

Workability of mRNA Sequencing for Predicting Protein Abundance
 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
1. The Attractiveness of Transcriptomic Methods for Assessing Gene Expression
- the absence of an analogue of direct amplification of protein sequences, in contrast to genes and transcripts [20];
- the high diversity of proteinogenic amino acids (23 proteinogenic blocks and modified amino acids [21]) compared to the four-letter nucleotide composition of RNAs (even considering possible PTMs);
- the lack of high-specific enzyme selection to cleave protein sequences (compared to a rich set of polymerases, DNases, RNases, and ligases).
2. Key Points of Transcriptome-to-Proteome Research
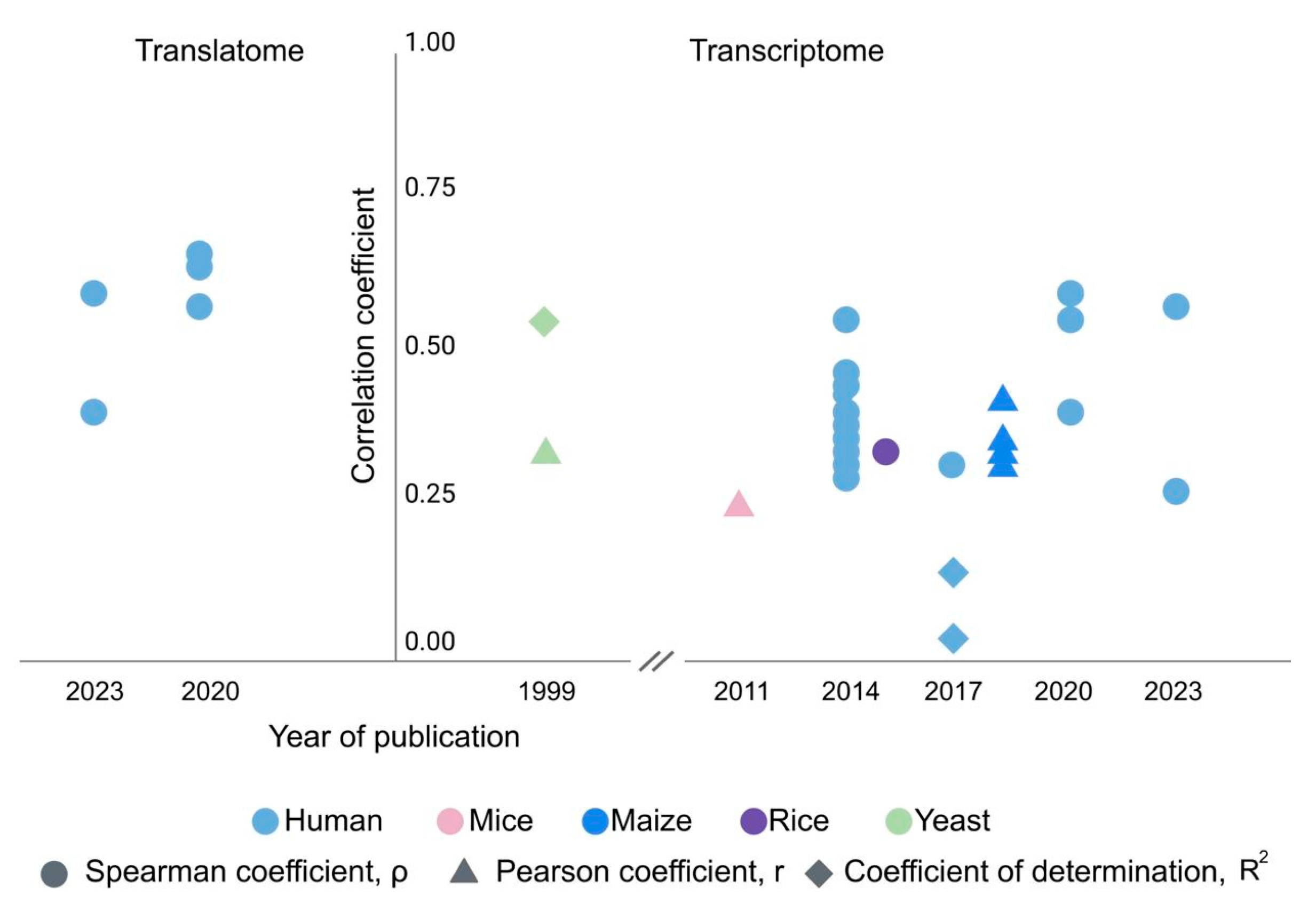
3. Gene-Centric Approach: The mRNA-Protein Ratio Varies Greatly between Different Genes but Is Conserved in Different Tissues and Cell Types
- (a)
- constant gene-specific and context-independent (i.e., codon selection, secondary structures of transcript and mRNA, protein tertiary structure, molecular weight, tRNA repertoire);
- (b)
- depending on the specific state of the cell (the number of available ribosomes and translation initiation factors, the availability of tRNA, the rate of protein degradation).
4. Regulation of Gene Expression
5. Translatome
6. Single-Cell Transcriptomics–Proteomics
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aebersold, R.; Blattmann, P. Mass Spectrometric Exploration of the Biochemical Basis of Living Systems. Chimia 2019, 73, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Vitrinel, B.; Koh, H.W.L.; Mujgan Kar, F.; Maity, S.; Rendleman, J.; Choi, H.; Vogel, C. Exploiting Interdata Relationships in Next-Generation Proteomics Analysis. Mol. Cell Proteom. MCP 2019, 18, S5–S14. [Google Scholar] [CrossRef] [PubMed]
- Edfors, F.; Danielsson, F.; Hallström, B.M.; Käll, L.; Lundberg, E.; Pontén, F.; Forsström, B.; Uhlén, M. Gene-Specific Correlation of RNA and Protein Levels in Human Cells and Tissues. Mol. Syst. Biol. 2016, 12, 883. [Google Scholar] [CrossRef] [PubMed]
- Spainhour, J.C.; Lim, H.S.; Yi, S.V.; Qiu, P. Correlation Patterns Between DNA Methylation and Gene Expression in The Cancer Genome Atlas. Cancer Inform. 2019, 18, 1176935119828776. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Smyczynska, U.; Stanczak, M.; Kuljanin, M.; Włodarczyk, A.; Stoczynska-Fidelus, E.; Taha, J.; Pawlik, B.; Borowiec, M.; Mancias, J.D.; Mlynarski, W.; et al. Proteomic and Transcriptomic Landscapes of Alström and Bardet–Biedl Syndromes. Genes 2022, 13, 2370. [Google Scholar] [CrossRef] [PubMed]
- Archakov, A.; Aseev, A.; Bykov, V.; Grigoriev, A.; Govorun, V.; Ivanov, V.; Khlunov, A.; Lisitsa, A.; Mazurenko, S.; Makarov, A.A.; et al. Gene-Centric View on the Human Proteome Project: The Example of the Russian Roadmap for Chromosome 18. Proteomics 2011, 11, 1853–1856. [Google Scholar] [CrossRef]
- Poverennaya, E.V.; Ilgisonis, E.V.; Ponomarenko, E.A.; Kopylov, A.T.; Zgoda, V.G.; Radko, S.P.; Lisitsa, A.V.; Archakov, A.I. Why Are the Correlations between mRNA and Protein Levels so Low among the 275 Predicted Protein-Coding Genes on Human Chromosome 18? J. Proteome Res. 2017, 16, 4311–4318. [Google Scholar] [CrossRef]
- Zgoda, V.G.; Kopylov, A.T.; Tikhonova, O.V.; Moisa, A.A.; Pyndyk, N.V.; Farafonova, T.E.; Novikova, S.E.; Lisitsa, A.V.; Ponomarenko, E.A.; Poverennaya, E.V.; et al. Chromosome 18 Transcriptome Profiling and Targeted Proteome Mapping in Depleted Plasma, Liver Tissue and HepG2 Cells. J. Proteome Res. 2013, 12, 123–134. [Google Scholar] [CrossRef]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallström, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.-H.; Meng, C.; et al. A Deep Proteome and Transcriptome Abundance Atlas of 29 Healthy Human Tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell Proteom. MCP 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Ramsköld, D.; Wang, E.T.; Burge, C.B.; Sandberg, R. An Abundance of Ubiquitously Expressed Genes Revealed by Tissue Transcriptome Sequence Data. PLoS Comput. Biol. 2009, 5, e1000598. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cui, Y.; Jin, J.; Guo, J.; Wang, G.; Yin, X.; He, Q.-Y.; Zhang, G. Translating mRNAs Strongly Correlate to Proteins in a Multivariate Manner and Their Translation Ratios Are Phenotype Specific. Nucleic Acids Res. 2013, 41, 4743–4754. [Google Scholar] [CrossRef] [PubMed]
- Kiseleva, O.; Ponomarenko, E.; Poverennaya, E. Empowering Shotgun Mass Spectrometry with 2DE: A HepG2 Study. Int. J. Mol. Sci. 2020, 21, 3813. [Google Scholar] [CrossRef]
- Lisitsa, A.; Moshkovskii, S.; Chernobrovkin, A.; Ponomarenko, E.; Archakov, A. Profiling Proteoforms: Promising Follow-up of Proteomics for Biomarker Discovery. Expert Rev. Proteom. 2014, 11, 121–129. [Google Scholar] [CrossRef]
- Song, C.; Wang, F.; Cheng, K.; Wei, X.; Bian, Y.; Wang, K.; Tan, Y.; Wang, H.; Ye, M.; Zou, H. Large-Scale Quantification of Single Amino-Acid Variations by a Variation-Associated Database Search Strategy. J. Proteome Res. 2014, 13, 241–248. [Google Scholar] [CrossRef]
- Cao, R.; Shi, Y.; Chen, S.; Ma, Y.; Chen, J.; Yang, J.; Chen, G.; Shi, T. dbSAP: Single Amino-Acid Polymorphism Database for Protein Variation Detection. Nucleic Acids Res. 2017, 45, D827–D832. [Google Scholar] [CrossRef]
- Yang, Y.; Nan, Y.; Cai, J.; Xu, J.; Huang, Z.; Cai, X. The Thr to Met Substitution of Amino Acid 118 in Hepatitis B Virus Surface Antigen Escapes from Immune-Assay-Based Screening of Blood Donors. J. Gen. Virol. 2016, 97, 1210–1217. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Archakov, A.; Zgoda, V.; Kopylov, A.; Naryzhny, S.; Chernobrovkin, A.; Ponomarenko, E.; Lisitsa, A. Chromosome-Centric Approach to Overcoming Bottlenecks in the Human Proteome Project. Expert Rev. Proteom. 2012, 9, 667–676. [Google Scholar] [CrossRef]
- Yuan, Z.; Liu, X.; Liu, C.; Zhang, Y.; Rao, Y. Recent Advances in Rapid Synthesis of Non-Proteinogenic Amino Acids from Proteinogenic Amino Acids Derivatives via Direct Photo-Mediated C-H Functionalization. Molecules 2020, 25, 5270. [Google Scholar] [CrossRef] [PubMed]
- Giansanti, P.; Tsiatsiani, L.; Low, T.Y.; Heck, A.J.R. Six Alternative Proteases for Mass Spectrometry-Based Proteomics beyond Trypsin. Nat. Protoc. 2016, 11, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Ilgisonis, E.V.; Kopylov, A.T.; Ponomarenko, E.A.; Poverennaya, E.V.; Tikhonova, O.V.; Farafonova, T.E.; Novikova, S.; Lisitsa, A.V.; Zgoda, V.G.; Archakov, A.I. Increased Sensitivity of Mass Spectrometry by Alkaline Two-Dimensional Liquid Chromatography: Deep Cover of the Human Proteome in Gene-Centric Mode. J. Proteome Res. 2018, 17, 4258–4266. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. Central Dogma of Molecular Biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Chick, J.M.; Munger, S.C.; Simecek, P.; Huttlin, E.L.; Choi, K.; Gatti, D.M.; Raghupathy, N.; Svenson, K.L.; Churchill, G.A.; Gygi, S.P. Defining the Consequences of Genetic Variation on a Proteome-Wide Scale. Nature 2016, 534, 500–505. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into the Regulation of Protein Abundance from Proteomic and Transcriptomic Analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Cheng, Z.; Teo, G.; Krueger, S.; Rock, T.M.; Koh, H.W.L.; Choi, H.; Vogel, C. Differential Dynamics of the Mammalian mRNA and Protein Expression Response to Misfolding Stress. Mol. Syst. Biol. 2016, 12, 855. [Google Scholar] [CrossRef]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing Protein Abundance and mRNA Expression Levels on a Genomic Scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef]
- Gygi, S.P.; Rochon, Y.; Franza, B.R.; Aebersold, R. Correlation between Protein and mRNA Abundance in Yeast. Mol. Cell Biol. 1999, 19, 1720–1730. [Google Scholar] [CrossRef]
- Futcher, B.; Latter, G.I.; Monardo, P.; McLaughlin, C.S.; Garrels, J.I. A Sampling of the Yeast Proteome. Mol. Cell Biol. 1999, 19, 7357–7368. [Google Scholar] [CrossRef]
- Lu, P.; Vogel, C.; Wang, R.; Yao, X.; Marcotte, E.M. Absolute Protein Expression Profiling Estimates the Relative Contributions of Transcriptional and Translational Regulation. Nat. Biotechnol. 2007, 25, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Ghazalpour, A.; Bennett, B.; Petyuk, V.A.; Orozco, L.; Hagopian, R.; Mungrue, I.N.; Farber, C.R.; Sinsheimer, J.; Kang, H.M.; Furlotte, N.; et al. Comparative Analysis of Proteome and Transcriptome Variation in Mouse. PLoS Genet. 2011, 7, e1001393. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Qin, Z.; Zhang, G.; Guo, Y.; Huang, J. Integration of the Proteome and Transcriptome Reveals Multiple Levels of Gene Regulation in the Rice Dl2 Mutant. Front. Plant Sci. 2015, 6, 351. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Sun, W.; Li, M.; Zhang, Z. Integrated Analysis of Protein Abundance, Transcript Level, and Tissue Diversity to Reveal Developmental Regulation of Maize. J. Proteome Res. 2018, 17, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Torres-García, W.; Zhang, W.; Runger, G.C.; Johnson, R.H.; Meldrum, D.R. Integrative Analysis of Transcriptomic and Proteomic Data of Desulfovibrio Vulgaris: A Non-Linear Model to Predict Abundance of Undetected Proteins. Bioinformatics 2009, 25, 1905–1914. [Google Scholar] [CrossRef]
- Lundberg, E.; Fagerberg, L.; Klevebring, D.; Matic, I.; Geiger, T.; Cox, J.; Algenäs, C.; Lundeberg, J.; Mann, M.; Uhlen, M. Defining the Transcriptome and Proteome in Three Functionally Different Human Cell Lines. Mol. Syst. Biol. 2010, 6, 450. [Google Scholar] [CrossRef]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-Spectrometry-Based Draft of the Human Proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef]
- Fortelny, N.; Overall, C.M.; Pavlidis, P.; Freue, G.V.C. Can We Predict Protein from mRNA Levels? Nature 2017, 547, E19–E20. [Google Scholar] [CrossRef]
- Nie, L.; Wu, G.; Zhang, W. Correlation between mRNA and Protein Abundance in Desulfovibrio Vulgaris: A Multiple Regression to Identify Sources of Variations. Biochem. Biophys. Res. Commun. 2006, 339, 603–610. [Google Scholar] [CrossRef]
- Santos, F.B.; Del-Bem, L.-E. The Evolution of tRNA Copy Number and Repertoire in Cellular Life. Genes 2023, 14, 27. [Google Scholar] [CrossRef]
- Silva, G.M.; Vogel, C. Quantifying Gene Expression: The Importance of Being Subtle. Mol. Syst. Biol. 2016, 12, 885. [Google Scholar] [CrossRef] [PubMed]
- Franks, A.; Airoldi, E.; Slavov, N. Post-Transcriptional Regulation across Human Tissues. PLoS Comput. Biol. 2017, 13, e1005535. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A Draft Map of the Human Proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Hershey, J.W.B.; Sonenberg, N.; Mathews, M.B. Principles of Translational Control. Cold Spring Harb. Perspect. Biol. 2019, 11, a032607. [Google Scholar] [CrossRef]
- Teo, G.; Vogel, C.; Ghosh, D.; Kim, S.; Choi, H. PECA: A Novel Statistical Tool for Deconvoluting Time-Dependent Gene Expression Regulation. J. Proteome Res. 2014, 13, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.K.; Hammond, D.E.; Clague, M.J.; Gaskell, S.J.; Beynon, R.J. Turnover of the Human Proteome: Determination of Protein Intracellular Stability by Dynamic SILAC. J. Proteome Res. 2009, 8, 104–112. [Google Scholar] [CrossRef]
- Eraslan, B.; Wang, D.; Gusic, M.; Prokisch, H.; Hallström, B.M.; Uhlén, M.; Asplund, A.; Pontén, F.; Wieland, T.; Hopf, T.; et al. Quantification and Discovery of Sequence Determinants of Protein-per-mRNA Amount in 29 Human Tissues. Mol. Syst. Biol. 2019, 15, e8513. [Google Scholar] [CrossRef]
- Besser, D.; Götz, F.; Schulze-Forster, K.; Wagner, H.; Kröger, H.; Simon, D. DNA Methylation Inhibits Transcription by RNA Polymerase III of a tRNA Gene, but Not of a 5S rRNA Gene. FEBS Lett. 1990, 269, 358–362. [Google Scholar] [CrossRef]
- Arzumanian, V.A.; Dolgalev, G.V.; Kurbatov, I.Y.; Kiseleva, O.I.; Poverennaya, E.V. Epitranscriptome: Review of Top 25 Most-Studied RNA Modifications. Int. J. Mol. Sci. 2022, 23, 13851. [Google Scholar] [CrossRef]
- Jimeno-González, S.; Payán-Bravo, L.; Muñoz-Cabello, A.M.; Guijo, M.; Gutierrez, G.; Prado, F.; Reyes, J.C. Defective Histone Supply Causes Changes in RNA Polymerase II Elongation Rate and Cotranscriptional Pre-mRNA Splicing. Proc. Natl. Acad. Sci. USA 2015, 112, 14840–14845. [Google Scholar] [CrossRef]
- Chambeyron, S.; Bickmore, W.A. Chromatin Decondensation and Nuclear Reorganization of the HoxB Locus upon Induction of Transcription. Genes Dev. 2004, 18, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Transcription Factors—ScienceDirect. Available online: https://www.sciencedirect.com/science/article/abs/pii/B9780128012383054660 (accessed on 6 October 2023).
- Alternative RNA Splicing and Editing: A Functional Molecular Tool Directed to Successful Protein Synthesis in Plants|SpringerLink. Available online: https://link.springer.com/chapter/10.1007/978-3-030-68828-8_5 (accessed on 6 October 2023).
- Hildyard, J.C.W.; Piercy, R.J. When Size Really Matters: The Eccentricities of Dystrophin Transcription and the Hazards of Quantifying mRNA from Very Long Genes. Biomedicines 2023, 11, 2082. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-E.; Yi, H.; Kim, Y.; Chang, H.; Kim, V.N. Regulation of Poly(A) Tail and Translation during the Somatic Cell Cycle. Mol. Cell 2016, 62, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Lim, J.; Ha, M.; Kim, V.N. TAIL-Seq: Genome-Wide Determination of Poly(A) Tail Length and 3’ End Modifications. Mol. Cell 2014, 53, 1044–1052. [Google Scholar] [CrossRef]
- Floor, S.N.; Doudna, J.A. Tunable Protein Synthesis by Transcript Isoforms in Human Cells. eLife 2016, 5, e10921. [Google Scholar] [CrossRef]
- Salovska, B.; Zhu, H.; Gandhi, T.; Frank, M.; Li, W.; Rosenberger, G.; Wu, C.; Germain, P.-L.; Zhou, H.; Hodny, Z.; et al. Isoform-Resolved Correlation Analysis between mRNA Abundance Regulation and Protein Level Degradation. Mol. Syst. Biol. 2020, 16, e9170. [Google Scholar] [CrossRef]
- Barbieri, I.; Kouzarides, T. Role of RNA Modifications in Cancer. Nat. Rev. Cancer 2020, 20, 303–322. [Google Scholar] [CrossRef]
- Fernandez Rodriguez, G.; Cesaro, B.; Fatica, A. Multiple Roles of m6A RNA Modification in Translational Regulation in Cancer. Int. J. Mol. Sci. 2022, 23, 8971. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, J.-Z.; Xu, Z.; Cao, M.; Hu, Q.; Pan, C.; Guo, M.; Wei, J.-F.; Yang, H. Detection of N6-methyladenosine Modification Residues (Review). Int. J. Mol. Med. 2019, 43, 2267–2278. [Google Scholar] [CrossRef]
- Zhong, Z.-D.; Xie, Y.-Y.; Chen, H.-X.; Lan, Y.-L.; Liu, X.-H.; Ji, J.-Y.; Wu, F.; Jin, L.; Chen, J.; Mak, D.W.; et al. Systematic Comparison of Tools Used for m6A Mapping from Nanopore Direct RNA Sequencing. Nat. Commun. 2023, 14, 1906. [Google Scholar] [CrossRef]
- Williams, A.E. Functional Aspects of Animal microRNAs. Cell Mol. Life Sci. 2008, 65, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yin, G.; Zhang, Y.; Zhu, L.; Huang, H.; Lv, K. Recent Advances in the Functional Explorations of Nuclear microRNAs. Front. Immunol. 2023, 14, 1097491. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Rossi, J.J. Uncoupling of RNAi from Active Translation in Mammalian Cells. RNA 2005, 11, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Neumeier, J.; Meister, G. siRNA Specificity: RNAi Mechanisms and Strategies to Reduce Off-Target Effects. Front. Plant Sci. 2020, 11, 526455. [Google Scholar] [CrossRef]
- Mullenbrock, S.; Liu, F.; Szak, S.; Hronowski, X.; Gao, B.; Juhasz, P.; Sun, C.; Liu, M.; McLaughlin, H.; Xiao, Q.; et al. Systems Analysis of Transcriptomic and Proteomic Profiles Identifies Novel Regulation of Fibrotic Programs by miRNAs in Pulmonary Fibrosis Fibroblasts. Genes 2018, 9, 588. [Google Scholar] [CrossRef]
- Pantaleão, L.C.; Ozanne, S.E. Small RNA Sequencing: A Technique for miRNA Profiling. Methods Mol. Biol. 2018, 1735, 321–330. [Google Scholar] [CrossRef]
- Hücker, S.M.; Fehlmann, T.; Werno, C.; Weidele, K.; Lüke, F.; Schlenska-Lange, A.; Klein, C.A.; Keller, A.; Kirsch, S. Single-Cell microRNA Sequencing Method Comparison and Application to Cell Lines and Circulating Lung Tumor Cells. Nat. Commun. 2021, 12, 4316. [Google Scholar] [CrossRef]
- Suhre, K.; McCarthy, M.I.; Schwenk, J.M. Genetics Meets Proteomics: Perspectives for Large Population-Based Studies. Nat. Rev. Genet. 2021, 22, 19–37. [Google Scholar] [CrossRef]
- Sjaarda, J.; Gerstein, H.C.; Kutalik, Z.; Mohammadi-Shemirani, P.; Pigeyre, M.; Hess, S.; Paré, G. Influence of Genetic Ancestry on Human Serum Proteome. Am. J. Hum. Genet. 2020, 106, 303–314. [Google Scholar] [CrossRef]
- Zhao, J.; Qin, B.; Nikolay, R.; Spahn, C.M.T.; Zhang, G. Translatomics: The Global View of Translation. Int. J. Mol. Sci. 2019, 20, 212. [Google Scholar] [CrossRef]
- Ingolia, N.T. Ribosome Footprint Profiling of Translation throughout the Genome. Cell 2016, 165, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.K.; Lehmann, R. Translational Control during Developmental Transitions. Cold Spring Harb. Perspect. Biol. 2019, 11, a032987. [Google Scholar] [CrossRef] [PubMed]
- Buszczak, M.; Signer, R.A.J.; Morrison, S.J. Cellular Differences in Protein Synthesis Regulate Tissue Homeostasis. Cell 2014, 159, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Snee, M.; Benz, D.; Jen, J.; Macdonald, P.M. Two Distinct Domains of Bruno Bind Specifically to the Oskar mRNA. RNA Biol. 2008, 5, 49–57. [Google Scholar] [CrossRef]
- Chang, J.S.; Tan, L.; Schedl, P. The Drosophila CPEB Homolog, Orb, Is Required for Oskar Protein Expression in Oocytes. Dev. Biol. 1999, 215, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.; Fire, A. Conserved Translatome Remodeling in Nematode Species Executing a Shared Developmental Transition. PLoS Genet. 2013, 9, e1003739. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Y.; Yang, J.; Zhao, Q.; Tang, N.; Chen, C.; Li, H.; Cheng, C.; Xie, M.; Yang, Y.; et al. Tissue- and Stage-Specific Landscape of the Mouse Translatome. Nucleic Acids Res. 2021, 49, 6165–6180. [Google Scholar] [CrossRef]
- Khan, Z.; Ford, M.J.; Cusanovich, D.A.; Mitrano, A.; Pritchard, J.K.; Gilad, Y. Primate Transcript and Protein Expression Levels Evolve under Compensatory Selection Pressures. Science 2013, 342, 1100–1104. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.S.; Weissman, J.S. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef]
- Smircich, P.; Eastman, G.; Bispo, S.; Duhagon, M.A.; Guerra-Slompo, E.P.; Garat, B.; Goldenberg, S.; Munroe, D.J.; Dallagiovanna, B.; Holetz, F.; et al. Ribosome Profiling Reveals Translation Control as a Key Mechanism Generating Differential Gene Expression in Trypanosoma Cruzi. BMC Genom. 2015, 16, 443. [Google Scholar] [CrossRef]
- Thomas, A.; Lee, P.-J.; Dalton, J.E.; Nomie, K.J.; Stoica, L.; Costa-Mattioli, M.; Chang, P.; Nuzhdin, S.; Arbeitman, M.N.; Dierick, H.A. A Versatile Method for Cell-Specific Profiling of Translated mRNAs in Drosophila. PLoS ONE 2012, 7, e40276. [Google Scholar] [CrossRef]
- Inada, T.; Winstall, E.; Tarun, S.Z.; Yates, J.R.; Schieltz, D.; Sachs, A.B. One-Step Affinity Purification of the Yeast Ribosome and Its Associated Proteins and mRNAs. RNA 2002, 8, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.Y.; Xiao, C. An Integrated Polysome Profiling and Ribosome Profiling Method to Investigate In Vivo Translatome. Methods Mol. Biol. 2018, 1712, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ruiz Cuevas, M.V.; Hardy, M.P.; Hollý, J.; Bonneil, É.; Durette, C.; Courcelles, M.; Lanoix, J.; Côté, C.; Staudt, L.M.; Lemieux, S.; et al. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Rep. 2021, 34, 108815. [Google Scholar] [CrossRef] [PubMed]
- Blevins, W.R.; Tavella, T.; Moro, S.G.; Blasco-Moreno, B.; Closa-Mosquera, A.; Díez, J.; Carey, L.B.; Albà, M.M. Extensive Post-Transcriptional Buffering of Gene Expression in the Response to Severe Oxidative Stress in Baker’s Yeast. Sci. Rep. 2019, 9, 11005. [Google Scholar] [CrossRef]
- Buccitelli, C.; Selbach, M. mRNAs, Proteins and the Emerging Principles of Gene Expression Control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Leushkin, E.; Liechti, A.; Ovchinnikova, S.; Mößinger, K.; Brüning, T.; Rummel, C.; Grützner, F.; Cardoso-Moreira, M.; Janich, P.; et al. Transcriptome and Translatome Co-Evolution in Mammals. Nature 2020, 588, 642–647. [Google Scholar] [CrossRef]
- Gebauer, F.; Hentze, M.W. Molecular Mechanisms of Translational Control. Nat. Rev. Mol. Cell Biol. 2004, 5, 827–835. [Google Scholar] [CrossRef]
- Shen, Z.; Zeng, L.; Zhang, Z. Translatome and Transcriptome Profiling of Hypoxic-Induced Rat Cardiomyocytes. Mol. Ther. Nucleic Acids 2020, 22, 1016–1024. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-Inducible Factor 1 Is a Basic-Helix-Loop-Helix-PAS Heterodimer Regulated by Cellular O2 Tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Hu, W.; Zeng, H.; Shi, Y.; Zhou, C.; Huang, J.; Jia, L.; Xu, S.; Feng, X.; Zeng, Y.; Xiong, T.; et al. Single-Cell Transcriptome and Translatome Dual-Omics Reveals Potential Mechanisms of Human Oocyte Maturation. Nat. Commun. 2022, 13, 5114. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Guo, J.; Gu, W.; Cui, Y.; Zhong, J.; Jin, J.; He, Q.-Y.; Wang, T.; Zhang, G. Genome-Wide and Experimental Resolution of Relative Translation Elongation Speed at Individual Gene Level in Human Cells. PLoS Genet. 2016, 12, e1005901. [Google Scholar] [CrossRef]
- Aldridge, S.; Teichmann, S.A. Single Cell Transcriptomics Comes of Age. Nat. Commun. 2020, 11, 4307. [Google Scholar] [CrossRef]
- Rodriguez, J.; Larson, D.R. Transcription in Living Cells: Molecular Mechanisms of Bursting. Annu. Rev. Biochem. 2020, 89, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Martin-Rufino, J.D.; Jones, T.R.; Subramanian, V.; Qiu, X.; Grody, E.I.; Bloemendal, A.; Weng, C.; Niu, S.-Y.; Min, K.H.; et al. Inferring Gene Regulation from Stochastic Transcriptional Variation across Single Cells at Steady State. Proc. Natl. Acad. Sci. USA 2022, 119, e2207392119. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhong, Y.; Hu, J.; Shang, X. SCC: An Accurate Imputation Method for scRNA-Seq Dropouts Based on a Mixture Model. BMC Bioinform. 2021, 22, 5. [Google Scholar] [CrossRef]
- Arzalluz-Luque, Á.; Conesa, A. Single-Cell RNAseq for the Study of Isoforms-How Is That Possible? Genome Biol. 2018, 19, 110. [Google Scholar] [CrossRef]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven Grand Challenges in Single-Cell Data Science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Tabula Muris Consortium; Overall Coordination; Logistical Coordination; Organ Collection and Processing; Library Preparation and Sequencing; Computational Data Analysis; Cell Type Annotation; Writing Group; Supplemental Text Writing Group; Principal Investigators. Single-Cell Transcriptomics of 20 Mouse Organs Creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef]
- Park, J.-E.; Botting, R.A.; Domínguez Conde, C.; Popescu, D.-M.; Lavaert, M.; Kunz, D.J.; Goh, I.; Stephenson, E.; Ragazzini, R.; Tuck, E.; et al. A Cell Atlas of Human Thymic Development Defines T Cell Repertoire Formation. Science 2020, 367, eaay3224. [Google Scholar] [CrossRef]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-Cell Profiling of Tumor Heterogeneity and the Microenvironment in Advanced Non-Small Cell Lung Cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Carpp, L.N.; Miller, H.E.R.; Zager, M.; Newell, E.W.; Gottardo, R. Single-Cell Immunology of SARS-CoV-2 Infection. Nat. Biotechnol. 2022, 40, 30–41. [Google Scholar] [CrossRef]
- Petrosius, V.; Schoof, E.M. Recent Advances in the Field of Single-Cell Proteomics. Transl. Oncol. 2023, 27, 101556. [Google Scholar] [CrossRef] [PubMed]
- Singh, A. Towards Resolving Proteomes in Single Cells. Nat. Methods 2021, 18, 856. [Google Scholar] [CrossRef] [PubMed]
- Specht, H.; Emmott, E.; Petelski, A.A.; Huffman, R.G.; Perlman, D.H.; Serra, M.; Kharchenko, P.; Koller, A.; Slavov, N. Single-Cell Proteomic and Transcriptomic Analysis of Macrophage Heterogeneity Using SCoPE2. Genome Biol. 2021, 22, 50. [Google Scholar] [CrossRef]
- Brunner, A.-D.; Thielert, M.; Vasilopoulou, C.; Ammar, C.; Coscia, F.; Mund, A.; Hoerning, O.B.; Bache, N.; Apalategui, A.; Lubeck, M.; et al. Ultra-High Sensitivity Mass Spectrometry Quantifies Single-Cell Proteome Changes upon Perturbation. Mol. Syst. Biol. 2022, 18, e10798. [Google Scholar] [CrossRef]
- Chai, J.; Song, Q. Multiple-Protein Detections of Single-Cells Reveal Cell-Cell Heterogeneity in Human Cells. IEEE Trans. Biomed. Eng. 2015, 62, 30–38. [Google Scholar] [CrossRef]
- Emmert-Buck, M.R.; Bonner, R.F.; Smith, P.D.; Chuaqui, R.F.; Zhuang, Z.; Goldstein, S.R.; Weiss, R.A.; Liotta, L.A. Laser Capture Microdissection. Science 1996, 274, 998–1001. [Google Scholar] [CrossRef]
- Hu, P.; Zhang, W.; Xin, H.; Deng, G. Single Cell Isolation and Analysis. Front. Cell Dev. Biol. 2016, 4, 116. [Google Scholar] [CrossRef]
- Yin, H.; Marshall, D. Microfluidics for Single Cell Analysis. Curr. Opin. Biotechnol. 2012, 23, 110–119. [Google Scholar] [CrossRef]
- Quantifying, E. Coli Proteome and Transcriptome with Single-Molecule Sensitivity in Single Cells | Science. Available online: https://www.science.org/doi/10.1126/science.1188308 (accessed on 6 October 2023).
- Adan, A.; Alizada, G.; Kiraz, Y.; Baran, Y.; Nalbant, A. Flow Cytometry: Basic Principles and Applications. Crit. Rev. Biotechnol. 2017, 37, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, C.; Ruff, D.; Kirvell, S.; Johnson, G.; Dhillon, H.S.; Bustin, S.A. Proximity Assays for Sensitive Quantification of Proteins. Biomol. Detect. Quantif. 2015, 4, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Balashova, E.E.; Lokhov, P.G.; Ponomarenko, E.A.; Markin, S.S.; Lisitsa, A.V.; Archakov, A.I. Metabolomic Diagnostics and Human Digital Image. Pers. Med. 2019, 16, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Tsepilov, Y.A.; Sharapov, S.Z.; Zaytseva, O.O.; Krumsiek, J.; Prehn, C.; Adamski, J.; Kastenmüller, G.; Wang-Sattler, R.; Strauch, K.; Gieger, C.; et al. A Network-Based Conditional Genetic Association Analysis of the Human Metabolome. GigaScience 2018, 7, giy137. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponomarenko, E.A.; Krasnov, G.S.; Kiseleva, O.I.; Kryukova, P.A.; Arzumanian, V.A.; Dolgalev, G.V.; Ilgisonis, E.V.; Lisitsa, A.V.; Poverennaya, E.V. Workability of mRNA Sequencing for Predicting Protein Abundance. Genes 2023, 14, 2065. https://doi.org/10.3390/genes14112065
Ponomarenko EA, Krasnov GS, Kiseleva OI, Kryukova PA, Arzumanian VA, Dolgalev GV, Ilgisonis EV, Lisitsa AV, Poverennaya EV. Workability of mRNA Sequencing for Predicting Protein Abundance. Genes. 2023; 14(11):2065. https://doi.org/10.3390/genes14112065
Chicago/Turabian StylePonomarenko, Elena A., George S. Krasnov, Olga I. Kiseleva, Polina A. Kryukova, Viktoriia A. Arzumanian, Georgii V. Dolgalev, Ekaterina V. Ilgisonis, Andrey V. Lisitsa, and Ekaterina V. Poverennaya. 2023. "Workability of mRNA Sequencing for Predicting Protein Abundance" Genes 14, no. 11: 2065. https://doi.org/10.3390/genes14112065
APA StylePonomarenko, E. A., Krasnov, G. S., Kiseleva, O. I., Kryukova, P. A., Arzumanian, V. A., Dolgalev, G. V., Ilgisonis, E. V., Lisitsa, A. V., & Poverennaya, E. V. (2023). Workability of mRNA Sequencing for Predicting Protein Abundance. Genes, 14(11), 2065. https://doi.org/10.3390/genes14112065