
NUP85 as a Neurodevelopmental Gene: From Podocyte to Neuron

 , , , , , , ,
, , , , , , ,  ,
,
Abstract
:1. Introduction
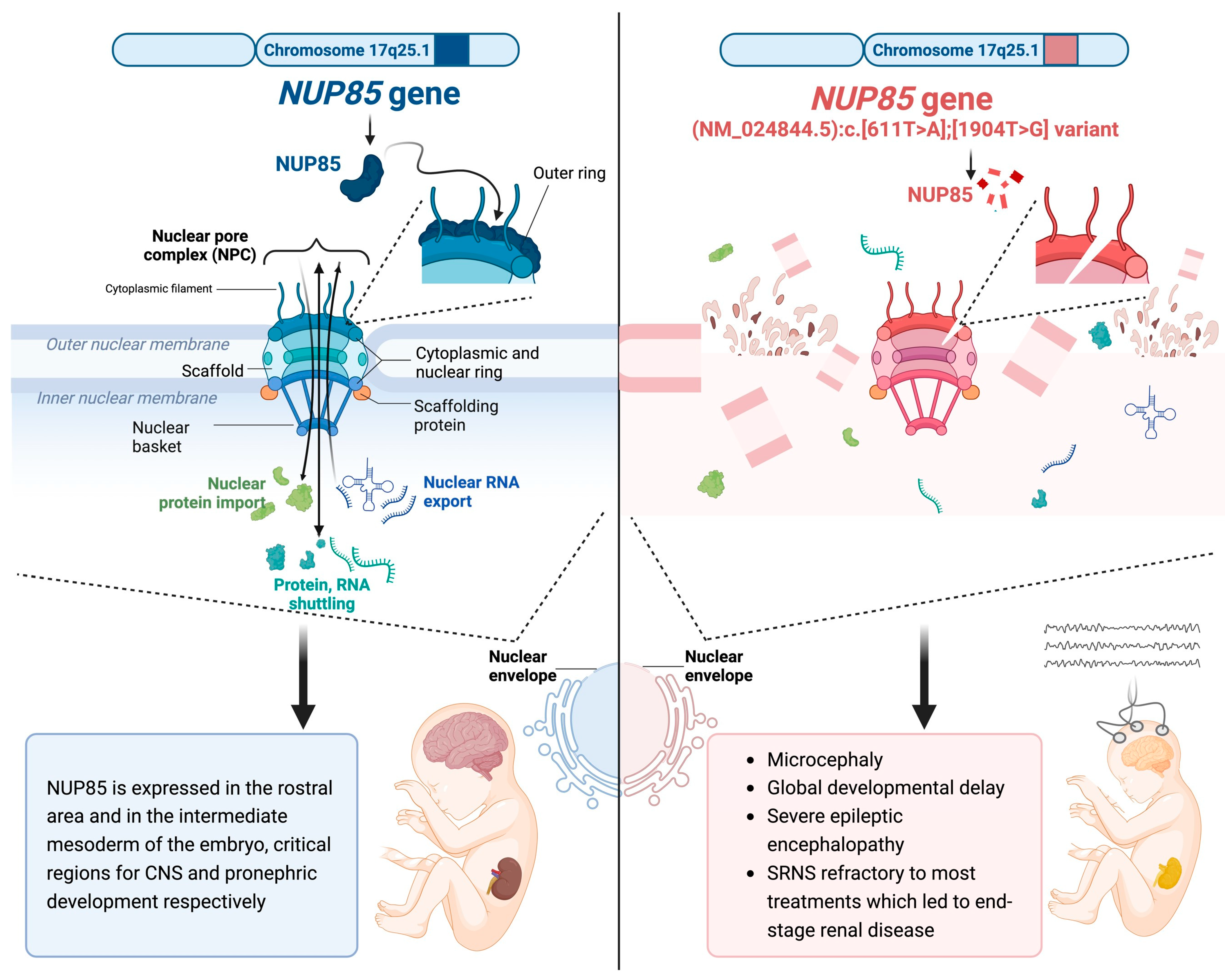
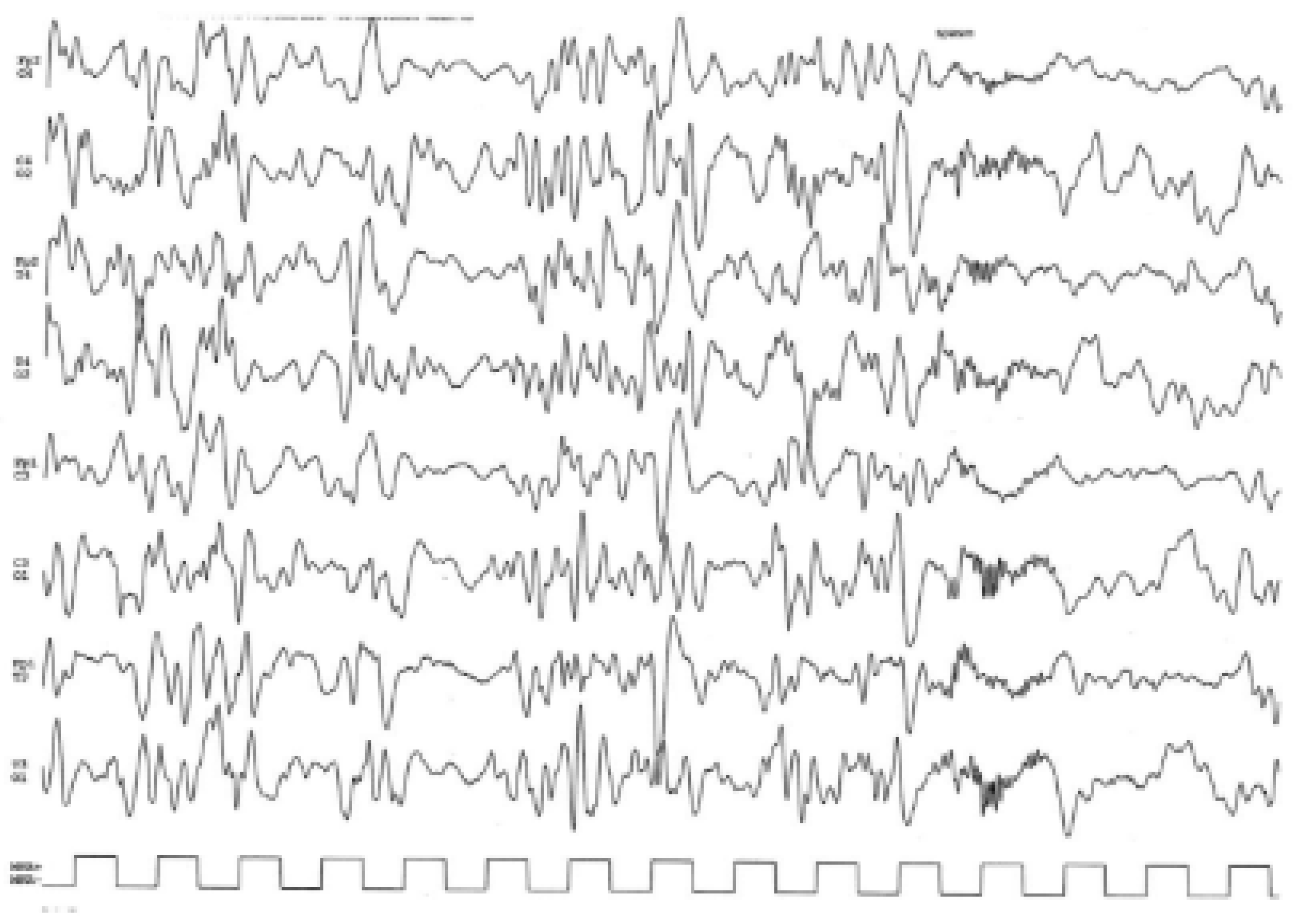
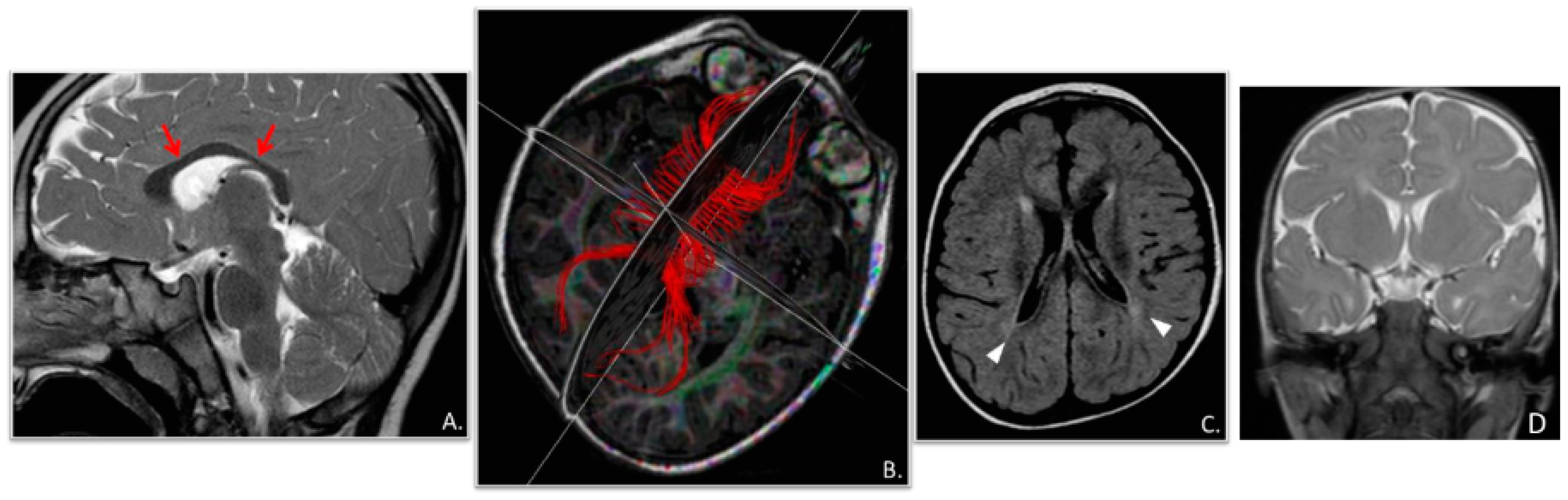
2. Clinical Report
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Downie, M.L.; Gallibois, C.; Parekh, R.S.; Noone, D.G. Nephrotic syndrome in infants and children: Pathophysiology and management. Paediatr. Int. Child Health 2017, 37, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, A.; Vivarelli, M.; Samuel, S.; Gipson, D.; Sinha, A.; Schaefer, F.; Hui, N.K.; Boyer, O.; Saleem, M.A.; Feltran, L.; et al. IPNA clinical practice recommendations for the diagnosis and management of children with steroid-resistant nephrotic syndrome. Pediatr. Nephrol. 2020, 35, 1529–1561. [Google Scholar] [CrossRef] [PubMed]
- Tullus, K.; Webb, H.; Bagga, A. Management of steroid-resistant nephrotic syndrome in children and adolescents. Lancet Child Adolesc. Health 2018, 2, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Iacomino, M.; Baldassari, S.; Tochigi, Y.; Kośla, K.; Buffelli, F.; Torella, A.; Severino, M.; Paladini, D.; Mandarà, L.; Riva, A.; et al. Loss of Wwox Perturbs Neuronal Migration and Impairs Early Cortical Development. Front. Neurosci. 2020, 14, 644. [Google Scholar] [CrossRef] [PubMed]
- Manole, A.; Efthymiou, S.; O’connor, E.; Mendes, M.I.; Jennings, M.; Maroofian, R.; Davagnanam, I.; Mankad, K.; Lopez, M.R.; Salpietro, V.; et al. De Novo and Bi-allelic Pathogenic Variants in NARS1 Cause Neurodevelopmental Delay Due to Toxic Gain-of-Function and Partial Loss-of-Function Effects. Am. J. Hum. Genet. 2020, 107, 311–324. [Google Scholar] [CrossRef]
- Dias, C.M.; Punetha, J.; Zheng, C.; Mazaheri, N.; Rad, A.; Efthymiou, S.; Petersen, A.; Dehghani, M.; Pehlivan, D.; Partlow, J.N.; et al. Homozygous Missense Variants in NTNG2, Encoding a Presynaptic Netrin-G2 Adhesion Protein, Lead to a Distinct Neurodevelopmental Disorder. Am. J. Hum. Genet. 2019, 105, 1048–1056. [Google Scholar] [CrossRef]
- Steel, D.; Salpietro, V.; Phadke, R.; Pitt, M.; Gentile, G.; Massoud, A.; Batten, L.; Bashamboo, A.; Mcelreavey, K.; Saggar, A.; et al. Whole exome sequencing reveals a MLL de novo mutation associated with mild developmental delay and without ‘hairy elbows’: Expanding the phenotype of Wiedemann–Steiner syndrome. J. Genet. 2015, 94, 755–758. [Google Scholar] [CrossRef]
- Jordan, P.; Dorval, G.; Arrondel, C.; Morinière, V.; Tournant, C.; Audrezet, M.; Michel-Calemard, L.; Putoux, A.; Lesca, G.; Labalme, A.; et al. Targeted next-generation sequencing in a large series of fetuses with severe renal diseases. Hum. Mutat. 2022, 43, 347–361. [Google Scholar] [CrossRef]
- Beck, M.; Hurt, E. The nuclear pore complex: Understanding its function through structural insight. Nat. Rev. Mol. Cell Biol. 2017, 18, 73–89. [Google Scholar] [CrossRef]
- Jühlen, R.; Fahrenkrog, B. Moonlighting nuclear pore proteins: Tissue-specific nucleoporin function in health and disease. Histochem. Cell Biol. 2018, 150, 593–605. [Google Scholar] [CrossRef]
- Guglielmi, V.; Sakuma, S.; D’Angelo, M.A. Nuclear pore complexes in development and tissue homeostasis. Development 2020, 147, dev183442. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, R.; Burke, B.; Doye, V. Nuclear transport and the mitotic apparatus: An evolving relationship. Cell. Mol. Life Sci. 2010, 67, 2215–2230. [Google Scholar] [CrossRef] [PubMed]
- Chatel, G.; Fahrenkrog, B. Nucleoporins: Leaving the nuclear pore complex for a successful mitosis. Cell. Signal. 2011, 23, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- del Viso, F.; Huang, F.; Myers, J.; Chalfant, M.; Zhang, Y.; Reza, N.; Bewersdorf, J.; Lusk, C.P.; Khokha, M.K. Congenital Heart Disease Genetics Uncovers Context-Dependent Organization and Function of Nucleoporins at Cilia. Dev. Cell 2016, 38, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Yang, S.; Han, Y.; Zhao, X.; Zhao, L.; Tian, T.; Tong, J.; Xu, P.; Xiong, C.; Meng, A. Loss of Zygotic NUP107 Protein Causes Missing of Pharyngeal Skeleton and Other Tissue Defects with Impaired Nuclear Pore Function in Zebrafish Embryos. J. Biol. Chem. 2012, 287, 38254–38264. [Google Scholar] [CrossRef]
- Lupu, F.; Alves, A.; Anderson, K.; Doye, V.; Lacy, E. Nuclear Pore Composition Regulates Neural Stem/Progenitor Cell Differentiation in the Mouse Embryo. Dev. Cell 2008, 14, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Reza, N.; Khokha, M.K.; Del Viso, F. Nucleoporin gene expression in Xenopus tropicalis embryonic development. Int. J. Dev. Biol. 2016, 60, 181–188. [Google Scholar] [CrossRef]
- Braun, D.A.; Lovric, S.; Schapiro, D.; Schneider, R.; Marquez, J.; Asif, M.; Hussain, M.S.; Daga, A.; Widmeier, E.; Rao, J.; et al. Mutations in multiple components of the nuclear pore complex cause nephrotic syndrome. J. Clin. Investig. 2018, 128, 4313–4328. [Google Scholar] [CrossRef]
- Ravindran, E.; Jühlen, R.; Vieira-Vieira, C.H.; Ha, T.; Salzberg, Y.; Fichtman, B.; Luise-Becker, L.; Martins, N.; Picker-Minh, S.; Bessa, P.; et al. Expanding the phenotype of NUP85 mutations beyond nephrotic syndrome to primary autosomal recessive microcephaly and Seckel syndrome spectrum disorders. Hum. Mol. Genet. 2021, 30, 2068–2081. [Google Scholar] [CrossRef]
- Niestroj, L.-M.; Perez-Palma, E.; Howrigan, D.P.; Zhou, Y.; Cheng, F.; Saarentaus, E.; Stevelink, R.; Daly, M.J.; Palotie, A.; Lal, D.; et al. Epilepsy subtype-specific copy number burden observed in a genome-wide study of 17 458 subjects. Brain 2020, 143, 2106–2118. [Google Scholar] [CrossRef]
- Wiessner, M.; Maroofian, R.; Ni, M.-Y.; Pedroni, A.; Müller, J.S.; Stucka, R.; Beetz, C.; Efthymiou, S.; Santorelli, F.M.; Alfares, A.; et al. Biallelic variants in HPDL cause pure and complicated hereditary spastic paraplegia. Brain 2021, 144, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Dworschak, G.C.; Punetha, J.; Kalanithy, J.C.; Mingardo, E.; Erdem, H.B.; Akdemir, Z.C.; Karaca, E.; Mitani, T.; Marafi, D.; Fatih, J.M.; et al. Biallelic and monoallelic variants in PLXNA1 are implicated in a novel neurodevelopmental disorder with variable cerebral and eye anomalies. Anesthesia Analg. 2021, 23, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Donkervoort, S.; Kutzner, C.E.; Hu, Y.; Lornage, X.; Rendu, J.; Stojkovic, T.; Baets, J.; Neuhaus, S.B.; Tanboon, J.; Maroofian, R.; et al. Pathogenic Variants in the Myosin Chaperone UNC-45B Cause Progressive Myopathy with Eccentric Cores. Am. J. Hum. Genet. 2020, 107, 1078–1095. [Google Scholar] [CrossRef] [PubMed]
- Granata, F.; Morabito, R.; Mormina, E.; Alafaci, C.; Marino, S.; Laganà, A.; Vinci, S.L.; Briguglio, M.; Calamuneri, A.; Gaeta, M.; et al. 3T Double Inversion Recovery Magnetic Resonance Imaging: Diagnostic advantages in the evaluation of cortical development anomalies. Eur. J. Radiol. 2016, 85, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Colin, E.; Cong, E.H.; Mollet, G.; Guichet, A.; Gribouval, O.; Arrondel, C.; Boyer, O.; Daniel, L.; Gubler, M.-C.; Ekinci, Z.; et al. Loss-of-Function Mutations in WDR73 Are Responsible for Microcephaly and Steroid-Resistant Nephrotic Syndrome: Galloway-Mowat Syndrome. Am. J. Hum. Genet. 2014, 95, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Ben-Omran, T.; Fahiminiya, S.; Sorfazlian, N.; Almuriekhi, M.; Nawaz, Z.; Nadaf, J.; Abu Khadija, K.; Zaineddin, S.; Kamel, H.; Majewski, J.; et al. Nonsense mutation in the WDR73 gene is associated with Galloway-Mowat syndrome. J. Med. Genet. 2015, 52, 381–390. [Google Scholar] [CrossRef]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.; et al. Accelerating Novel Candidate Gene Discovery in Neurogenetic Disorders via Whole-Exome Sequencing of Prescreened Multiplex Consanguineous Families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef]
- Rosti, R.O.; Sotak, B.N.; Bielas, S.L.; Bhat, G.; Silhavy, J.L.; Aslanger, A.D.; Altunoglu, U.; Bilge, I.; Tasdemir, M.; Yzaguirrem, A.D.; et al. Homozygous mutation in NUP107 leads to microcephaly with steroid-resistant nephrotic condition similar to Galloway-Mowat syndrome. J. Med. Genet. 2017, 54, 399–403. [Google Scholar] [CrossRef]
- Ravindran, E.; Lesca, G.; Januel, L.; Goldgruber, L.; Dickmanns, A.; Margot, H.; Kaindl, A.M. Case report: Compound heterozygous NUP85 variants cause autosomal recessive primary microcephaly. Front. Neurol. 2023, 14, 1124886. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Our Study | Ravindran et al., 2023 [29] | Ravindran et al., 2021 [19] | Braun et al., 2018 [18] | |||||
|---|---|---|---|---|---|---|---|---|
| Our Individual | Individual 1 | Individual 2 | Individual 3 | Individual 4 | Individual 5 | Individual 6 | Individual 7 | |
| NUP85 variant (NM_024844.5) | c.611T>A, c.1904T>G | c.454C>A, c.487C>A | c.932G>A | c.1109A>G, c.1589T>C | c.1430C>T | c.1933C>T | c.405+1G>A | c.1741G>C |
| Amino acid sequence changes | p.Val204Glu, p.Leu635Arg | p.Leu152Ile, p.Leu163Ile | p.Arg311Gln | p.Asn370Ser, p.Met530Thr | p.Ala477Val | p.Arg645Trp | Donor splice site | p.Ala581Pro |
| Parents’ consanguinity | − | − | + | − | + | − | − | − |
| Sex | Male | Male | Female | Female | Female | Male | Female | Male |
| Age at last assessment | 4 years | 3.6 years | 9 years | 27 GW | 8 years | 11 years | 7 years | 4 years |
| Age at onset | birth | birth | birth | prenatal | 8 years | 11 years | 7 years | 4 years |
| Congenital microcephaly | + | + | + | + | NC | NC | NC | NC |
| Intrauterine growth retardation | − | − | + | − | NC | NC | NC | NC |
| Short stature | − | + | + | − | + | − | − | + |
| Underweight | − | − | + | NC | NC | NC | NC | |
| Upslanted palpebral fissures | − | − | + | − | NC | NC | NC | NC |
| Short philtrum | − | + | + | − | NC | NC | NC | NC |
| High nasal bridge | − | − | + | − | NC | NC | NC | NC |
| Reduced vision | − | − | + | Unknown | NC | NC | NC | NC |
| Optic nerve atrophy | − | − | + | Unknown | NC | NC | NC | NC |
| Astigmatism | − | + | + | Unknown | NC | NC | NC | NC |
| Esophoria | − | + | + | Unknown | NC | NC | NC | NC |
| Long, skinny fingers | − | − | − | NC | NC | NC | NC | |
| Syndactyly | − | − | + | − | NC | NC | NC | NC |
| Pes adductus | − | + | + | − | NC | NC | NC | NC |
| Epilepsy | + | − | + | N/A | NC | NC | NC | NC |
| Intellectual disability, moderate | + | + | + | N/A | − | − | + | + |
| Delayed speech and language development | + | + | + | N/A | NC | NC | NC | NC |
| SRNS | + | − | − | N/A | + | + | + | + |
| Muscular hypotonia | + | + | + | N/A | NC | NC | NC | NC |
| Cranial MRI abnormalities | + | + | − | + | − | − | − | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambadauro, A.; Mangano, G.D.; Galletta, K.; Granata, F.; Riva, A.; Massella, L.; Guzzo, I.; Farello, G.; Scorrano, G.; Di Francesco, L.; et al. NUP85 as a Neurodevelopmental Gene: From Podocyte to Neuron. Genes 2023, 14, 2143. https://doi.org/10.3390/genes14122143
Gambadauro A, Mangano GD, Galletta K, Granata F, Riva A, Massella L, Guzzo I, Farello G, Scorrano G, Di Francesco L, et al. NUP85 as a Neurodevelopmental Gene: From Podocyte to Neuron. Genes. 2023; 14(12):2143. https://doi.org/10.3390/genes14122143
Chicago/Turabian StyleGambadauro, Antonella, Giuseppe Donato Mangano, Karol Galletta, Francesca Granata, Antonella Riva, Laura Massella, Isabella Guzzo, Giovanni Farello, Giovanna Scorrano, Ludovica Di Francesco, and et al. 2023. "NUP85 as a Neurodevelopmental Gene: From Podocyte to Neuron" Genes 14, no. 12: 2143. https://doi.org/10.3390/genes14122143
APA StyleGambadauro, A., Mangano, G. D., Galletta, K., Granata, F., Riva, A., Massella, L., Guzzo, I., Farello, G., Scorrano, G., Di Francesco, L., Di Donato, G., Ianni, C., Di Ludovico, A., La Bella, S., Striano, P., Efthymiou, S., Houlden, H., Nardello, R., & Chimenz, R. (2023). NUP85 as a Neurodevelopmental Gene: From Podocyte to Neuron. Genes, 14(12), 2143. https://doi.org/10.3390/genes14122143