
The Role of IgLON Cell Adhesion Molecules in Neurodegenerative Diseases

 , ,
, , 
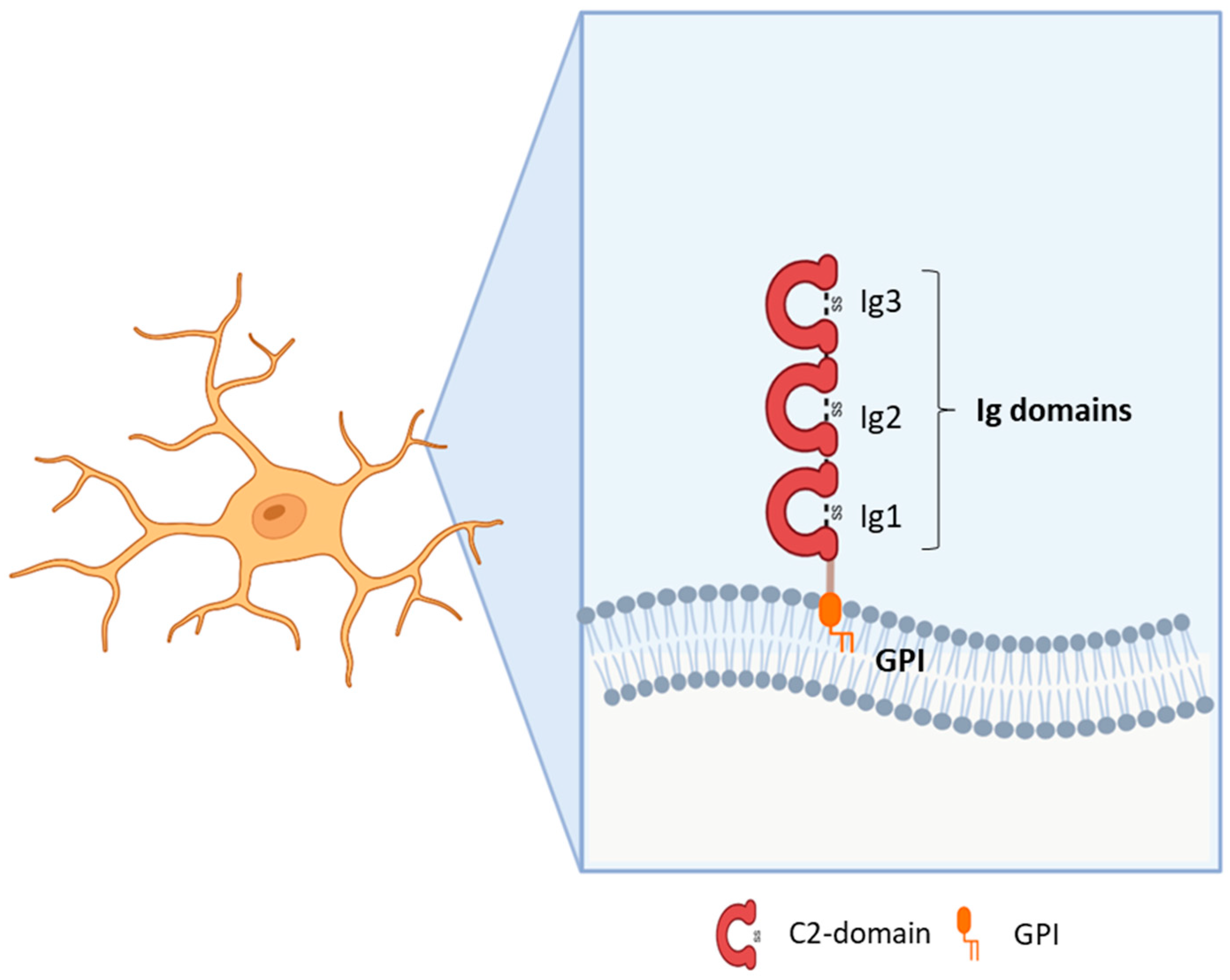{kind=link}
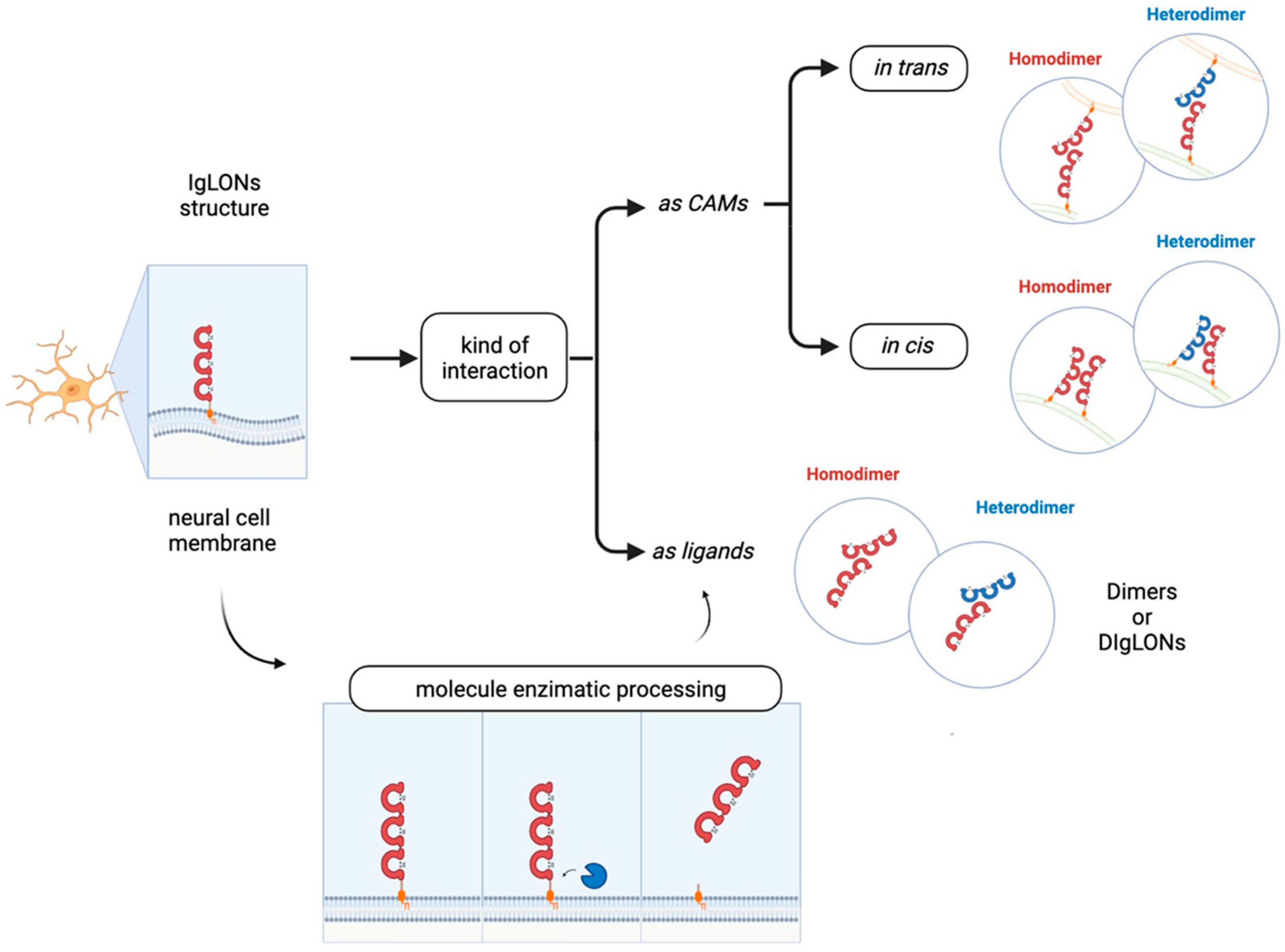{kind=link}
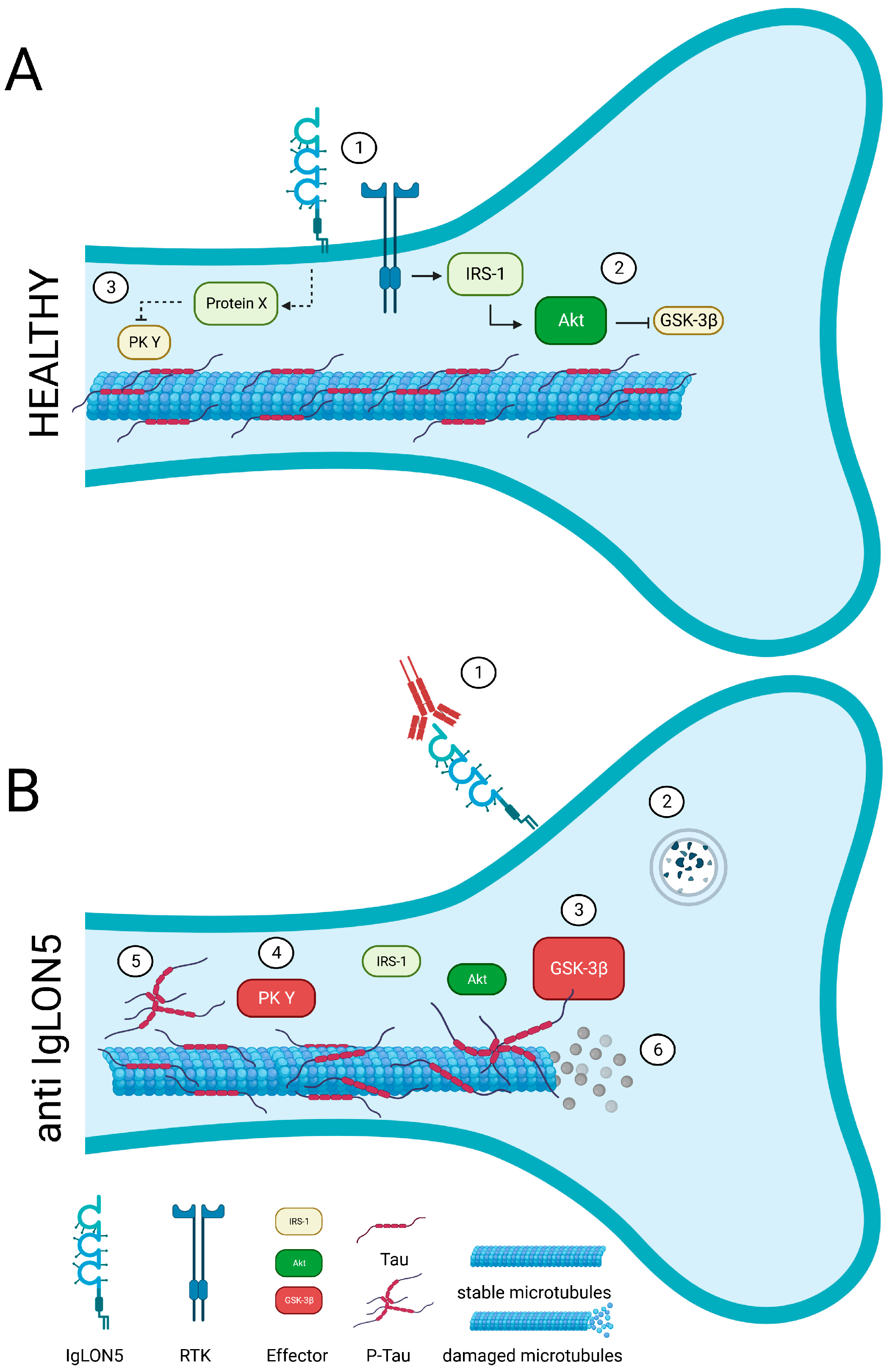{kind=link}
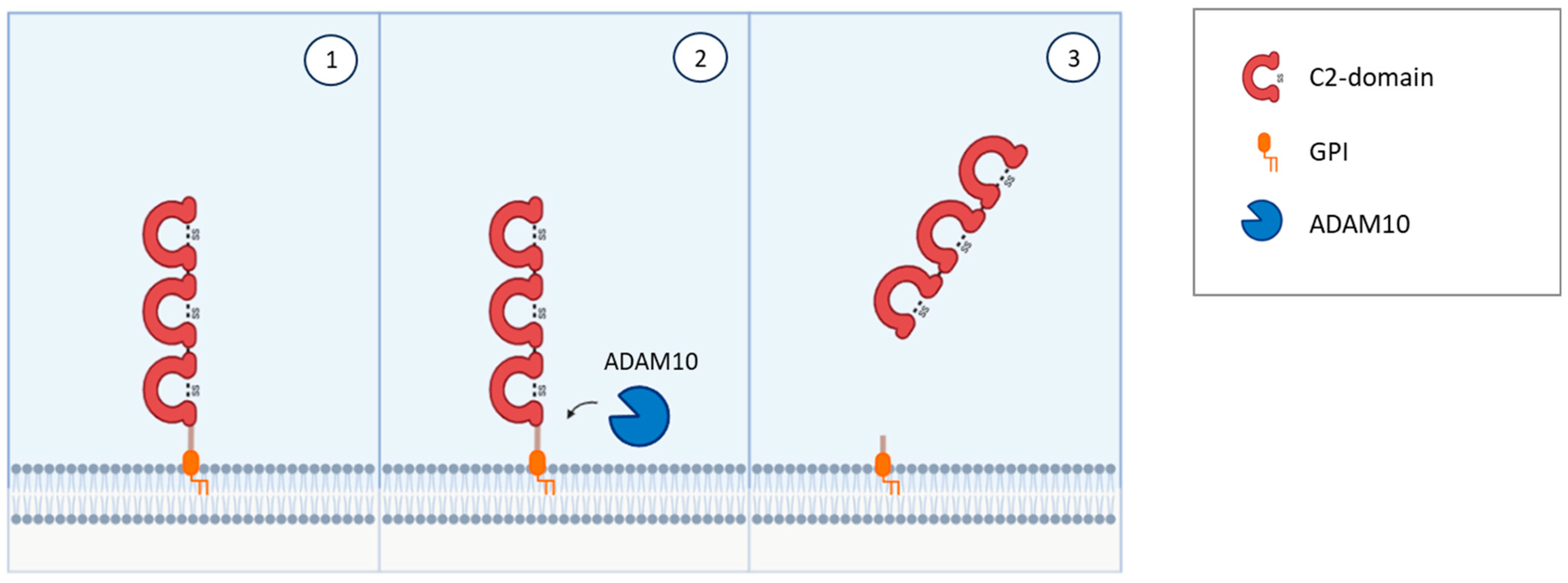{kind=link}
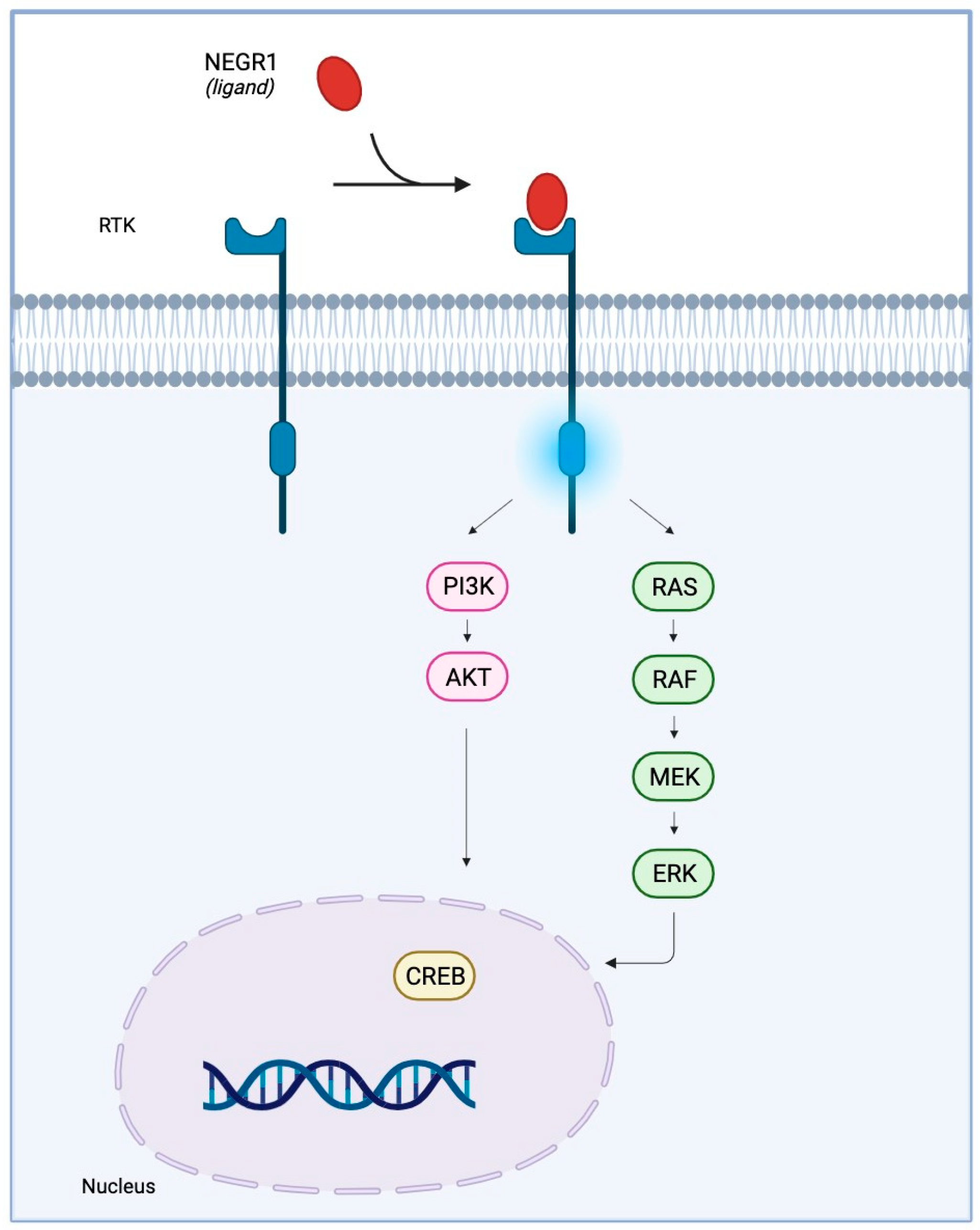{kind=link}
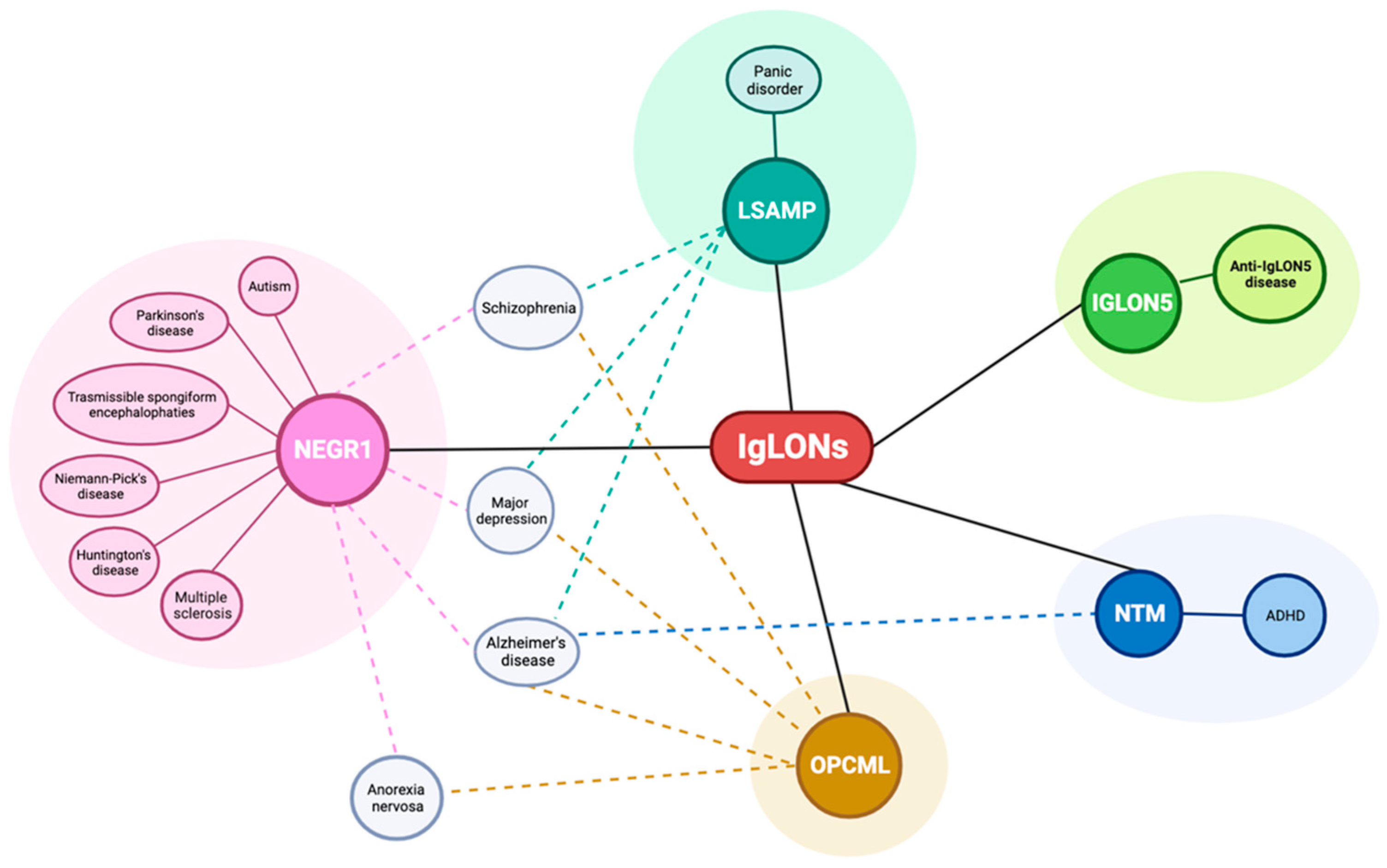{kind=link}
Abstract
:1. Introduction
2. IgLON5 (IgLON Family Member 5)
3. NEGR1 (Neuronal Growth Regulator 1)
4. OPCML (Opioid Binding Protein/Cell Adhesion Molecule Like)
5. LSAMP (Limbic System Associated Membrane Protein)
6. NTM (Neurotrimin)
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shapiro, L.; Love, J.; Colman, D.R. Adhesion Molecules in the Nervous System: Structural Insights into Function and Diversity. Annu. Rev. Neurosci. 2007, 30, 451–474. [Google Scholar] [CrossRef] [PubMed]
- Stachowicz, K. Physicochemical Principles of Adhesion Mechanisms in the Brain. Int. J. Mol. Sci. 2023, 24, 5070. [Google Scholar] [CrossRef] [PubMed]
- Kadry, Y.A.; Calderwood, D.A. Chapter 22: Structural and signaling functions of integrins. Biochim. Biophys. Acta—Biomembr. 2020, 1862, 183206. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [PubMed]
- McEver, R.P.; Zhu, C. Rolling cell adhesion. Annu. Rev. Cell Dev. Biol. 2010, 26, 363–396. [Google Scholar] [CrossRef]
- Ley, K.; Kansas, G.S. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat. Rev. Immunol. 2004, 4, 325–335. [Google Scholar] [CrossRef]
- Siew, J.J.; Chern, Y. Microglial Lectins in Health and Neurological Diseases. Front. Mol. Neurosci. 2018, 11, 158. [Google Scholar] [CrossRef]
- Angiari, S. Selectin-mediated leukocyte trafficking during the development of autoimmune disease. Autoimmun. Rev. 2015, 14, 984–995. [Google Scholar] [CrossRef]
- Redies, C. Cadherins in the central nervous system. Prog. Neurobiol. 2000, 61, 611–648. [Google Scholar] [CrossRef]
- Takeichi, M. The cadherin superfamily in neuronal connections and interactions. Nat. Rev. Neurosci. 2007, 8, 11–20. [Google Scholar] [CrossRef]
- Cameron, S.; McAllister, A.K. Immunoglobulin-Like Receptors and Their Impact on Wiring of Brain Synapses. Annu. Rev. Genet. 2018, 52, 567–590. [Google Scholar] [CrossRef] [PubMed]
- Barclay, A.N. Membrane proteins with immunoglobulin-like domains—A master superfamily of interaction molecules. Semin. Immunol. 2003, 15, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Gelfand, I.; Kister, A. Structural determinants in the sequences of immunoglobulin variable domain. J. Mol. Biol. 1998, 278, 457–479. [Google Scholar] [CrossRef]
- Walsh, F.S.; Doherty, P. Neural cell adhesion molecules of the immunoglobulin superfamily: Role in axon growth and guidance. Annu. Rev. Cell Dev. Biol. 1997, 13, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T. The role of cell adhesion molecules in brain wiring and neuropsychiatric disorders. Mol. Cell. Neurosci. 2017, 81, 4–11. [Google Scholar] [CrossRef]
- Zinn, K.; Özkan, E. Neural immunoglobulin superfamily interaction networks. Curr. Opin. Neurobiol. 2017, 45, 99–105. [Google Scholar] [CrossRef]
- Tan, R.P.A.; Leshchyns’ka, I.; Sytnyk, V. Glycosylphosphatidylinositol-Anchored Immunoglobulin Superfamily Cell Adhesion Molecules and Their Role in Neuronal Development and Synapse Regulation. Front. Mol. Neurosci. 2017, 10, 378. [Google Scholar] [CrossRef]
- Leshchyns’ka, I.; Sytnyk, V. Reciprocal Interactions between Cell Adhesion Molecules of the Immunoglobulin Superfamily and the Cytoskeleton in Neurons. Front. Cell Dev. Biol. 2016, 4, 9. [Google Scholar] [CrossRef]
- Maness, P.F.; Schachner, M. Neural recognition molecules of the immunoglobulin superfamily: Signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 2007, 10, 19–26. [Google Scholar] [CrossRef]
- Karagogeos, D. Neural GPI-anchored cell adhesion molecules. Front. Biosci. 2003, 8, s1304–s1320. [Google Scholar] [CrossRef]
- Levitt, P. A monoclonal antibody to limbic system neurons. Science 1984, 223, 299–301. [Google Scholar] [CrossRef]
- Schofield, P.R.; McFarland, K.C.; Hayflick, J.S.; Wilcox, J.N.; Cho, T.M.; Roy, S.; Lee, N.M.; Loh, H.H.; Seeburg, P.H. Molecular characterization of a new immunoglobulin superfamily protein with potential roles in opioid binding and cell contact. EMBO J. 1989, 8, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Struyk, A.F.; Canoll, P.D.; Wolfgang, M.J.; Rosen, C.L.; D’Eustachio, P.; Salzer, J.L. Cloning of neurotrimin defines a new subfamily of differentially expressed neural cell adhesion molecules. J. Neurosci. 1995, 15, 2141–2156. [Google Scholar] [CrossRef]
- Funatsu, N.; Miyata, S.; Kumanogoh, H.; Shigeta, M.; Hamada, K.; Endo, Y.; Sokawa, Y.; Maekawa, S. Characterization of a novel rat brain glycosylphosphatidylinositol- anchored protein (Kilon), a member of the IgLON cell adhesion molecule family. J. Biol. Chem. 1999, 274, 8224–8230. [Google Scholar] [CrossRef]
- Marg, A.; Sirim, P.; Spaltmann, F.; Plagge, A.; Kauselmann, G.; Buck, F.; Rathjen, F.G.; Brümmendorf, T. Neurotractin, a novel neurite outgrowth-promoting Ig-like protein that interacts with CEPU-1 and LAMP. J. Cell Biol. 1999, 145, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Sabater, L.; Gaig, C.; Gelpi, E.; Bataller, L.; Lewerenz, J.; Torres-Vega, E.; Contreras, A.; Giometto, B.; Compta, Y.; Embid, C.; et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: A case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014, 13, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Grønborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brügger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Pischedda, F.; Piccoli, G. The IgLON Family Member Negr1 Promotes Neuronal Arborization Acting as Soluble Factor via FGFR2. Front. Mol. Neurosci. 2016, 8, 89. [Google Scholar] [CrossRef]
- Szczurkowska, J.; Pischedda, F.; Pinto, B.; Managò, F.; Haas, C.A.; Summa, M.; Bertorelli, R.; Papaleo, F.; Schäfer, M.K.; Piccoli, G.; et al. NEGR1 and FGFR2 cooperatively regulate cortical development and core behaviours related to autism disorders in mice. Brain 2018, 141, 2772–2794. [Google Scholar] [CrossRef]
- Fearnley, S.; Raja, R.; Cloutier, J.F. Spatiotemporal expression of IgLON family members in the developing mouse nervous system. Sci. Rep. 2021, 11, 19536. [Google Scholar] [CrossRef]
- Kubick, N.; Brösamle, D.; Mickael, M.E. Molecular Evolution and Functional Divergence of the IgLON Family. Evol. Bioinforma. 2018, 14, 1176934318775081. [Google Scholar] [CrossRef] [PubMed]
- Leshchyns’ka, I.; Sytnyk, V. Synaptic Cell Adhesion Molecules in Alzheimer’s Disease. Neural Plast. 2016, 2016, 6427537. [Google Scholar] [CrossRef] [PubMed]
- Wennström, M.; Nielsen, H.M. Cell adhesion molecules in Alzheimer’s disease. Degener. Neurol. Neuromuscul. Dis. 2012, 2, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Leshchyns’ka, I.; Liew, H.T.; Shepherd, C.; Halliday, G.M.; Stevens, C.H.; Ke, Y.D.; Ittner, L.M.; Sytnyk, V. Aβ-dependent reduction of NCAM2-mediated synaptic adhesion contributes to synapse loss in Alzheimer’s disease. Nat. Commun. 2015, 6, 8836. [Google Scholar] [CrossRef]
- Tang, X.; Tena, J.; Di Lucente, J.; Maezawa, I.; Harvey, D.J.; Jin, L.-W.; Lebrilla, C.B.; Zivkovic, A.M. Transcriptomic and glycomic analyses highlight pathway-specific glycosylation alterations unique to Alzheimer’s disease. Sci. Rep. 2023, 13, 7816. [Google Scholar] [CrossRef]
- Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Neural Cell Adhesion Molecules of the Immunoglobulin Superfamily Regulate Synapse Formation, Maintenance, and Function. Trends Neurosci. 2017, 40, 295–308. [Google Scholar] [CrossRef]
- Gaig, C.; Graus, F.; Compta, Y.; Högl, B.; Bataller, L.; Brüggemann, N.; Giordana, C.; Heidbreder, A.; Kotschet, K.; Lewerenz, J.; et al. Clinical manifestations of the anti-IgLON5 disease. Neurology 2017, 88, 1736–1743. [Google Scholar] [CrossRef]
- Nissen, M.S.; Blaabjerg, M. Anti-IgLON5 Disease: A Case With 11-Year Clinical Course and Review of the Literature. Front. Neurol. 2019, 10, 1056. [Google Scholar] [CrossRef]
- Werner, J.; Jelcic, I.; Schwarz, E.I.; Probst-Müller, E.; Nilsson, J.; Schwizer, B.; Bloch, K.E.; Lutterotti, A.; Jung, H.-H.; Schreiner, B. Anti-IgLON5 Disease: A New Bulbar-Onset Motor Neuron Mimic Syndrome. Neurol.-Neuroimmunol. Neuroinflamm. 2021, 8, e962. [Google Scholar] [CrossRef]
- Gelpi, E.; Höftberger, R.; Graus, F.; Ling, H.; Holton, J.L.; Dawson, T.; Popovic, M.; Pretnar-Oblak, J.; Högl, B.; Schmutzhard, E.; et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol. 2016, 132, 531–543. [Google Scholar] [CrossRef]
- Ranaivoson, F.M.; Turk, L.S.; Ozgul, S.; Kakehi, S.; von Daake, S.; Lopez, N.; Trobiani, L.; De Jaco, A.; Denissova, N.; Demeler, B.; et al. A Proteomic Screen of Neuronal Cell-Surface Molecules Reveals IgLONs as Structurally Conserved Interaction Modules at the Synapse. Structure 2019, 27, 893–906.e9. [Google Scholar] [CrossRef] [PubMed]
- Sabater, L.; Planagumà, J.; Dalmau, J.; Graus, F. Cellular investigations with human antibodies associated with the anti-IgLON5 syndrome. J. Neuroinflamm. 2016, 13, 226. [Google Scholar] [CrossRef] [PubMed]
- Landa, J.; Gaig, C.; Plagumà, J.; Saiz, A.; Antonell, A.; Sanchez-Valle, R.; Dalmau, J.; Graus, F.; Sabater, L. Effects of IgLON5 Antibodies on Neuronal Cytoskeleton: A Link between Autoimmunity and Neurodegeneration. Ann. Neurol. 2020, 88, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Ryding, M.; Gamre, M.; Nissen, M.S.; Nilsson, A.C.; Okarmus, J.; Poulsen, A.A.E.; Meyer, M.; Blaabjerg, M. Neurodegeneration Induced by Anti-IgLON5 Antibodies Studied in Induced Pluripotent Stem Cell-Derived Human Neurons. Cells 2021, 10, 837. [Google Scholar] [CrossRef] [PubMed]
- Honorat, J.A.; Komorowski, L.; Josephs, K.A.; Fechner, K.; St Louis, E.K.; Hinson, S.R.; Lederer, S.; Kumar, N.; Gadoth, A.; Lennon, V.A.; et al. IgLON5 antibody: Neurological accompaniments and outcomes in 20 patients. Neurol.-Neuroimmunol. Neuroinflamm. 2017, 4, e385. [Google Scholar] [CrossRef] [PubMed]
- Erro, M.E.; Sabater, L.; Martínez, L.; Herrera, M.; Ostolaza, A.; García de Gurtubay, I.; Tuñón, T.; Graus, F.; Gelpi, E. Anti-IGLON5 disease: A new case without neuropathologic evidence of brainstem tauopathy. Neurol.-Neuroimmunol. Neuroinflamm. 2020, 7, e651. [Google Scholar] [CrossRef]
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol. Neurodegener. 2021, 16, 37. [Google Scholar] [CrossRef]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), S141–S144. [Google Scholar] [CrossRef]
- Kanno, T.; Tsuchiya, A.; Tanaka, A.; Nishizaki, T. Combination of PKCε Activation and PTP1B Inhibition Effectively Suppresses Aβ-Induced GSK-3β Activation and Tau Phosphorylation. Mol. Neurobiol. 2016, 53, 4787–4797. [Google Scholar] [CrossRef]
- Landa, J.; Serafim, A.B.; Gaig, C.; Saiz, A.; Koneczny, I.; Hoftberger, R.; Santamaria, J.; Dalmau, J.; Graus, F.; Sabater, L. Patients’ IgLON5 autoantibodies interfere with IgLON5-protein interactions. Front. Immunol. 2023, 14, 1151574. [Google Scholar] [CrossRef]
- Itoh, S.; Hachisuka, A.; Kawasaki, N.; Hashii, N.; Teshima, R.; Hayakawa, T.; Kawanishi, T.; Yamaguchi, T. Glycosylation analysis of IgLON family proteins in rat brain by liquid chromatography and multiple-stage mass spectrometry. Biochemistry 2008, 47, 10132–10154. [Google Scholar] [CrossRef] [PubMed]
- Sim, G.; Jeong, M.; Seo, H.; Kim, J.; Lee, S. The Role of N-Glycosylation in the Intracellular Trafficking and Functionality of Neuronal Growth Regulator 1. Cells 2022, 11, 1242. [Google Scholar] [CrossRef] [PubMed]
- Venkannagari, H.; Kasper, J.M.; Misra, A.; Rush, S.A.; Fan, S.; Lee, H.; Sun, H.; Seshadrinathan, S.; Machius, M.; Hommel, J.D.; et al. Highly Conserved Molecular Features in IgLONs Contrast Their Distinct Structural and Biological Outcomes. J. Mol. Biol. 2020, 432, 5287–5303. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Funatsu, N.; Matsunaga, W.; Kiyohara, T.; Sokawa, Y.; Maekawa, S. Expression of the IgLON cell adhesion molecules Kilon and OBCAM in hypothalamic magnocellular neurons. J. Comp. Neurol. 2000, 424, 74–85. [Google Scholar] [CrossRef]
- Miyata, S.; Matsumoto, N.; Taguchi, K.; Akagi, A.; Iino, T.; Funatsu, N.; Maekawa, S. Biochemical and ultrastructural analyses of IgLON cell adhesion molecules, Kilon and OBCAM in the rat brain. Neuroscience 2003, 117, 645–658. [Google Scholar] [CrossRef]
- Bräuer, A.U.; Savaskan, N.E.; Plaschke, M.; Prehn, S.; Ninnemann, O.; Nitsch, R. IG-molecule kilon shows differential expression pattern from LAMP in the developing and adult rat hippocampus. Hippocampus 2000, 10, 632–644. [Google Scholar] [CrossRef]
- Vanaveski, T.; Singh, K.; Narvik, J.; Eskla, K.-L.L.; Visnapuu, T.; Heinla, I.; Jayaram, M.; Innos, J.; Lilleväli, K.; Philips, M.-A.A.; et al. Promoter-Specific Expression and Genomic Structure of IgLON Family Genes in Mouse. Front. Neurosci. 2017, 11, 38. [Google Scholar] [CrossRef]
- Schäfer, M.; Bräuer, A.U.; Savaskan, N.E.; Rathjen, F.G.; Brümmendorf, T. Neurotractin/kilon promotes neurite outgrowth and is expressed on reactive astrocytes after entorhinal cortex lesion. Mol. Cell. Neurosci. 2005, 29, 580–590. [Google Scholar] [CrossRef]
- Singh, K.; Loreth, D.; Pöttker, B.; Hefti, K.; Innos, J.; Schwald, K.; Hengstler, H.; Menzel, L.; Sommer, C.J.; Radyushkin, K.; et al. Neuronal growth and behavioral alterations in mice deficient for the psychiatric disease-associated negr1 gene. Front. Mol. Neurosci. 2018, 11, 30. [Google Scholar] [CrossRef]
- Sanz, R.; Ferraro, G.B.; Fournier, A.E. IgLON Cell Adhesion Molecules Are Shed from the Cell Surface of Cortical Neurons to Promote Neuronal Growth. J. Biol. Chem. 2015, 290, 4330–4342. [Google Scholar] [CrossRef]
- Hashimoto, T.; Yamada, M.; Maekawa, S.; Nakashima, T.; Miyata, S. IgLON cell adhesion molecule Kilon is a crucial modulator for synapse number in hippocampal neurons. Brain Res. 2008, 1224, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pischedda, F.; Szczurkowska, J.; Cirnaru, M.D.; Giesert, F.; Vezzoli, E.; Ueffing, M.; Sala, C.; Francolini, M.; Hauck, S.M.; Cancedda, L.; et al. A cell surface biotinylation assay to reveal membrane-associated neuronal cues: Negr1 regulates dendritic arborization. Mol. Cell. Proteom. 2014, 13, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.; Lee, H.; Choi, T.-Y.; Joo, Y.; Kim, S.-J.; Kim, H.; Kim, J.Y.; Jahng, J.W.; Lee, S.; Choi, S.-Y.; et al. Negr1 controls adult hippocampal neurogenesis and affective behaviors. Mol. Psychiatry 2019, 24, 1189–1205. [Google Scholar] [CrossRef] [PubMed]
- Kaare, M.; Jayaram, M.; Jagomäe, T.; Singh, K.; Kilk, K.; Mikheim, K.; Leevik, M.; Leidmaa, E.; Varul, J.; Nõmm, H.; et al. Depression-Associated Negr1 Gene-Deficiency Induces Alterations in the Monoaminergic Neurotransmission Enhancing Time-Dependent Sensitization to Amphetamine in Male Mice. Brain Sci. 2022, 12, 1696. [Google Scholar] [CrossRef]
- Willer, C.; Speliotes, E.; Loos, R.; Li, S.; Lindgren, C.; Heid, I.; Berndt, S.; Elliott, A.; Jackson, A.; Lamina, C.; et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 2009, 41, 25–34. [Google Scholar] [CrossRef]
- Renström, F.; Payne, F.; Nordström, A.; Brito, E.C.; Rolandsson, O.; Hallmans, G.; Barroso, I.; Nordström, P.; Franks, P.W. Replication and extension of genome-wide association study results for obesity in 4923 adults from northern Sweden. Hum. Mol. Genet. 2009, 18, 1489–1496. [Google Scholar] [CrossRef]
- Zhao, J.; Bradfield, J.P.; Li, M.; Wang, K.; Zhang, H.; Kim, C.E.; Annaiah, K.; Glessner, J.T.; Thomas, K.; Garris, M.; et al. The role of obesity-associated loci identified in genome-wide association studies in the determination of pediatric BMI. Obesity 2009, 17, 2254–2257. [Google Scholar] [CrossRef]
- Schmid, P.M.; Heid, I.; Buechler, C.; Steege, A.; Resch, M.; Birner, C.; Endemann, D.H.; Riegger, G.A.; Luchner, A. Expression of fourteen novel obesity-related genes in zucker diabetic fatty rats. Cardiovasc. Diabetol. 2012, 11, 48. [Google Scholar] [CrossRef]
- Boender, A.J.; Van Rozen, A.J.; Adan, R.A.H. Nutritional state affects the expression of the obesity-associated genes Etv5, faim2, Fto, and negr1. Obesity 2012, 20, 2420–2425. [Google Scholar] [CrossRef]
- Boender, A.J.; van Gestel, M.A.; Garnerv, K.M.; Luijendijk, M.C.M.; Adan, R.A.H. The Obesity-Associated gene NEGR1 regulates aspects of energy balance in rat hypothalamic areas. Physiol. Rep. 2014, 2, e12083. [Google Scholar] [CrossRef]
- Lee, A.W.S.; Hengstler, H.; Schwald, K.; Berriel-Diaz, M.; Loreth, D.; Kirsch, M.; Kretz, O.; Haas, C.A.; de Angelis, M.H.; Herzig, S.; et al. Functional inactivation of the genome-wide association study obesity gene neuronal growth regulator 1 in mice causes a body mass phenotype. PLoS ONE 2012, 7, e41537. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Ouyang, M.; Wang, J.; Xie, M.; Huang, Y.; Yuan, F.; Jia, Y.; Zhang, X.; Liu, N.; Zhang, N. Shared genetics between classes of obesity and psychiatric disorders: A large-scale genome-wide cross-trait analysis. J. Psychosom. Res. 2022, 162, 111032. [Google Scholar] [CrossRef] [PubMed]
- Hyde, C.L.; Nagle, M.W.; Tian, C.; Chen, X.; Paciga, S.A.; Wendland, J.R.; Tung, J.Y.; Hinds, D.A.; Perlis, R.H.; Winslow, A.R. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 2016, 48, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef]
- Howard, D.M.; Adams, M.J.; Clarke, T.-K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef]
- Levey, D.F.; Stein, M.B.; Wendt, F.R.; Pathak, G.A.; Zhou, H.; Aslan, M.; Quaden, R.; Harrington, K.M.; Nuñez, Y.Z.; Overstreet, C.; et al. Bi-ancestral depression GWAS in the Million Veteran Program and meta-analysis in >1.2 million individuals highlight new therapeutic directions. Nat. Neurosci. 2021, 24, 954–963. [Google Scholar] [CrossRef]
- Dall’Aglio, L.; Lewis, C.M.; Pain, O. Delineating the Genetic Component of Gene Expression in Major Depression. Biol. Psychiatry 2021, 89, 627–636. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, W.; Zhu, J.; Yin, H.; Chang, S.; Yue, W.; Yu, H. Integrating genome-wide association study and expression quantitative trait loci data identifies NEGR1 as a causal risk gene of major depression disorder. J. Affect. Disord. 2020, 265, 679–686. [Google Scholar] [CrossRef]
- Deng, Y.T.; Ou, Y.N.; Wu, B.S.; Yang, Y.X.; Jiang, Y.; Huang, Y.Y.; Liu, Y.; Tan, L.; Dong, Q.; Suckling, J.; et al. Identifying causal genes for depression via integration of the proteome and transcriptome from brain and blood. Mol. Psychiatry 2022, 27, 2849–2857. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Li, X.; Liu, J.; Huo, Y.; Wang, J.; Liu, Z.; Li, M.; Luo, X.-J. Regulatory mechanisms of major depressive disorder risk variants. Mol. Psychiatry 2020, 25, 1926–1945. [Google Scholar] [CrossRef]
- Maccarrone, G.; Ditzen, C.; Yassouridis, A.; Rewerts, C.; Uhr, M.; Uhlen, M.; Holsboer, F.; Turck, C.W. Psychiatric patient stratification using biosignatures based on cerebrospinal fluid protein expression clusters. J. Psychiatr. Res. 2013, 47, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Tamási, V.; Petschner, P.; Adori, C.; Kirilly, E.; Ando, R.D.; Tothfalusi, L.; Juhasz, G.; Bagdy, G. Transcriptional evidence for the role of chronic venlafaxine treatment in neurotrophic signaling and neuroplasticity including also glutatmatergic- and insulin-mediated neuronal processes. PLoS ONE 2014, 9, e113662. [Google Scholar] [CrossRef] [PubMed]
- Carboni, L.; Pischedda, F.; Piccoli, G.; Lauria, M.; Musazzi, L.; Popoli, M.; Mathé, A.A.; Domenici, E. Depression-Associated Gene Negr1-Fgfr2 Pathway Is Altered by Antidepressant Treatment. Cells 2020, 9, 1818. [Google Scholar] [CrossRef] [PubMed]
- Amare, A.T.; Schubert, K.O.; Tekola-Ayele, F.; Hsu, Y.H.; Sangkuhl, K.; Jenkins, G.; Whaley, R.M.; Barman, P.; Batzler, A.; Altman, R.B.; et al. The association of obesity and coronary artery disease genes with response to SSRIs treatment in major depression. J. Neural Transm. 2019, 126, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Karis, K.; Eskla, K.L.; Kaare, M.; Täht, K.; Tuusov, J.; Visnapuu, T.; Innos, J.; Jayaram, M.; Timmusk, T.; Weickert, C.S.; et al. Altered expression profile of igLON family of neural cell adhesion molecules in the dorsolateral prefrontal cortex of schizophrenic patients. Front. Mol. Neurosci. 2018, 11, 8. [Google Scholar] [CrossRef]
- Cox, D.A.; Gottschalk, M.G.; Wesseling, H.; Ernst, A.; Cooper, J.D.; Bahn, S. Proteomic systems evaluation of the molecular validity of preclinical psychosis models compared to schizophrenia brain pathology. Schizophr. Res. 2016, 177, 98–107. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, G. A logical relationship for schizophrenia, bipolar, and major depressive disorder. Part 1: Evidence from chromosome 1 high density association screen. J. Comp. Neurol. 2020, 528, 2620–2635. [Google Scholar] [CrossRef]
- Steiger, H.; Booij, L.; Thaler, L.; St-Hilaire, A.; Israël, M.; Casey, K.F.; Oliverio, S.; Crescenzi, O.; Lee, V.; Turecki, G.; et al. DNA methylation in people with anorexia nervosa: Epigenome-wide patterns in actively ill, long-term remitted, and healthy-eater women. World J. Biol. Psychiatry 2023, 24, 254–259. [Google Scholar] [CrossRef]
- Ni, H.; Xu, M.; Zhan, G.-L.; Fan, Y.; Zhou, H.; Jiang, H.-Y.; Lu, W.-H.; Tan, L.; Zhang, D.-F.; Yao, Y.-G.; et al. The GWAS Risk Genes for Depression May Be Actively Involved in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 1149–1161. [Google Scholar] [CrossRef]
- Raghavan, N.S.; Vardarajan, B.; Mayeux, R. Genomic variation in educational attainment modifies Alzheimer disease risk. Neurol. Genet. 2019, 5, e310. [Google Scholar] [CrossRef]
- Harrison, J.R.; Bhatia, S.; Tan, Z.X.; Mirza-Davies, A.; Benkert, H.; Tax, C.M.W.; Jones, D.K. Imaging Alzheimer’s genetic risk using diffusion MRI: A systematic review. NeuroImage Clin. 2020, 27, 102359. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Pan, P.L.; Song, W.; Huang, R.; Chen, K.; Shang, H.F. A meta-analysis of diffusion tensor imaging studies in amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 1833–1838. [Google Scholar] [CrossRef]
- Ciccarelli, O.; Catani, M.; Johansen-Berg, H.; Clark, C.; Thompson, A. Diffusion-based tractography in neurological disorders: Concepts, applications, and future developments. Lancet Neurol. 2008, 7, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Lachén-Montes, M.; González-Morales, A.; Fernández-Irigoyen, J.; Santamaría, E. Deployment of Label-Free Quantitative Olfactory Proteomics to Detect Cerebrospinal Fluid Biomarker Candidates in Synucleinopathies. Methods Mol. Biol. 2019, 2044, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Abdi, I.Y.; Bartl, M.; Dakna, M.; Abdesselem, H.; Majbour, N.; Trenkwalder, C.; El-Agnaf, O.; Mollenhauer, B. Cross-sectional proteomic expression in Parkinson’s disease-related proteins in drug-naïve patients vs healthy controls with longitudinal clinical follow-up. Neurobiol. Dis. 2023, 177, 105997. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, L.; Simon, S.L.R.; Waitt, B.; Knox, J.D. Proteomic Screen of Brain Glycoproteome Reveals Prion Specific Marker of Pathogenesis. Proteomics 2018, 18, 1700296. [Google Scholar] [CrossRef]
- Kaltenbach, L.S.; Romero, E.; Becklin, R.R.; Chettier, R.; Bell, R.; Phansalkar, A.; Strand, A.; Torcassi, C.; Savage, J.; Hurlburt, A.; et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007, 3, 689–708. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chun, Y.; Che, L.; Kim, J.; Lee, S.; Lee, S. The new obesity-associated protein, neuronal growth regulator 1 (NEGR1), is implicated in Niemann-Pick disease Type C (NPC2)-mediated cholesterol trafficking. Biochem. Biophys. Res. Commun. 2017, 482, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural Variation of Chromosomes in Autism Spectrum Disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef]
- Veerappa, A.M.; Saldanha, M.; Padakannaya, P.; Ramachandra, N.B. Family-based genome-wide copy number scan identifies five new genes of dyslexia involved in dendritic spinal plasticity. J. Hum. Genet. 2013, 58, 539–547. [Google Scholar] [CrossRef]
- Genovese, A.; Cox, D.; Butler, M. Partial Deletion of Chromosome 1p31.1 Including only the Neuronal Growth Regulator 1 Gene in Two Siblings. J. Pediatr. Genet. 2015, 4, 023–028. [Google Scholar] [CrossRef]
- Biswal, S.; Parida, P.; Dubbudu, A.; Sharawat, I.K.; Panda, P.K. Chromosome 1p31.1 Deletion Syndrome: Limited Expression. Ann. Indian Acad. Neurol. 2021, 24, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Tassano, E.; Gamucci, A.; Celle, M.E.; Ronchetto, P.; Cuoco, C.; Gimelli, G. Clinical and Molecular Cytogenetic Characterization of a de novo Interstitial 1p31.1p31.3 Deletion in a Boy with Moderate Intellectual Disability and Severe Language Impairment. Cytogenet. Genome Res. 2015, 146, 39–43. [Google Scholar] [CrossRef]
- Katiyar, A.; Sharma, S.; Singh, T.P.; Kaur, P. Identification of shared molecular signatures indicate the susceptibility of endometriosis to multiple sclerosis. Front. Genet. 2018, 9, 42. [Google Scholar] [CrossRef]
- Brugger, S.W.; Gardner, M.C.; Beales, J.T.; Briggs, F.; Davis, M.F. Depression in multiple sclerosis patients associated with risk variant near NEGR1. Mult. Scler. Relat. Disord. 2020, 46, 102537. [Google Scholar] [CrossRef] [PubMed]
- Lippman, D.A.; Lee, N.M.; Loh, H.H. Opioid-binding cell adhesion molecule (OBCAM)-related clones from a rat brain cDNA library. Gene 1992, 117, 249–254. [Google Scholar] [CrossRef]
- Shark, K.B.; Lee, N.M. Cloning, sequencing and localization to chromosome 11 of a cDNA encoding a human opioid-binding cell adhesion molecule (OBCAM). Gene 1995, 155, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Ying, Y.; van Hasselt, A.; Ng, K.M.; Yu, J.; Zhang, Q.; Jin, J.; Liu, D.; Rhim, J.S.; Rha, S.Y.; et al. OPCML is a broad tumor suppressor for multiple carcinomas and lymphomas with frequently epigenetic inactivation. PLoS ONE 2008, 3, e2990. [Google Scholar] [CrossRef]
- Cho, T.M.; Hasegawa, J.; Ge, B.L.; Loh, H.H. Purification to apparent homogeneity of a mu-type opioid receptor from rat brain. Proc. Natl. Acad. Sci. USA 1986, 83, 4138–4142. [Google Scholar] [CrossRef]
- Hachisuka, A.; Yamazaki, T.; Sawada, J.; Terao, T. Characterization and tissue distribution of opioid-binding cell adhesion molecule (OBCAM) using monoclonal antibodies. Neurochem. Int. 1996, 28, 373–379. [Google Scholar] [CrossRef]
- Reed, J.; McNamee, C.; Rackstraw, S.; Jenkins, J.; Moss, D. Diglons are heterodimeric proteins composed of IgLON subunits, and Diglon-CO inhibits neurite outgrowth from cerebellar granule cells. J. Cell Sci. 2004, 117, 3961–3973. [Google Scholar] [CrossRef]
- Hachisuka, A.; Nakajima, O.; Yamazaki, T.; Sawada, J. Developmental expression of opioid-binding cell adhesion molecule (OBCAM) in rat brain. Dev. Brain Res. 2000, 122, 183–191. [Google Scholar] [CrossRef]
- Yamada, M.; Hashimoto, T.; Hayashi, N.; Higuchi, M.; Murakami, A.; Nakashima, T.; Maekawa, S.; Miyata, S. Synaptic adhesion molecule OBCAM; synaptogenesis and dynamic internalization. Brain Res. 2007, 1165, 5–14. [Google Scholar] [CrossRef]
- Athanasiu, L.; Mattingsdal, M.; Kähler, A.K.; Brown, A.; Gustafsson, O.; Agartz, I.; Giegling, I.; Muglia, P.; Cichon, S.; Rietschel, M.; et al. Gene variants associated with schizophrenia in a Norwegian genome-wide study are replicated in a large European cohort. J. Psychiatr. Res. 2010, 44, 748–753. [Google Scholar] [CrossRef]
- Panichareon, B.; Nakayama, K.; Thurakitwannakarn, W.; Iwamoto, S.; Sukhumsirichart, W. OPCML gene as a schizophrenia susceptibility locus in Thai population. J. Mol. Neurosci. 2012, 46, 373–377. [Google Scholar] [CrossRef]
- O’Donovan, M.C.; Craddock, N.; Norton, N.; Williams, H.; Peirce, T.; Moskvina, V.; Nikolov, I.; Hamshere, M.; Carroll, L.; Georgieva, L.; et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 2008, 40, 1053–1055. [Google Scholar] [CrossRef]
- Zhang, Z.; Ye, M.; Li, Q.; You, Y.; Yu, H.; Ma, Y.; Mei, L.; Sun, X.; Wang, L.; Yue, W.; et al. The Schizophrenia Susceptibility Gene OPCML Regulates Spine Maturation and Cognitive Behaviors through Eph-Cofilin Signaling. Cell Rep. 2019, 29, 49–61.e7. [Google Scholar] [CrossRef]
- Umeda-Yano, S.; Hashimoto, R.; Yamamori, H.; Weickert, C.S.; Yasuda, Y.; Ohi, K.; Fujimoto, M.; Ito, A.; Takeda, M. Expression analysis of the genes identified in GWAS of the postmortem brain tissues from patients with schizophrenia. Neurosci. Lett. 2014, 568, 12–16. [Google Scholar] [CrossRef]
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef]
- Schol-Gelok, S.; Janssens, A.C.J.W.; Tiemeier, H.; Liu, F.; Lopez-Leon, S.; Zorkoltseva, I.V.; Axenovich, T.I.; Van Swieten, J.C.; Uitterlinden, A.G.; Hofman, A.; et al. A genome-wide screen for depression in two independent dutch populations. Biol. Psychiatry 2010, 68, 187–196. [Google Scholar] [CrossRef]
- Huckins, L.M.; Hatzikotoulas, K.; Southam, L.; Thornton, L.M.; Steinberg, J.; Aguilera-McKay, F.; Treasure, J.; Schmidt, U.; Gunasinghe, C.; Romero, A.; et al. Investigation of common, low-frequency and rare genome-wide variation in anorexia nervosa. Mol. Psychiatry 2018, 23, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Minhas, H.M.; Pescosolido, M.F.; Schwede, M.; Piasecka, J.; Gaitanis, J.; Tantravahi, U.; Morrow, E.M. An unbalanced translocation involving loss of 10q26.2 and gain of 11q25 in a pedigree with autism spectrum disorder and cerebellar juvenile pilocytic astrocytoma. Am. J. Med. Genet. A 2013, 161A, 787–791. [Google Scholar] [CrossRef]
- Liu, F.; Arias-Vásquez, A.; Sleegers, K.; Aulchenko, Y.S.; Kayser, M.; Sanchez-Juan, P.; Feng, B.-J.J.; Bertoli-Avella, A.M.; van Swieten, J.; Axenovich, T.I.; et al. A genomewide screen for late-onset Alzheimer disease in a genetically isolated Dutch population. Am. J. Hum. Genet. 2007, 81, 17–31. [Google Scholar] [CrossRef]
- Weller, A.E.; Ferraro, T.N.; Doyle, G.A.; Reiner, B.C.; Crist, R.C.; Berrettini, W.H. Single Nucleus Transcriptome Data from Alzheimer’s Disease Mouse Models Yield New Insight into Pathophysiology. J. Alzheimer’s Dis. 2022, 90, 1233–1247. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, C.; Chin, L.-S.; Li, L. Integrative glycoproteomics reveals protein N-glycosylation aberrations and glycoproteomic network alterations in Alzheimer’s disease. Sci. Adv. 2020, 6, eabc5802. [Google Scholar] [CrossRef]
- Zacco, A.; Cooper, V.; Chantler, P.; Fisher-Hyland, S.; Horton, H.; Levitt, P. Isolation, biochemical characterization and ultrastructural analysis of the limbic system-associated membrane protein (LAMP), a protein expressed by neurons comprising functional neural circuits. J. Neurosci. 1990, 10, 73–90. [Google Scholar] [CrossRef]
- Pimenta, A.F.; Zhukareva, V.; Barbe, M.F.; Reinoso, B.S.; Grimley, C.; Henzel, W.; Fischer, I.; Levitt, P. The limbic system-associated membrane protein is an Ig superfamily member that mediates selective neuronal growth and axon targeting. Neuron 1995, 15, 287–297. [Google Scholar] [CrossRef]
- Pimenta, A.F.; Reinoso, B.S.; Levitt, P.; Pimenta, A.F.; Levitt, P. Expression of the mRNAs encoding the limbic system-associated membrane protein (LAMP): II. Fetal rat brain. J. Comp. Neurol. 1996, 375, 274–288. [Google Scholar] [CrossRef]
- Zhukareva, V.; Levitt, P. The limbic system-associated membrane protein (LAMP) selectively mediates interactions with specific central neuron populations. Development 1995, 121, 1161–1172. [Google Scholar] [CrossRef]
- Keller, F.; Levitt, P. Developmental and regeneration-associated regulation of the limbic system associated membrane protein in explant cultures of the rat brain. Neuroscience 1989, 28, 455–474. [Google Scholar] [CrossRef]
- Horton, H.; Levitt, P. A unique membrane protein is expressed on early developing limbic system axons and cortical targets. J. Neurosci. 1988, 8, 4653–4661. [Google Scholar] [CrossRef] [PubMed]
- Barbe, M.F.; Levitt, P. The early commitment of fetal neurons to the limbic cortex. J. Neurosci. 1991, 11, 519–533. [Google Scholar] [CrossRef]
- Barbe, M.F.; Levitt, P. Attraction of specific thalamic input by cerebral grafts depends on the molecular identity of the implant. Proc. Natl. Acad. Sci. USA 1992, 89, 3706–3710. [Google Scholar] [CrossRef]
- Ferri, R.T.; Levitt, P. Cerebral Cortical Progenitors Are Fated to Produce Region-specific Neuronal Populations. Cereb. Cortex 1993, 3, 187–198. [Google Scholar] [CrossRef]
- Mann, F.; Zhukareva, V.; Pimenta, A.; Levitt, P.; Bolz, J. Membrane-Associated Molecules Guide Limbic and Nonlimbic Thalamocortical Projections. J. Neurosci. 1998, 18, 9409–9419. [Google Scholar] [CrossRef] [PubMed]
- Lodge, A.P.; Howard, M.R.; McNamee, C.J.; Moss, D.J. Co-localisation, heterophilic interactions and regulated expression of IgLON family proteins in the chick nervous system. Mol. Brain Res. 2000, 82, 84–94. [Google Scholar] [CrossRef]
- Gil, O.D.; Zhang, L.; Chen, S.; Ren, Y.Q.Q.; Pimenta, A.; Zanazzi, G.; Hillman, D.; Levitt, P.; Salzer, J.L. Complementary expression and heterophilic interactions between igLON family members neurotrimin and LAMP. J. Neurobiol. 2002, 51, 190–204. [Google Scholar] [CrossRef]
- McNamee, C.J.; Youssef, S.; Moss, D. IgLONs form heterodimeric complexes on forebrain neurons. Cell Biochem. Funct. 2011, 29, 114–119. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maekawa, S.; Miyata, S. IgLON cell adhesion molecules regulate synaptogenesis in hippocampal neurons. Cell Biochem. Funct. 2009, 27, 496–498. [Google Scholar] [CrossRef]
- Eagleson, K.L.; Pimenta, A.F.; Burns, M.M.; Fairfull, L.D.; Cornuet, P.K.; Zhang, L.; Levitt, P. Distinct domains of the limbic system-associated membrane protein (LAMP) mediate discrete effects on neurite outgrowth. Mol. Cell. Neurosci. 2003, 24, 725–740. [Google Scholar] [CrossRef]
- Sanz, R.L.; Ferraro, G.B.; Girouard, M.-P.P.; Fournier, A.E. Ectodomain shedding of Limbic System-Associated Membrane Protein (LSAMP) by ADAM Metallopeptidases promotes neurite outgrowth in DRG neurons. Sci. Rep. 2017, 7, 7961. [Google Scholar] [CrossRef] [PubMed]
- Nelovkov, A.; Philips, M.A.; Kõks, S.; Vasar, E. Rats with low exploratory activity in the elevated plus-maze have the increased expression of limbic system-associated membrane protein gene in the periaqueductal grey. Neurosci. Lett. 2003, 352, 179–182. [Google Scholar] [CrossRef]
- Nelovkov, A.; Areda, T.; Innos, J.; Kõks, S.; Vasar, E. Rats displaying distinct exploratory activity also have different expression patterns of γ-aminobutyric acid- and cholecystokinin-related genes in brain regions. Brain Res. 2006, 1100, 21–31. [Google Scholar] [CrossRef]
- Catania, E.H.; Pimenta, A.; Levitt, P. Genetic deletion of Lsamp causes exaggerated behavioral activation in novel environments. Behav. Brain Res. 2008, 188, 380–390. [Google Scholar] [CrossRef]
- Innos, J.; Philips, M.-A.A.; Leidmaa, E.; Heinla, I.; Raud, S.; Reemann, P.; Plaas, M.; Nurk, K.; Kurrikoff, K.; Matto, V.; et al. Lower anxiety and a decrease in agonistic behaviour in Lsamp-deficient mice. Behav. Brain Res. 2011, 217, 21–31. [Google Scholar] [CrossRef]
- Bregin, A.; Mazitov, T.; Aug, I.; Philips, M.-A.; Innos, J.; Vasar, E. Increased sensitivity to psychostimulants and GABAergic drugs in Lsamp-deficient mice. Pharmacol. Biochem. Behav. 2019, 183, 87–97. [Google Scholar] [CrossRef]
- Innos, J.; Leidmaa, E.; Philips, M.-A.A.; Sütt, S.; Alttoa, A.; Harro, J.; Kõks, S.; Vasar, E. Lsamp−/− mice display lower sensitivity to amphetamine and have elevated 5-HT turnover. Biochem. Biophys. Res. Commun. 2013, 430, 413–418. [Google Scholar] [CrossRef]
- Innos, J.; Philips, M.-A.; Raud, S.; Lilleväli, K.; Kõks, S.; Vasar, E. Deletion of the Lsamp gene lowers sensitivity to stressful environmental manipulations in mice. Behav. Brain Res. 2012, 228, 74–81. [Google Scholar] [CrossRef]
- Qiu, S.; Champagne, D.L.; Peters, M.; Catania, E.H.; Weeber, E.J.; Levitt, P.; Pimenta, A.F. Loss of limbic system-associated membrane protein leads to reduced hippocampal mineralocorticoid receptor expression, impaired synaptic plasticity, and spatial memory deficit. Biol. Psychiatry 2010, 68, 197–204. [Google Scholar] [CrossRef]
- Must, A.; Tasa, G.; Lang, A.; Vasar, E.; Kõks, S.; Maron, E.; Väli, M. Association of limbic system-associated membrane protein (LSAMP) to male completed suicide. BMC Med. Genet. 2008, 9, 34. [Google Scholar] [CrossRef]
- Miret, M.; Ayuso-Mateos, J.L.; Sanchez-Moreno, J.; Vieta, E. Depressive disorders and suicide: Epidemiology, risk factors, and burden. Neurosci. Biobehav. Rev. 2013, 37, 2372–2374. [Google Scholar] [CrossRef] [PubMed]
- Koido, K.; Traks, T.; Balõtšev, R.; Eller, T.; Must, A.; Koks, S.; Maron, E.; Tõru, I.; Shlik, J.; Vasar, V.; et al. Associations between LSAMP gene polymorphisms and major depressive disorder and panic disorder. Transl. Psychiatry 2012, 2, e152. [Google Scholar] [CrossRef] [PubMed]
- Behan, A.T.; Byrne, C.; Dunn, M.J.; Cagney, G.; Cotter, D.R.; Behan, Á.; Byrne, C.; Dunn, M.J.; Cagney, G.; Cotter, D.R. Proteomic analysis of membrane microdomain-associated proteins in the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder reveals alterations in LAMP, STXBP1 and BASP1 protein expression. Mol. Psychiatry 2009, 14, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Koido, K.; Janno, S.; Traks, T.; Parksepp, M.; Ljubajev, Ü.; Veiksaar, P.; Must, A.; Shlik, J.; Vasar, V.; Vasar, E. Associations between polymorphisms of LSAMP gene and schizophrenia. Psychiatry Res. 2014, 215, 797–798. [Google Scholar] [CrossRef]
- Heywood, W.E.; Galimberti, D.; Bliss, E.; Sirka, E.; Paterson, R.W.; Magdalinou, N.K.; Carecchio, M.; Reid, E.; Heslegrave, A.; Fenoglio, C.; et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Mol. Neurodegener. 2015, 10, 64. [Google Scholar] [CrossRef]
- Spaltmann, F.; Brümmendorf, T. CEPU-1, a novel immunoglobulin superfamily molecule, is expressed by developing cerebellar Purkinje cells. J. Neurosci. 1996, 16, 1770–1779. [Google Scholar] [CrossRef]
- Gil, O.D.; Zanazzi, G.; Struyk, A.F.; Salzer, J.L. Neurotrimin mediates bifunctional effects on neurite outgrowth via homophilic and heterophilic interactions. J. Neurosci. 1998, 18, 9312–9325. [Google Scholar] [CrossRef]
- McNamee, C.J.; Reed, J.E.; Howard, M.R.; Lodge, A.P.; Moss, D.J. Promotion of neuronal cell adhesion by members of the IgLON family occurs in the absence of either support or modification of neurite outgrowth. J. Neurochem. 2002, 80, 941–948. [Google Scholar] [CrossRef]
- Chen, S.; Gil, O.; Ren, Y.Q.; Zanazzi, G.; Salzer, J.L.; Hillman, D.E. Neurotrimin expression during cerebellar development suggests roles in axon fasciculation and synaptogenesis. J. Neurocytol. 2001, 30, 927–937. [Google Scholar] [CrossRef]
- Singh, K.; Lilleväli, K.; Gilbert, S.F.; Bregin, A.; Narvik, J.; Jayaram, M.; Rahi, M.; Innos, J.; Kaasik, A.; Vasar, E.; et al. The combined impact of IgLON family proteins Lsamp and Neurotrimin on developing neurons and behavioral profiles in mouse. Brain Res. Bull. 2018, 140, 5–18. [Google Scholar] [CrossRef]
- Lodge, A.P.; McNamee, C.J.; Howard, M.R.; Reed, J.E.; Moss, D.J. Identification and characterization of CEPU-Se-A secreted isoform of the IgLON family protein, CEPU-1. Mol. Cell. Neurosci. 2001, 17, 746–760. [Google Scholar] [CrossRef]
- Mazitov, T.; Bregin, A.; Philips, M.A.; Innos, J.; Vasar, E. Deficit in emotional learning in neurotrimin knockout mice. Behav. Brain Res. 2017, 317, 311–318. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, K.S.; Aragam, N. NTM and NR3C2 polymorphisms influencing intelligence: Family-based association studies. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Maruani, A.; Huguet, G.; Beggiato, A.; ElMaleh, M.; Toro, R.; Leblond, C.S.; Mathieu, A.; Amsellem, F.; Lemière, N.; Verloes, A.; et al. 11q24.2-25 micro-rearrangements in autism spectrum disorders: Relation to brain structures. Am. J. Med. Genet. A 2015, 167A, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Brevik, E.J.; van Donkelaar, M.M.J.; Weber, H.; Sánchez-Mora, C.; Jacob, C.; Rivero, O.; Kittel-Schneider, S.; Garcia-Martínez, I.; Aebi, M.; van Hulzen, K.; et al. Genome-wide analyses of aggressiveness in attention-deficit hyperactivity disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Herms, J.; Dorostkar, M.M. Dendritic Spine Pathology in Neurodegenerative Diseases. Annu. Rev. Pathol. 2016, 11, 221–250. [Google Scholar] [CrossRef]
- Kayser, M.S.; Dalmau, J. The emerging link between autoimmune disorders and neuropsychiatric disease. J. Neuropsychiatry Clin. Neurosci. 2011, 23, 90–97. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.B.; Vaughan, S.; Zanini, E.; Okon, I.S.; Louis, L.; de Sousa, C.; Greene, M.I.; Wang, Q.; Agarwal, R.; Shaposhnikov, D.; et al. The OPCML tumor suppressor functions as a cell surface repressor-adaptor, negatively regulating receptor tyrosine kinases in epithelial ovarian cancer. Cancer Discov. 2012, 2, 156–171. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salluzzo, M.; Vianello, C.; Abdullatef, S.; Rimondini, R.; Piccoli, G.; Carboni, L. The Role of IgLON Cell Adhesion Molecules in Neurodegenerative Diseases. Genes 2023, 14, 1886. https://doi.org/10.3390/genes14101886
Salluzzo M, Vianello C, Abdullatef S, Rimondini R, Piccoli G, Carboni L. The Role of IgLON Cell Adhesion Molecules in Neurodegenerative Diseases. Genes. 2023; 14(10):1886. https://doi.org/10.3390/genes14101886
Chicago/Turabian StyleSalluzzo, Marco, Clara Vianello, Sandra Abdullatef, Roberto Rimondini, Giovanni Piccoli, and Lucia Carboni. 2023. "The Role of IgLON Cell Adhesion Molecules in Neurodegenerative Diseases" Genes 14, no. 10: 1886. https://doi.org/10.3390/genes14101886
APA StyleSalluzzo, M., Vianello, C., Abdullatef, S., Rimondini, R., Piccoli, G., & Carboni, L. (2023). The Role of IgLON Cell Adhesion Molecules in Neurodegenerative Diseases. Genes, 14(10), 1886. https://doi.org/10.3390/genes14101886