam 10 [26,80] |
| 19 | g.12313delG | c.3179delG | p.(Ser1060Thrfs*254) | [84] |
| 20 | g.12572delC | c.3438delC | p.(Asp1146Glufs*168) | [83] |
| 21 | g.12595delG | c.3461delG | p.(Ser1154Metfs*160) | Family 11 |
| 22 | g.12614_12615insCTGCT | c.3480_3481insCTGCT | p.(Asp1161Leufs*155) | [84] |
| 23 | g.12638_12642dupCAGCG | c.3504_3508dupCAGCG | p.(Asp1170Alafs*146) | [26] |
| 24 | g.12643_12655del13 | c.3509_3521delACAGCAGCGATAG | p.(Asp1170Alafs*140) | [68] |
| 25 | g.12680_12684delinsG | c.3546_3550delTAGCAinsG | p.(Asp1182Glufs*131) | [83] |
| 26 | g.12694delG | c.3560delG | p.(Ser1187Metfs*127) | [85] |
| 27 | g.12716_12725del | c.3582_3591delCAGCAGCGAT | p.(Asp1194Glufs*117) | [81] |
| 28 | g.12810delA | c.3676delA | p.(Ser1226Alafs*88) | [86] |
| 29 | g.12759_12834del76 | c.3625-3700del76 | p.(Asp1209Alafs*80) | [81] |
| 30 | g.12834delA | c.3700delA | p.(Ser1234Alafs*80) | Family 12 |
| # | Sequencing Platform | Causative Mutations | Defect | WES Mean DSPP Sequence Depth |
|---|---|---|---|---|
| 1 | Illumina HiSeq 2500 (WES) | NG_011595.1:g.7430C>T; NM_014208.3:c.50C>T; NP_055023.2:p.(Pro17Leu) | Missense | II:6, unaffected mother: 136.75× III:5, affected 1st child: 159.3× III:6, affected 2nd child: 129.86× |
| 2 | Illumina HiSeq 2500 (WES) | NG_011595.1:g.8576G>A; NM_014208.3:c.52-1G>A | Splice Acceptor | IV:2, affected 2nd child: 177.72× |
| 3 | Illumina HiSeq 2500 (WES) | NG_011595.1:g.8578T>C; NM_014208.3:c.53T>C; NP_055023.2:p.(Val18Ala) | Missense | III:2, affected mother: 285.78× IV:2, affected child: 189.42× |
| 4 | Sanger Sequencing | NG_011595.1:g.8663A>G; NM_014208.3:c.135+3A>G | Splice Donor | |
| 5 | Illumina HiSeq 2500 (WES) | NG_011595.1:g.11049_11052delAAGT; NM_014208.3:c.1915_1918delAAGT; NP_055023.2: p.(Lys639Glnfs*674) | −1 Frameshift | I:1, affected father: 233.91× I:2, unaffected mother: 165.19× II:2, affected 2nd child: 178.45× |
| 6 | Illumina HiSeq 2500 (WES) | NG_011595.1:g.11052_11055delTCAG; NM_014208.3:c.1918_1921delTCAG; NP_055023.2: p.(Ser640Thrfs*673) | −1 Frameshift | II:1, unaffected father: 193.27× II:2, affected mother: 178.71× III:2, affected 2nd child: 192.59× |
| 7 | Illumina HiSeq 2500 (WES) | NG_011595.1:g.11268delA; NM_014208.3:c.2134delA; NP_055023.2: p.(Ser712Alafs*602) | −1 Frameshift | III:1, affected 1st child: 203.26× |
| 8 | PacBio SMRT | NG_011595.1:g.11659delG; NM_014208.3:c.2525delG; NP_055023.2:p.(Ser842Thrfs*472) | −1 Frameshift | |
| 9 | PacBio SMRT | NG_011595.1:g.11659delG; NM_014208.3:c.2525delG; NP_055023.2:p.(Ser842Thrfs*472) | −1 Frameshift | |
| 10 | PacBio SMRT | NG_011595.1:g.12269delC; NM_014208.3:c.3135delC; NP_055023.2:p.(Ser1045Argfs*269) | −1 Frameshift | |
| 11 | PacBio SMRT Illumina HiSeq 2500 (WES) | NG_011595.1:g.12595delG; NM_014208.3: c.3461delG; NP_055023.2:p.(Ser1154Metfs*160) | −1 Frameshift | II:6, unaffected grandfather: 188.54× II:3, affected grandmother: 197.2× III2, affected mother: 217.83× |
| 12 | PacBio SMRT | NG_011595.1:g.12834delA; NM_014208.3:c.3700delA; NP_055023.2:p.(Ser1234Alafs*80) | −1 Frameshift |
| Family Number | Primer Name and Sequence | Annealing T | Size (bp) |
|---|---|---|---|
| 1 Exon 2 | PCR DSP2F: TAGTGCTGAGCCTGGTGATG DSP2R: CTCCATGACTTCTGGGCATT | 58 °C | 610 |
| 2&3 Exon 3–4 | PCR DSP34F1: CAAGCCCTGTAAGAAGCCACT DSP34R1: TCTGCCCACTTAGAGCCATT | 58 °C | 530 |
| 4 Exon 3–4 | PCR DSP34F2: TCAAAGCAAACCACTGGAGA DSP34R2: TCCTCATTGTGACCTGCATC | 58 °C | 601 |
| 5–12 Exon 5 | PCR DPP F: AGTCCATGCAAGGAGATGATCC DPP R: CTAATCATCACTGGTTGAGTGG | 60 °C | ~2534 |
| 5–7 | Sequencing For: CAGTAGCCGAGGAGATGCTTCTTATAACTC | ||
| 8–9 | Sequencing For: AGCAAATCAGAGAGCGACAGCAG | ||
| 10 | Sequencing Rev: TACCAGACTTGCTCTGGCTGTCACTCTCAT | ||
| 11–12 | Sequencing Rev: ACCAGACTTGCTCTGGCTGT |
| Family | Primer Name and Sequence | Annealing T |
|---|---|---|
| 8 | DPP BF4: ATCACACTGCATCTGAAGTCCATGCAAGGAGATGATCC DPP BR4: ATCACACTGCATCTGACTAATCATCACTGGTTGAGTGG | 72 °C |
| 9 | DPP BF2: TCATGAGTCGACACTAAGTCCATGCAAGGAGATGATCC DPP BR2: TCATGAGTCGACACTACTAATCATCACTGGTTGAGTGG | 72 °C |
| 10 | DPP BF1: GCGCTCTGTGTGCAGCAGTCCATGCAAGGAGATGATCC DPP BR1: GCGCTCTGTGTGCAGCCTAATCATCACTGGTTGAGTGG | 72 °C |
| 11 | DPP BF7: AGAGACACGATACTCAAGTCCATGCAAGGAGATGATCC DPP BR7: AGAGACACGATACTCACTAATCATCACTGGTTGAGTGG | 72 °C |
| 12 | DPP BF6: TGTGAGTCAGTACGCGAGTCCATGCAAGGAGATGATCC DPP BR6: TGTGAGTCAGTACGCGCTAATCATCACTGGTTGAGTGG | 72 °C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simmer, J.P.; Zhang, H.; Moon, S.J.H.; Donnelly, L.A.-J.; Lee, Y.-L.; Seymen, F.; Koruyucu, M.; Chan, H.-C.; Lee, K.Y.; Wu, S.; et al. The Modified Shields Classification and 12 Families with Defined DSPP Mutations. Genes 2022, 13, 858. https://doi.org/10.3390/genes13050858
Simmer JP, Zhang H, Moon SJH, Donnelly LA-J, Lee Y-L, Seymen F, Koruyucu M, Chan H-C, Lee KY, Wu S, et al. The Modified Shields Classification and 12 Families with Defined DSPP Mutations. Genes. 2022; 13(5):858. https://doi.org/10.3390/genes13050858
Chicago/Turabian StyleSimmer, James P., Hong Zhang, Sophie J. H. Moon, Lori A-J. Donnelly, Yuan-Ling Lee, Figen Seymen, Mine Koruyucu, Hui-Chen Chan, Kevin Y. Lee, Suwei Wu, and et al. 2022. "The Modified Shields Classification and 12 Families with Defined DSPP Mutations" Genes 13, no. 5: 858. https://doi.org/10.3390/genes13050858
APA StyleSimmer, J. P., Zhang, H., Moon, S. J. H., Donnelly, L. A. -J., Lee, Y. -L., Seymen, F., Koruyucu, M., Chan, H. -C., Lee, K. Y., Wu, S., Hsiang, C. -L., Tsai, A. T. P., Slayton, R. L., Morrow, M., Wang, S. -K., Shields, E. D., & Hu, J. C. -C. (2022). The Modified Shields Classification and 12 Families with Defined DSPP Mutations. Genes, 13(5), 858. https://doi.org/10.3390/genes13050858