
Evaluation of Cell-Specific Alterations in Alzheimer’s Disease and Relevance of In Vitro Models

 , , ,
, , ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
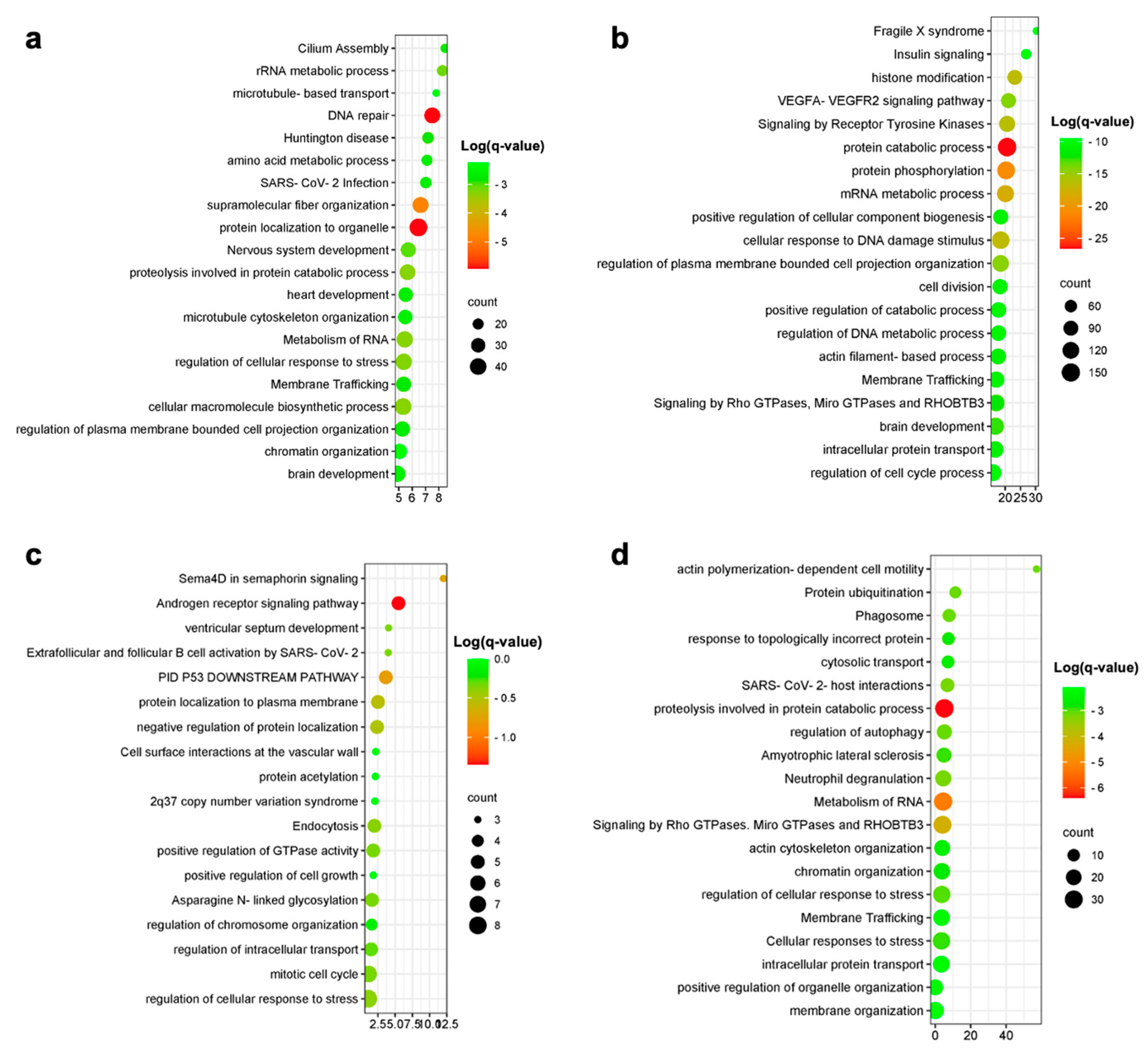
3. Results
3.1. Meta-Analysis of Human Alzheimer’s Disease Transcriptomic Data
3.2. Relevance of an In Vitro Model of AD Neurons
3.3. Relevance of an In Vitro Model of AD Astrocytes
3.4. Relevance of an In Vitro Model of AD Microglia
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grobler, C.; Van Tongeren, M.; Gettemans, J.; Kell, D.B.; Pretorius, E. Alzheimer’s Disease: A Systems View Provides a Unifying Explanation of Its Development. J. Alzheimers Dis. 2023, 91, 43–70. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 2017, 140, 1829–1850. [Google Scholar] [CrossRef] [PubMed]
- Chuang, E.; Hori, A.M.; Hesketh, C.D.; Shorter, J. Amyloid assembly and disassembly. J. Cell Sci. 2018, 131, jcs189928. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Zhang, X.Y.; Tan, L.; Yu, J.T. Tauopathies: Mechanisms and Therapeutic Strategies. J. Alzheimers Dis. 2018, 61, 487–508. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Staging of alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Vogel, J.W.; Initiative, T.A.D.N.; Young, A.L.; Oxtoby, N.P.; Smith, R.; Ossenkoppele, R.; Strandberg, O.T.; La Joie, R.; Aksman, L.M.; Grothe, M.J.; et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med. 2021, 27, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Bettcher, B.M.; Tansey, M.G.; Dorothée, G.; Heneka, M.T. Peripheral and central immune system crosstalk in Alzheimer disease—A research prospectus. Nat. Rev. Neurol. 2021, 17, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Fattorelli, N.; Martinez-Muriana, A.; Wolfs, L.; Geric, I.; De Strooper, B.; Mancuso, R. Stem-cell-derived human microglia transplanted into mouse brain to study human disease. Nat. Protoc. 2021, 16, 1013–1033. [Google Scholar] [CrossRef] [PubMed]
- Graff-Radford, J.; Yong, K.X.X.; Apostolova, L.G.; Bouwman, F.H.; Carrillo, M.; Dickerson, B.C.; Rabinovici, G.D.; Schott, J.M.; Jones, D.T.; Murray, M.E. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021, 20, 222–234. [Google Scholar] [CrossRef]
- Morris, J.C.; McDade, E.M. Alzheimer Disease. Continuum 2022, 28, 648–675. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Zhang, Y. Animal models of Alzheimer’s disease: Applications, evaluation, and perspectives. Zool. Res. 2022, 43, 1026–1040. [Google Scholar] [CrossRef]
- Golde, T.E. Alzheimer’s disease—The journey of a healthy brain into organ failure. Mol. Neurodegener. 2022, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Loera-Valencia, R.; Piras, A.; Ismail, M.A.M.; Manchanda, S.; Eyjolfsdottir, H.; Saido, T.C.; Johansson, J.; Eriksdotter, M.; Winblad, B.; Nilsson, P. Targeting Alzheimer’s disease with gene and cell therapies. J. Intern. Med. 2018, 284, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, J.W.; Victor, M.B.; Tsai, L.H. Dissecting the complexities of Alzheimer disease with in vitro models of the human brain. Nat. Rev. Neurol. 2022, 18, 25–39. [Google Scholar] [CrossRef]
- Saura, C.A.; Deprada, A.; Capilla-López, M.D.; Parra-Damas, A. Revealing cell vulnerability in Alzheimer’s disease by single-cell transcriptomics. Semin. Cell Dev. Biol. 2023, 139, 73–83. [Google Scholar] [CrossRef]
- Desale, S.E.; Chidambaram, H.; Chinnathambi, S. G-protein coupled receptor, PI3K and Rho signaling pathways regulate the cascades of Tau and amyloid-β in Alzheimer’s disease. Mol. Biomed. 2021, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.I.; Blaabjerg, M.; Freude, K.; Meyer, M. RhoA Signaling in Neurodegenerative Diseases. Cells 2022, 11, 1520. [Google Scholar] [CrossRef] [PubMed]
- Takada, E.; Okubo, K.; Yano, Y.; Iida, K.; Someda, M.; Hirasawa, A.; Yonehara, S.; Matsuzaki, K. Molecular Mechanism of Apoptosis by Amyloid β-Protein Fibrils Formed on Neuronal Cells. ACS Chem. Neurosci. 2020, 11, 796–805. [Google Scholar] [CrossRef]
- Smit, T.; Ormel, P.R.; Sluijs, J.A.; Hulshof, L.A.; Middeldorp, J.; de Witte, L.D.; Hol, E.M.; Donega, V. Transcriptomic and functional analysis of Aβ1-42 oligomer-stimulated human monocyte-derived microglia-like cells. Brain Behav. Immun. 2022, 100, 219–230. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Cavalier, A.N.; Roberts, C.M.; Lemieux, M.R.; Ramesh, P.; Garcia, M.A.; Link, C.D. Amyloid beta acts synergistically as a pro-inflammatory cytokine. Neurobiol. Dis. 2021, 159, 105493. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, J.; Hou, Y.; Bekris, L.; Leverenz, J.B.; Pieper, A.A.; Cummings, J.; Cheng, F. The Alzheimer’s Cell Atlas (TACA): A single-cell molecular map for translational therapeutics accelerator in Alzheimer’s disease. Alzheimers Dement. 2022, 8, e12350. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, W.; Liu, Q.; Yang, J.Y.; Li, J.; Yang, M.Q. A new statistical approach to combining p-values using gamma distribution and its application to genome-wide association study. BMC Bioinform. 2014, 15 (Suppl. S17), S3. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Mungenast, A.E.; Siegert, S.; Tsai, L.H. Modeling Alzheimer’s disease with human induced pluripotent stem (iPS) cells. Mol. Cell. Neurosci. 2016, 73, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y.; Harischandra, D.S.; Wang, R.; Ghaisas, S.; Zhao, J.Y.; McMonagle, T.P.; Zhu, G.; Lacuarta, K.D.; Song, J.; Trojanowski, J.Q.; et al. TRIM11 protects against tauopathies and is down-regulated in Alzheimer’s disease. Science 2023, 381, eadd6696. [Google Scholar] [CrossRef] [PubMed]
- Kheiri, G.; Dolatshahi, M.; Rahmani, F.; Rezaei, N. Role of p38/MAPKs in Alzheimer’s disease: Implications for amyloid beta toxicity targeted therapy. Rev. Neurosci. 2018, 30, 9–30. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, B.J.; Zhu, Y.; Lu, Q. Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 97. [Google Scholar] [CrossRef]
- Eichmüller, O.L.; Knoblich, J.A. Human cerebral organoids—A new tool for clinical neurology research. Nat. Rev. Neurol. 2022, 18, 661–680. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO Accession | Model Description |
|---|---|
| GSE139643 | Abeta-stimulated neuroblastoma SH-SY5Y cells |
| GSE187452 | Abeta-stimulated human monocyte-derived microglia cells |
| GSE157461 | Abeta-stimulated primary human astrocytes |
| GEO Accession | Brain Region | N. of Samples |
|---|---|---|
| GSE148822 | Occipital cortex (OC) and occipitotemporal cortex (OCT) | 10 AD, 8 CTR |
| GSE157827 | Prefrontal cortex (PFC) | 12 AD, 9 CTR |
| UP EXC. NEURONS | UP INIB. NEURONS | UP MICROGLIA | DOWN MICROGLIA | UP ASTROCYTES | DOWN ASTROCYTES |
|---|---|---|---|---|---|
| RNF180 | CLDN1 | SSBP1 | C7orf50 | GABBR1 | OSBPL9 |
| PIK3IP1 | RNF180 | ARHGEF37 | PRPSAP2 | SDC4 | ZNF91 |
| FOXP1 | SLC8A1-AS1 | TRIM25 | SP140 | BTRC | ZNF442 |
| SREK1 | COL12A1 | SLC8A1-AS1 | ARID3A | GTF2H2 | AUTS2 |
| MSC-AS1 | ZNF154 | FRA10AC1 | MAGT1 | ZNF654 | PCCA |
| MAPK1IP1L | C11orf1 | SLC7A6OS | SUFU | GBA | TRIM2 |
| RERG | PIK3IP1 | ARL17A | DHTKD1 | BCAP29 | PHIP |
| SESN3 | ARNT | FAM13A | PID1 | TMED9 | ARSG |
| TMEM232 | PIAS2 | OPRM1 | FOXO1 | STOML1 | |
| ZC3H15 | MEGF10 | TK2 | ARID1B | INTS2 | |
| CD109 | GNA13 | LINC02328 | PFKFB4 | SLC35F5 | |
| SYNJ1 | TRPC4 | DLST | GRIPAP1 | RPAP2 | |
| PDXDC1 | CCNL1 | MICU3 | MAPKAPK3 | KDSR | |
| GRM8 | SAV1 | DNAH14 | NEK6 | TRIM44 | |
| SLC48A1 | NT5E | DIAPH2-AS1 | RAB40C | RPP30 | |
| DENND6B | MTMR7 | EFHC1 | ARHGAP35 | REX1BD | |
| CFAP221 | TNXB | RNF217 | RCAN1 | CHST7 | |
| EMCN | SEC14L2 | AUTS2 | SEMA4D | PIAS1 | |
| PRKAA2 | PTPRD | G2E3 | PGPEP1 | DENR | |
| SERP2 | SREK1 | ZNF718 | ROCK2 | DYM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guido, G.; Mangano, K.; Tancheva, L.; Kalfin, R.; Leone, G.M.; Saraceno, A.; Fagone, P.; Nicoletti, F.; Petralia, M.C. Evaluation of Cell-Specific Alterations in Alzheimer’s Disease and Relevance of In Vitro Models. Genes 2023, 14, 2187. https://doi.org/10.3390/genes14122187
Guido G, Mangano K, Tancheva L, Kalfin R, Leone GM, Saraceno A, Fagone P, Nicoletti F, Petralia MC. Evaluation of Cell-Specific Alterations in Alzheimer’s Disease and Relevance of In Vitro Models. Genes. 2023; 14(12):2187. https://doi.org/10.3390/genes14122187
Chicago/Turabian StyleGuido, Giorgio, Katia Mangano, Lyubka Tancheva, Reni Kalfin, Gian Marco Leone, Andrea Saraceno, Paolo Fagone, Ferdinando Nicoletti, and Maria Cristina Petralia. 2023. "Evaluation of Cell-Specific Alterations in Alzheimer’s Disease and Relevance of In Vitro Models" Genes 14, no. 12: 2187. https://doi.org/10.3390/genes14122187
APA StyleGuido, G., Mangano, K., Tancheva, L., Kalfin, R., Leone, G. M., Saraceno, A., Fagone, P., Nicoletti, F., & Petralia, M. C. (2023). Evaluation of Cell-Specific Alterations in Alzheimer’s Disease and Relevance of In Vitro Models. Genes, 14(12), 2187. https://doi.org/10.3390/genes14122187