
INPP5D/SHIP1: Expression, Regulation and Roles in Alzheimer’s Disease Pathophysiology



Abstract
:1. Introduction
2. Expression and Regulation of INPP5D in AD
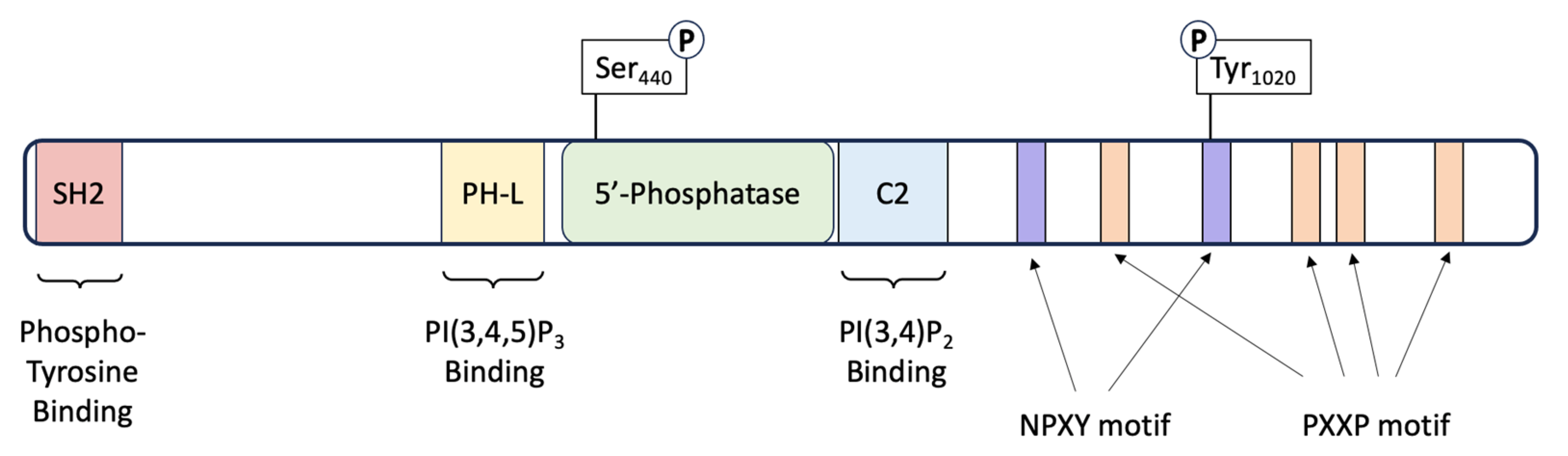
3. SHIP1: Structure
4. Actions of SHIP1 in Immune Cells, Including Microglia
5. Roles of SHIP1 in Alzheimer’s Disease Pathophysiology
6. SHIP1 as a Therapeutic Target for Alzheimer’s Disease
7. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Dementia-Social and Economic Impact. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 18 September 2023).
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef] [PubMed]
- Wightman, D.P.; Jansen, I.E.; Savage, J.E.; Shadrin, A.A.; Bahrami, S.; Holland, D.; Rongve, A.; Borte, S.; Winsvold, B.S.; Drange, O.K.; et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 2021, 53, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Olufunmilayo, E.O.; Holsinger, R.M.D. Variant TREM2 Signaling in Alzheimer’s Disease. J. Mol. Biol. 2022, 434, 167470. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Saxton, T.M.; van Oostveen, I.; Bowtell, D.; Aebersold, R.; Gold, M.R. B cell antigen receptor cross-linking induces phosphorylation of the p21ras oncoprotein activators SHC and mSOS1 as well as assembly of complexes containing SHC, GRB-2, mSOS1, and a 145-kDa tyrosine-phosphorylated protein. J. Immunol. 1994, 153, 623–636. [Google Scholar] [CrossRef]
- Chacko, G.W.; Tridandapani, S.; Damen, J.E.; Liu, L.; Krystal, G.; Coggeshall, K.M. Negative signaling in B lymphocytes induces tyrosine phosphorylation of the 145-kDa inositol polyphosphate 5-phosphatase, SHIP. J. Immunol. 1996, 157, 2234–2238. [Google Scholar] [CrossRef]
- Damen, J.E.; Liu, L.; Rosten, P.; Humphries, R.K.; Jefferson, A.B.; Majerus, P.W.; Krystal, G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc. Natl. Acad. Sci. USA 1996, 93, 1689–1693. [Google Scholar] [CrossRef]
- Ware, M.D.; Rosten, P.; Damen, J.E.; Liu, L.; Humphries, R.K.; Krystal, G. Cloning and characterization of human SHIP, the 145-kD inositol 5-phosphatase that associates with SHC after cytokine stimulation. Blood 1996, 88, 2833–2840. [Google Scholar] [CrossRef]
- Zajac, D.J.; Simpson, J.; Zhang, E.; Parikh, I.; Estus, S. Expression of INPP5D Isoforms in Human Brain: Impact of Alzheimer’s Disease Neuropathology and Genetics. Genes 2023, 14, 763. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Malhotra, S.; Torchia, J.A.; Kerr, W.G.; Coggeshall, K.M.; Humphrey, M.B. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci. Signal 2010, 3, ra38. [Google Scholar] [CrossRef] [PubMed]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hagg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.C.; Cobb, B.S.; Sabbattini, P.; Meixlsperger, S.; Parelho, V.; Liberg, D.; Taylor, B.; Dillon, N.; Georgopoulos, K.; Jumaa, H.; et al. Ikaros DNA-binding proteins as integral components of B cell developmental-stage-specific regulatory circuits. Immunity 2007, 26, 335–344. [Google Scholar] [CrossRef]
- Read, K.A.; Jones, D.M.; Freud, A.G.; Oestreich, K.J. Established and emergent roles for Ikaros transcription factors in lymphoid cell development and function. Immunol. Rev. 2021, 300, 82–99. [Google Scholar] [CrossRef]
- Nera, K.P.; Alinikula, J.; Terho, P.; Narvi, E.; Tornquist, K.; Kurosaki, T.; Buerstedde, J.M.; Lassila, O. Ikaros has a crucial role in regulation of B cell receptor signaling. Eur. J. Immunol. 2006, 36, 516–525. [Google Scholar] [CrossRef]
- Dhanyamraju, P.K.; Iyer, S.; Smink, G.; Bamme, Y.; Bhadauria, P.; Payne, J.L.; Dovat, E.; Klink, M.; Ding, Y. Transcriptional Regulation of Genes by Ikaros Tumor Suppressor in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2020, 21, 1377. [Google Scholar] [CrossRef]
- Alinikula, J.; Kohonen, P.; Nera, K.P.; Lassila, O. Concerted action of Helios and Ikaros controls the expression of the inositol 5-phosphatase SHIP. Eur. J. Immunol. 2010, 40, 2599–2607. [Google Scholar] [CrossRef]
- Hahm, K.; Cobb, B.S.; McCarty, A.S.; Brown, K.E.; Klug, C.A.; Lee, R.; Akashi, K.; Weissman, I.L.; Fisher, A.G.; Smale, S.T. Helios, a T cell-restricted Ikaros family member that quantitatively associates with Ikaros at centromeric heterochromatin. Genes Dev. 1998, 12, 782–796. [Google Scholar] [CrossRef]
- Xia, R.; Cheng, Y.; Han, X.; Wei, Y.; Wei, X. Ikaros Proteins in Tumor: Current Perspectives and New Developments. Front. Mol. Biosci. 2021, 8, 788440. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.P.; Lin, P.B.; Dong, C.; Moutinho, M.; Casali, B.T.; Liu, Y.; Lamb, B.T.; Landreth, G.E.; Oblak, A.L.; Nho, K. INPP5D expression is associated with risk for Alzheimer’s disease and induced by plaque-associated microglia. Neurobiol. Dis. 2021, 153, 105303. [Google Scholar] [CrossRef]
- Huang, K.L.; Marcora, E.; Pimenova, A.A.; Di Narzo, A.F.; Kapoor, M.; Jin, S.C.; Harari, O.; Bertelsen, S.; Fairfax, B.P.; Czajkowski, J.; et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Pauls, S.D.; Marshall, A.J. Regulation of immune cell signaling by SHIP1: A phosphatase, scaffold protein, and potential therapeutic target. Eur. J. Immunol. 2017, 47, 932–945. [Google Scholar] [CrossRef] [PubMed]
- March, M.E.; Ravichandran, K. Regulation of the immune response by SHIP. Semin. Immunol. 2002, 14, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Aman, M.J.; Walk, S.F.; March, M.E.; Su, H.P.; Carver, D.J.; Ravichandran, K.S. Essential role for the C-terminal noncatalytic region of SHIP in FcgammaRIIB1-mediated inhibitory signaling. Mol. Cell Biol. 2000, 20, 3576–3589. [Google Scholar] [CrossRef]
- Zhang, J.; Ravichandran, K.S.; Garrison, J.C. A key role for the phosphorylation of Ser440 by the cyclic AMP-dependent protein kinase in regulating the activity of the Src homology 2 domain-containing Inositol 5’-phosphatase (SHIP1). J. Biol. Chem. 2010, 285, 34839–34849. [Google Scholar] [CrossRef]
- Ong, C.J.; Ming-Lum, A.; Nodwell, M.; Ghanipour, A.; Yang, L.; Williams, D.E.; Kim, J.; Demirjian, L.; Qasimi, P.; Ruschmann, J.; et al. Small-molecule agonists of SHIP1 inhibit the phosphoinositide 3-kinase pathway in hematopoietic cells. Blood 2007, 110, 1942–1949. [Google Scholar] [CrossRef]
- Manno, B.; Oellerich, T.; Schnyder, T.; Corso, J.; Losing, M.; Neumann, K.; Urlaub, H.; Batista, F.D.; Engelke, M.; Wienands, J. The Dok-3/Grb2 adaptor module promotes inducible association of the lipid phosphatase SHIP with the BCR in a coreceptor-independent manner. Eur. J. Immunol. 2016, 46, 2520–2530. [Google Scholar] [CrossRef]
- D’Ambrosio, D.; Hippen, K.L.; Cambier, J.C. Distinct mechanisms mediate SHC association with the activated and resting B cell antigen receptor. Eur. J. Immunol. 1996, 26, 1960–1965. [Google Scholar] [CrossRef]
- Lemay, S.; Davidson, D.; Latour, S.; Veillette, A. Dok-3, a novel adapter molecule involved in the negative regulation of immunoreceptor signaling. Mol. Cell Biol. 2000, 20, 2743–2754. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Sheng, R.; Silkov, A.; Jung, D.J.; Wang, Z.G.; Xin, Y.; Kim, H.; Thiagarajan-Rosenkranz, P.; Song, S.; Yoon, Y.; et al. SH2 Domains Serve as Lipid-Binding Modules for pTyr-Signaling Proteins. Mol. Cell 2016, 62, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Ming-Lum, A.; Shojania, S.; So, E.; McCarrell, E.; Shaw, E.; Vu, D.; Wang, I.; McIntosh, L.P.; Mui, A.L. A pleckstrin homology-related domain in SHIP1 mediates membrane localization during Fcgamma receptor-induced phagocytosis. FASEB J. 2012, 26, 3163–3177. [Google Scholar] [CrossRef]
- Lamkin, T.D.; Walk, S.F.; Liu, L.; Damen, J.E.; Krystal, G.; Ravichandran, K.S. Shc interaction with Src homology 2 domain containing inositol phosphatase (SHIP) in vivo requires the Shc-phosphotyrosine binding domain and two specific phosphotyrosines on SHIP. J. Biol. Chem. 1997, 272, 10396–10401. [Google Scholar] [CrossRef]
- Tamir, I.; Stolpa, J.C.; Helgason, C.D.; Nakamura, K.; Bruhns, P.; Daeron, M.; Cambier, J.C. The RasGAP-binding protein p62dok is a mediator of inhibitory FcgammaRIIB signals in B cells. Immunity 2000, 12, 347–358. [Google Scholar] [CrossRef]
- Kerr, W.G. Inhibitor and activator: Dual functions for SHIP in immunity and cancer. Ann. N. Y Acad. Sci. 2011, 1217, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.; Krause, U.; Desmedt, F.; Tanti, J.F.; Behrends, J.; Pesesse, X.; Sasaki, T.; Penninger, J.; Doherty, M.; Malaisse, W.; et al. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature 2001, 409, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Salim, K.; Bottomley, M.J.; Querfurth, E.; Zvelebil, M.J.; Gout, I.; Scaife, R.; Margolis, R.L.; Gigg, R.; Smith, C.I.; Driscoll, P.C.; et al. Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton’s tyrosine kinase. EMBO J. 1996, 15, 6241–6250. [Google Scholar] [CrossRef]
- Scharenberg, A.M.; El-Hillal, O.; Fruman, D.A.; Beitz, L.O.; Li, Z.; Lin, S.; Gout, I.; Cantley, L.C.; Rawlings, D.J.; Kinet, J.P. Phosphatidylinositol-3,4,5-trisphosphate (PtdIns-3,4,5-P3)/Tec kinase-dependent calcium signaling pathway: A target for SHIP-mediated inhibitory signals. EMBO J. 1998, 17, 1961–1972. [Google Scholar] [CrossRef]
- Konishi, H.; Kiyama, H. Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Front. Cell Neurosci. 2018, 12, 206. [Google Scholar] [CrossRef]
- Han, J.; Luby-Phelps, K.; Das, B.; Shu, X.; Xia, Y.; Mosteller, R.D.; Krishna, U.M.; Falck, J.R.; White, M.A.; Broek, D. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science 1998, 279, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Kommaddi, R.P.; Das, D.; Karunakaran, S.; Nanguneri, S.; Bapat, D.; Ray, A.; Shaw, E.; Bennett, D.A.; Nair, D.; Ravindranath, V. Abeta mediates F-actin disassembly in dendritic spines leading to cognitive deficits in Alzheimer’s disease. J. Neurosci. 2018, 38, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Rey-Ladino, J.A.; Huber, M.; Liu, L.; Damen, J.E.; Krystal, G.; Takei, F. The SH2-containing inositol-5’-phosphatase enhances LFA-1-mediated cell adhesion and defines two signaling pathways for LFA-1 activation. J. Immunol. 1999, 162, 5792–5799. [Google Scholar] [CrossRef] [PubMed]
- Landego, I.; Jayachandran, N.; Wullschleger, S.; Zhang, T.T.; Gibson, I.W.; Miller, A.; Alessi, D.R.; Marshall, A.J. Interaction of TAPP adapter proteins with phosphatidylinositol (3,4)-bisphosphate regulates B-cell activation and autoantibody production. Eur. J. Immunol. 2012, 42, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Aman, M.J.; Lamkin, T.D.; Okada, H.; Kurosaki, T.; Ravichandran, K.S. The inositol phosphatase SHIP inhibits Akt/PKB activation in B cells. J. Biol. Chem. 1998, 273, 33922–33928. [Google Scholar] [CrossRef]
- Yao, H.; Coppola, K.; Schweig, J.E.; Crawford, F.; Mullan, M.; Paris, D. Distinct Signaling Pathways Regulate TREM2 Phagocytic and NFkappaB Antagonistic Activities. Front. Cell Neurosci. 2019, 13, 457. [Google Scholar] [CrossRef]
- Pradhan, M.; Coggeshall, K.M. Activation-induced bi-dentate interaction of SHIP and Shc in B lymphocytes. J. Cell. Biochem. 1997, 67, 32–42. [Google Scholar] [CrossRef]
- Tridandapani, S.; Chacko, G.W.; Van Brocklyn, J.R.; Coggeshall, K.M. Negative signaling in B cells causes reduced Ras activity by reducing Shc-Grb2 interactions. J. Immunol. 1997, 158, 1125–1132. [Google Scholar] [CrossRef]
- An, H.; Xu, H.; Zhang, M.; Zhou, J.; Feng, T.; Qian, C.; Qi, R.; Cao, X. Src homology 2 domain-containing inositol-5-phosphatase 1 (SHIP1) negatively regulates TLR4-mediated LPS response primarily through a phosphatase activity- and PI-3K-independent mechanism. Blood 2005, 105, 4685–4692. [Google Scholar] [CrossRef]
- Conde, C.; Rambout, X.; Lebrun, M.; Lecat, A.; Di Valentin, E.; Dequiedt, F.; Piette, J.; Gloire, G.; Legrand, S. The inositol phosphatase SHIP-1 inhibits NOD2-induced NF-kappaB activation by disturbing the interaction of XIAP with RIP2. PLoS ONE 2012, 7, e41005. [Google Scholar] [CrossRef]
- Colonna, M.; Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Amode, M.R.; Barrell, D.; Beal, K.; Billis, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fitzgerald, S.; et al. Ensembl 2015. Nucleic Acids Res 2015, 43, D662–D669. [Google Scholar] [CrossRef]
- Iguchi, A.; Takatori, S.; Kimura, S.; Muneto, H.; Wang, K.; Etani, H.; Ito, G.; Sato, H.; Hori, Y.; Sasaki, J.; et al. INPP5D modulates TREM2 loss-of-function phenotypes in a β-amyloidosis mouse model. iScience 2023, 26, 106375. [Google Scholar] [CrossRef]
- Lin, P.B.; Tsai, A.P.; Soni, D.; Lee-Gosselin, A.; Moutinho, M.; Puntambekar, S.S.; Landreth, G.E.; Lamb, B.T.; Oblak, A.L. INPP5D deficiency attenuates amyloid pathology in a mouse model of Alzheimer’s disease. Alzheimers Dement. 2023, 19, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- Castranio, E.L.; Hasel, P.; Haure-Mirande, J.V.; Ramirez Jimenez, A.V.; Hamilton, B.W.; Kim, R.D.; Glabe, C.G.; Wang, M.; Zhang, B.; Gandy, S.; et al. Microglial INPP5D limits plaque formation and glial reactivity in the PSAPP mouse model of Alzheimer’s disease. Alzheimers Dement. 2023, 19, 2239–2252. [Google Scholar] [CrossRef]
- Malik, M.; Parikh, I.; Vasquez, J.B.; Smith, C.; Tai, L.; Bu, G.; LaDu, M.J.; Fardo, D.W.; Rebeck, G.W.; Estus, S. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 2015, 10, 52. [Google Scholar] [CrossRef]
- Huang, Z.Y.; Hunter, S.; Kim, M.K.; Indik, Z.K.; Schreiber, A.D. The effect of phosphatases SHP-1 and SHIP-1 on signaling by the ITIM- and ITAM-containing Fcgamma receptors FcgammaRIIB and FcgammaRIIA. J. Leukoc. Biol. 2003, 73, 823–829. [Google Scholar] [CrossRef]
- Hodges, A.K.; Piers, T.M.; Collier, D.; Cousins, O.; Pocock, J.M. Pathways linking Alzheimer’s disease risk genes expressed highly in microglia. Neuroimmunol. Neuroinflam 2021, 8, 245. [Google Scholar] [CrossRef]
- Boucrot, E.; Ferreira, A.P.; Almeida-Souza, L.; Debard, S.; Vallis, Y.; Howard, G.; Bertot, L.; Sauvonnet, N.; McMahon, H.T. Endophilin marks and controls a clathrin-independent endocytic pathway. Nature 2015, 517, 460–465. [Google Scholar] [CrossRef]
- Massa, P.T.; Wu, C. Increased inducible activation of NF-kappaB and responsive genes in astrocytes deficient in the protein tyrosine phosphatase SHP-1. J. Interferon Cytokine Res. 1998, 18, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Khaled, A.R.; Butfiloski, E.J.; Sobel, E.S.; Schiffenbauer, J. Functional consequences of the SHP-1 defect in motheaten viable mice: Role of NF-kappa B. Cell Immunol. 1998, 185, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Bourne, K.Z.; Ferrari, D.C.; Lange-Dohna, C.; Rossner, S.; Wood, T.G.; Perez-Polo, J.R. Differential regulation of BACE1 promoter activity by nuclear factor-kappaB in neurons and glia upon exposure to β-amyloid peptides. J. Neurosci. Res. 2007, 85, 1194–1204. [Google Scholar] [CrossRef]
- Terai, K.; Matsuo, A.; McGeer, P.L. Enhancement of immunoreactivity for NF-kappa B in the hippocampal formation and cerebral cortex of Alzheimer’s disease. Brain Res. 1996, 735, 159–168. [Google Scholar] [CrossRef]
- Yan, S.D.; Yan, S.F.; Chen, X.; Fu, J.; Chen, M.; Kuppusamy, P.; Smith, M.A.; Perry, G.; Godman, G.C.; Nawroth, P.; et al. Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid β-peptide. Nat. Med. 1995, 1, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Marti, E.; Lopez, E.; Tortosa, A. NF-kB immunoreactivity is observed in association with β A4 diffuse plaques in patients with Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 1998, 24, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef]
- Kitamura, Y.; Shimohama, S.; Ota, T.; Matsuoka, Y.; Nomura, Y.; Taniguchi, T. Alteration of transcription factors NF-kappaB and STAT1 in Alzheimer’s disease brains. Neurosci. Lett. 1997, 237, 17–20. [Google Scholar] [CrossRef]
- Mostafavi, S.; Gaiteri, C.; Sullivan, S.E.; White, C.C.; Tasaki, S.; Xu, J.; Taga, M.; Klein, H.U.; Patrick, E.; Komashko, V.; et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 2018, 21, 811–819. [Google Scholar] [CrossRef]
- Obst, J.; Bradshaw, W.; Roberts, H.H.; Priestley, R.; Jimenez-Antunez, C.; Cowley, S.A.; Gileadi, O.; Mead, E.; Daniel, E.D.; Davis, J.B. Targeting SHIP1 for therapeutic intervention in Alzheimer’s disease. Alzheimers Dement. 2020, 16, e045839. [Google Scholar] [CrossRef]
- Suwa, A.; Yamamoto, T.; Sawada, A.; Minoura, K.; Hosogai, N.; Tahara, A.; Kurama, T.; Shimokawa, T.; Aramori, I. Discovery and functional characterization of a novel small molecule inhibitor of the intracellular phosphatase, SHIP2. Br. J. Pharmacol. 2009, 158, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Soeda, Y.; Tsuneki, H.; Muranaka, H.; Mori, N.; Hosoh, S.; Ichihara, Y.; Kagawa, S.; Wang, X.; Toyooka, N.; Takamura, Y.; et al. The inositol phosphatase SHIP2 negatively regulates insulin/IGF-I actions implicated in neuroprotection and memory function in mouse brain. Mol. Endocrinol. 2010, 24, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Pedicone, C.; Fernandes, S.; Dungan, O.M.; Dormann, S.M.; Viernes, D.R.; Adhikari, A.A.; Choi, L.B.; De Jong, E.P.; Chisholm, J.D.; Kerr, W.G. Pan-SHIP1/2 inhibitors promote microglia effector functions essential for CNS homeostasis. J. Cell Sci. 2020, 133, jcs238030. [Google Scholar] [CrossRef] [PubMed]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Abeta Plaques. Cell Rep. 2019, 27, 1293–1306 e1296. [Google Scholar] [CrossRef] [PubMed]
- Pedicone, C.; Fernandes, S.; Matera, A.; Meyer, S.T.; Loh, S.; Ha, J.H.; Bernard, D.; Chisholm, J.D.; Paolicelli, R.C.; Kerr, W.G. Discovery of a novel SHIP1 agonist that promotes degradation of lipid-laden phagocytic cargo by microglia. iScience 2022, 25, 104170. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| INPP5D/SHIP1 Deficiency | Effect | Reference |
|---|---|---|
| INPP5D haploinsufficiency (Tyrobp-deficient TREM2 loss-of-function mouse) | Restored microglial association with plaques, partially restored plaque compaction and reduced phosphorylated tau(+) dystrophic neurites | [55] |
| INPP5D haploinsufficiency | Increased dense-core plaques, microglial association with plaques and uptake of Aβ | [56] |
| INPP5D downregulation | Increased microglial association with plaques, but increased plaque burden | [57] |
| SHIP1—transducing inhibitory signals from FcγRIIB and ITIM-containing proteins | Inhibitory effects on monocyte and microglia activation | [59] |
| SHIP1—NF-κB pathway | SHIP1 downregulation leads to increased NF-κB activation, and an increase in BACE1 expression in AD brain | [62,63,64,65,66,67,68,69] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olufunmilayo, E.O.; Holsinger, R.M.D. INPP5D/SHIP1: Expression, Regulation and Roles in Alzheimer’s Disease Pathophysiology. Genes 2023, 14, 1845. https://doi.org/10.3390/genes14101845
Olufunmilayo EO, Holsinger RMD. INPP5D/SHIP1: Expression, Regulation and Roles in Alzheimer’s Disease Pathophysiology. Genes. 2023; 14(10):1845. https://doi.org/10.3390/genes14101845
Chicago/Turabian StyleOlufunmilayo, Edward O., and R. M. Damian Holsinger. 2023. "INPP5D/SHIP1: Expression, Regulation and Roles in Alzheimer’s Disease Pathophysiology" Genes 14, no. 10: 1845. https://doi.org/10.3390/genes14101845
APA StyleOlufunmilayo, E. O., & Holsinger, R. M. D. (2023). INPP5D/SHIP1: Expression, Regulation and Roles in Alzheimer’s Disease Pathophysiology. Genes, 14(10), 1845. https://doi.org/10.3390/genes14101845