
Mouse Models of Musculocontractural Ehlers-Danlos Syndrome



Abstract
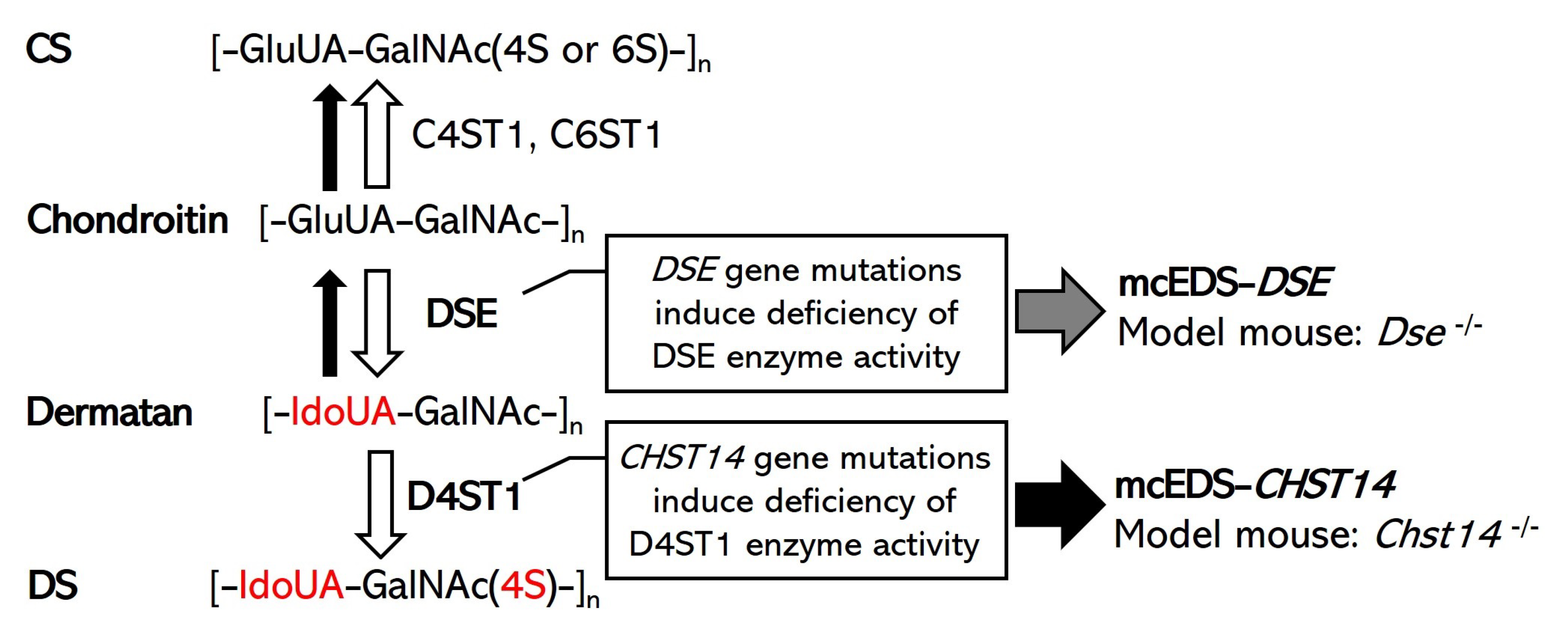
:1. Introduction

2. Model Mouse of mcEDS-DSE
2.1. Dse Gene-Deleted Mouse
2.2. Dsel Gene-Deleted Mouse
2.3. Double Knockout Mouse of Dse and Dsel
3. Model Mouse of mcEDS-CHST14
4. Phenotypic Similarities and Differences of Patients with mcEDS and the Mouse Models
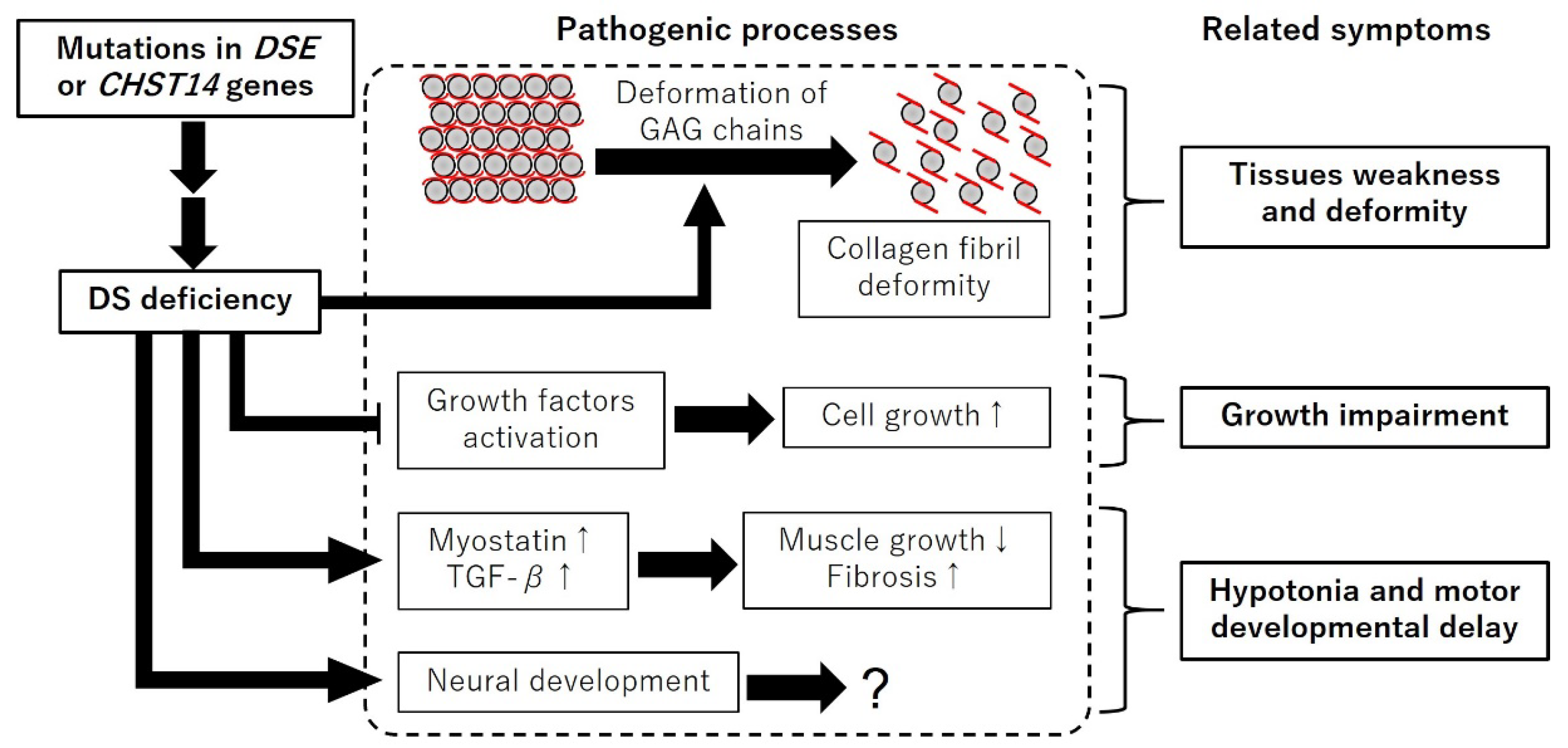
5. Consideration of the Pathogenic Processes of mcEDS
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef]
- Beighton, P.; De Paepe, A.; Steinmann, B.; Tsipouras, P.; Wenstrup, R.J. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am. J. Med. Genet. 1998, 77, 31–37. [Google Scholar] [CrossRef]
- Malfait, F.; Castori, M.; Francomano, C.A.; Giunta, C.; Kosho, T.; Byers, P.H. The Ehlers-Danlos syndromes. Nat. Rev. Dis. Prim. 2020, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Kosho, T.; Mizumoto, S.; Watanabe, T.; Yoshizawa, T.; Miyake, N.; Yamada, S. Recent Advances in the Pathophysiology of Musculocontractural Ehlers-Danlos Syndrome. Genes 2019, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Kosho, T. CHST14/D4ST1 deficiency: New form of Ehlers-Danlos syndrome. Pediatr. Int. 2016, 58, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Brady, A.F.; Demirdas, S.; Fournel-Gigleux, S.; Ghali, N.; Giunta, C.; Kapferer-Seebacher, I.; Kosho, T.; Mendoza-Londono, R.; Pope, M.F.; Rohrbach, M.; et al. The Ehlers-Danlos syndromes, rare types. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 70–115. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Mizumoto, S.; Suresh, I.; Komatsu, Y.; Vodopiutz, J.; Dundar, M.; Straub, V.; Lingenhel, A.; Melmer, A.; Lechner, S.; et al. Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers-Danlos syndrome. Hum. Mol. Genet. 2013, 22, 3761–3772. [Google Scholar] [CrossRef] [PubMed]
- Syx, D.; Delbaere, S.; Bui, C.; De Clercq, A.; Larson, G.; Mizumoto, S.; Kosho, T.; Fournel-Gigleux, S.; Malfait, F. Alterations in glycosaminoglycan biosynthesis associated with the Ehlers-Danlos syndromes. Am. J. Physiol. Cell Physiol. 2022, 323, C1843–C1859. [Google Scholar] [CrossRef] [PubMed]
- Dundar, M.; Muller, T.; Zhang, Q.; Pan, J.; Steinmann, B.; Vodopiutz, J.; Gruber, R.; Sonoda, T.; Krabichler, B.; Utermann, G.; et al. Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am. J. Hum. Genet. 2009, 85, 873–882. [Google Scholar] [CrossRef]
- Kosho, T.; Miyake, N.; Hatamochi, A.; Takahashi, J.; Kato, H.; Miyahara, T.; Igawa, Y.; Yasui, H.; Ishida, T.; Ono, K.; et al. A new Ehlers-Danlos syndrome with craniofacial characteristics, multiple congenital contractures, progressive joint and skin laxity, and multisystem fragility-related manifestations. Am. J. Med. Genet. A 2010, 152A, 1333–1346. [Google Scholar] [CrossRef]
- Miyake, N.; Kosho, T.; Mizumoto, S.; Furuichi, T.; Hatamochi, A.; Nagashima, Y.; Arai, E.; Takahashi, K.; Kawamura, R.; Wakui, K.; et al. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 2010, 31, 966–974. [Google Scholar] [CrossRef]
- Malfait, F.; Syx, D.; Vlummens, P.; Symoens, S.; Nampoothiri, S.; Hermanns-Le, T.; Van Laer, L.; De Paepe, A. Musculocontractural Ehlers-Danlos Syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum. Mutat. 2010, 31, 1233–1239. [Google Scholar] [CrossRef]
- Minatogawa, M.; Hirose, T.; Mizumoto, S.; Yamaguchi, T.; Nagae, C.; Taki, M.; Yamada, S.; Watanabe, T.; Kosho, T. Clinical and pathophysiological delineation of musculocontractural Ehlers-Danlos syndrome caused by dermatan sulfate epimerase deficiency (mcEDS-DSE): A detailed and comprehensive glycobiological and pathological investigation in a novel patient. Hum. Mutat. 2022, 8, e1197. [Google Scholar] [CrossRef] [PubMed]
- Minatogawa, M.; Unzaki, A.; Morisaki, H.; Syx, D.; Sonoda, T.; Janecke, A.R.; Slavotinek, A.; Voermans, N.C.; Lacassie, Y.; Mendoza-Londono, R.; et al. Clinical and molecular features of 66 patients with musculocontractural Ehlers-Danlos syndrome caused by pathogenic variants in CHST14 (mcEDS-CHST14). J. Med. Genet. 2022, 59, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Zhou, T.; Zheng, N.; Lu, Q.; Han, Y. Case report: Multiple gastrointestinal perforations in a rare musculocontractural Ehlers-Danlos syndrome with multiple organ dysfunction. Front. Genet. 2022, 13, 846529. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.R.; Xia, G.; Kang, H.G.; Schachner, M.; Baenziger, J.U. Molecular cloning and characterization of a dermatan-specific N-acetylgalactosamine 4-O-sulfotransferase. J. Biol. Chem. 2001, 276, 36344–36353. [Google Scholar] [CrossRef] [PubMed]
- Maccarana, M.; Olander, B.; Malmstrom, J.; Tiedemann, K.; Aebersold, R.; Lindahl, U.; Li, J.P.; Malmstrom, A. Biosynthesis of dermatan sulfate: Chondroitin-glucuronate C5-epimerase is identical to SART2. J. Biol. Chem. 2006, 281, 11560–11568. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Zhou, D.; Brown, J.R.; Crawford, B.E.; Hennet, T.; Esko, J.D. Biosynthesis of the linkage region of glycosaminoglycans: Cloning and activity of galactosyltransferase II, the sixth member of the β 1,3-galactosyltransferase family (β 3GalT6). J. Biol. Chem. 2001, 276, 48189–48195. [Google Scholar] [CrossRef] [PubMed]
- Gotting, C.; Kuhn, J.; Zahn, R.; Brinkmann, T.; Kleesiek, K. Molecular cloning and expression of human UDP-d-Xylose:proteoglycan core protein β-d-xylosyltransferase and its first isoform XT-II. J. Mol. Biol. 2000, 304, 517–528. [Google Scholar] [CrossRef]
- Kitagawa, H.; Tone, Y.; Tamura, J.; Neumann, K.W.; Ogawa, T.; Oka, S.; Kawasaki, T.; Sugahara, K. Molecular cloning and expression of glucuronyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 1998, 273, 6615–6618. [Google Scholar] [CrossRef] [Green Version]
- Okajima, T.; Yoshida, K.; Kondo, T.; Furukawa, K. Human homolog of Caenorhabditis elegans sqv-3 gene is galactosyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J. Biol. Chem. 1999, 274, 22915–22918. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Yamada, S. The Specific Role of Dermatan Sulfate as an Instructive Glycosaminoglycan in Tissue Development. Int. J. Mol. Sci. 2022, 23, 7845. [Google Scholar] [CrossRef] [PubMed]
- Thelin, M.A.; Bartolini, B.; Axelsson, J.; Gustafsson, R.; Tykesson, E.; Pera, E.; Oldberg, A.; Maccarana, M.; Malmstrom, A. Biological functions of iduronic acid in chondroitin/dermatan sulfate. FEBS J. 2013, 280, 2431–2446. [Google Scholar] [CrossRef]
- Trowbridge, J.M.; Gallo, R.L. Dermatan sulfate: New functions from an old glycosaminoglycan. Glycobiology 2002, 12, 117R–125R. [Google Scholar] [CrossRef] [PubMed]
- Mikami, T.; Mizumoto, S.; Kago, N.; Kitagawa, H.; Sugahara, K. Specificities of three distinct human chondroitin/dermatan N-acetylgalactosamine 4-O-sulfotransferases demonstrated using partially desulfated dermatan sulfate as an acceptor: Implication of differential roles in dermatan sulfate biosynthesis. J. Biol. Chem. 2003, 278, 36115–36127. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Kosho, T.; Matsumoto, N. Ehlers-Danlos syndrome associated with glycosaminoglycan abnormalities. Adv. Exp. Med. Biol. 2014, 802, 145–159. [Google Scholar] [CrossRef]
- Schonherr, E.; Witsch-Prehm, P.; Harrach, B.; Robenek, H.; Rauterberg, J.; Kresse, H. Interaction of biglycan with type I collagen. J. Biol. Chem. 1995, 270, 2776–2783. [Google Scholar] [CrossRef] [PubMed]
- Sugars, R.V.; Milan, A.A.; Brown, J.O.; Waddington, R.J.; Hall, R.C.; Embery, G. Molecular interaction of recombinant decorin and biglycan with type I collagen influences crystal growth. Connect. Tissue Res. 2003, 44, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Pogany, G.; Hernandez, D.J.; Vogel, K.G. The in vitro interaction of proteoglycans with type I collagen is modulated by phosphate. Arch. Biochem. Biophys. 1994, 313, 102–111. [Google Scholar] [CrossRef]
- Janecke, A.R.; Li, B.; Boehm, M.; Krabichler, B.; Rohrbach, M.; Muller, T.; Fuchs, I.; Golas, G.; Katagiri, Y.; Ziegler, S.G.; et al. The phenotype of the musculocontractural type of Ehlers-Danlos syndrome due to CHST14 mutations. Am. J. Med. Genet. A 2016, 170A, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizumoto, S.; Kosho, T.; Hatamochi, A.; Honda, T.; Yamaguchi, T.; Okamoto, N.; Miyake, N.; Yamada, S.; Sugahara, K. Defect in dermatan sulfate in urine of patients with Ehlers-Danlos syndrome caused by a CHST14/D4ST1 deficiency. Clin. Biochem. 2017, 50, 670–677. [Google Scholar] [CrossRef]
- Lautrup, C.K.; Teik, K.W.; Unzaki, A.; Mizumoto, S.; Syx, D.; Sin, H.H.; Nielsen, I.K.; Markholt, S.; Yamada, S.; Malfait, F.; et al. Delineation of musculocontractural Ehlers-Danlos Syndrome caused by dermatan sulfate epimerase deficiency. Mol. Genet. Genom. Med. 2020, 8, e1197. [Google Scholar] [CrossRef]
- Pacheco, B.; Malmstrom, A.; Maccarana, M. Two dermatan sulfate epimerases form iduronic acid domains in dermatan sulfate. J. Biol. Chem. 2009, 284, 9788–9795. [Google Scholar] [CrossRef]
- Maccarana, M.; Kalamajski, S.; Kongsgaard, M.; Magnusson, S.P.; Oldberg, A.; Malmstrom, A. Dermatan sulfate epimerase 1-deficient mice have reduced content and changed distribution of iduronic acids in dermatan sulfate and an altered collagen structure in skin. Mol. Cell. Biol. 2009, 29, 5517–5528. [Google Scholar] [CrossRef]
- Gustafsson, R.; Stachtea, X.; Maccarana, M.; Grottling, E.; Eklund, E.; Malmstrom, A.; Oldberg, A. Dermatan sulfate epimerase 1 deficient mice as a model for human abdominal wall defects. Birth Defects Res. A Clin. Mol. Teratol. 2014, 100, 712–720. [Google Scholar] [CrossRef]
- Nadafi, R.; Koning, J.J.; Veninga, H.; Stachtea, X.N.; Konijn, T.; Zwiers, A.; Malmstrom, A.; den Haan, J.M.M.; Mebius, R.E.; Maccarana, M.; et al. Dendritic Cell Migration to Skin-Draining Lymph Nodes Is Controlled by Dermatan Sulfate and Determines Adaptive Immunity Magnitude. Front. Immunol. 2018, 9, 206. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, B.; Thelin, M.A.; Rauch, U.; Feinstein, R.; Oldberg, A.; Malmstrom, A.; Maccarana, M. Mouse development is not obviously affected by the absence of dermatan sulfate epimerase 2 in spite of a modified brain dermatan sulfate composition. Glycobiology 2012, 22, 1007–1016. [Google Scholar] [CrossRef]
- Akatsu, C.; Mizumoto, S.; Kaneiwa, T.; Maccarana, M.; Malmstrom, A.; Yamada, S.; Sugahara, K. Dermatan sulfate epimerase 2 is the predominant isozyme in the formation of the chondroitin sulfate/dermatan sulfate hybrid structure in postnatal developing mouse brain. Glycobiology 2011, 21, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Stachtea, X.N.; Tykesson, E.; van Kuppevelt, T.H.; Feinstein, R.; Malmstrom, A.; Reijmers, R.M.; Maccarana, M. Dermatan Sulfate-Free Mice Display Embryological Defects and Are Neonatal Lethal Despite Normal Lymphoid and Non-Lymphoid Organogenesis. PLoS ONE 2015, 10, e0140279. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Li, L.; Tang, J.; Li, Y.; Lin, W.Y.; Martin, F.; Grant, D.; Solloway, M.; Parker, L.; Ye, W.; et al. A mouse knockout library for secreted and transmembrane proteins. Nat. Biotechnol. 2010, 28, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Akyuz, N.; Bernreuther, C.; Loers, G.; Laczynska, E.; Jakovcevski, I.; Schachner, M. Dermatan sulfotransferase Chst14/D4st1, but not chondroitin sulfotransferase Chst11/C4st1, regulates proliferation and neurogenesis of neural progenitor cells. J. Cell Sci. 2011, 124, 4051–4063. [Google Scholar] [CrossRef]
- Nitahara-Kasahara, Y.; Mizumoto, S.; Inoue, Y.U.; Saka, S.; Posadas-Herrera, G.; Nakamura-Takahashi, A.; Takahashi, Y.; Hashimoto, A.; Konishi, K.; Miyata, S.; et al. A new mouse model of Ehlers-Danlos syndrome generated using CRISPR/Cas9-mediated genomic editing. Dis. Model. Mech. 2021, 14, dmm048963. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, T.; Mizumoto, S.; Takahashi, Y.; Shimada, S.; Sugahara, K.; Nakayama, J.; Takeda, S.; Nomura, Y.; Nitahara-Kasahara, Y.; Okada, T.; et al. Vascular abnormalities in the placenta of Chst14-/- fetuses: Implications in the pathophysiology of perinatal lethality of the murine model and vascular lesions in human CHST14/D4ST1 deficiency. Glycobiology 2018, 28, 80–89. [Google Scholar] [CrossRef]
- Akyuz, N.; Rost, S.; Mehanna, A.; Bian, S.; Loers, G.; Oezen, I.; Mishra, B.; Hoffmann, K.; Guseva, D.; Laczynska, E.; et al. Dermatan 4-O-sulfotransferase1 ablation accelerates peripheral nerve regeneration. Exp. Neurol. 2013, 247, 517–530. [Google Scholar] [CrossRef]
- Shimada, S.; Yoshizawa, T.; Takahashi, Y.; Nitahara-Kasahara, Y.; Okada, T.; Nomura, Y.; Yamanaka, H.; Kosho, T.; Matsumoto, K. Backcrossing to an appropriate genetic background improves the birth rate of carbohydrate sulfotransferase 14 gene-deleted mice. Exp. Anim. 2020, 69, 407–413. [Google Scholar] [CrossRef]
- Hirose, T.; Mizumoto, S.; Hashimoto, A.; Takahashi, Y.; Yoshizawa, T.; Nitahara-Kasahara, Y.; Takahashi, N.; Nakayama, J.; Takehana, K.; Okada, T.; et al. Systematic investigation of the skin in Chst14-/- mice: A model for skin fragility in musculocontractural Ehlers-Danlos syndrome caused by CHST14 variants (mcEDS-CHST14). Glycobiology 2021, 31, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Nitahara-Kasahara, Y.; Posadas-Herrera, G.; Mizumoto, S.; Nakamura-Takahashi, A.; Inoue, Y.U.; Inoue, T.; Nomura, Y.; Takeda, S.; Yamada, S.; Kosho, T.; et al. Myopathy Associated With Dermatan Sulfate-Deficient Decorin and Myostatin in Musculocontractural Ehlers-Danlos Syndrome: A Mouse Model Investigation. Front. Cell Dev. Biol. 2021, 9, 695021. [Google Scholar] [CrossRef] [PubMed]
- Rost, S.; Akyuz, N.; Martinovic, T.; Huckhagel, T.; Jakovcevski, I.; Schachner, M. Germline ablation of dermatan-4O-sulfotransferase1 reduces regeneration after mouse spinal cord injury. Neuroscience 2016, 312, 74–85. [Google Scholar] [CrossRef]
- Li, Q.; Wu, X.; Na, X.; Ge, B.; Wu, Q.; Guo, X.; Ntim, M.; Zhang, Y.; Sun, Y.; Yang, J.; et al. Impaired Cognitive Function and Altered Hippocampal Synaptic Plasticity in Mice Lacking Dermatan Sulfotransferase Chst14/D4st1. Front. Mol. Neurosci. 2019, 12, 26. [Google Scholar] [CrossRef]
- Goossens, D.; Van Gestel, S.; Claes, S.; De Rijk, P.; Souery, D.; Massat, I.; Van den Bossche, D.; Backhovens, H.; Mendlewicz, J.; Van Broeckhoven, C.; et al. A novel CpG-associated brain-expressed candidate gene for chromosome 18q-linked bipolar disorder. Mol. Psychiatry 2003, 8, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Okamoto, N.; Miyake, N.; Taira, K.; Sato, Y.; Matsuda, K.; Akimaru, N.; Ohashi, H.; Wakui, K.; Fukushima, Y.; et al. Delineation of dermatan 4-O-sulfotransferase 1 deficient Ehlers-Danlos syndrome: Observation of two additional patients and comprehensive review of 20 reported patients. Am. J. Med. Genet. A 2011, 155A, 1949–1958. [Google Scholar] [CrossRef] [PubMed]
- Schirwani, S.; Metcalfe, K.; Wagner, B.; Berry, I.; Sobey, G.; Jewell, R. DSE associated musculocontractural EDS, a milder phenotype or phenotypic variability. Eur. J. Med. Genet. 2020, 63, 103798. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Bernfield, M.; Gotte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Penc, S.F.; Pomahac, B.; Winkler, T.; Dorschner, R.A.; Eriksson, E.; Herndon, M.; Gallo, R.L. Dermatan sulfate released after injury is a potent promoter of fibroblast growth factor-2 function. J. Biol. Chem. 1998, 273, 28116–28121. [Google Scholar] [CrossRef]
- Plichta, J.K.; Radek, K.A. Sugar-coating wound repair: A review of FGF-10 and dermatan sulfate in wound healing and their potential application in burn wounds. J. Burn Care Res. 2012, 33, 299–310. [Google Scholar] [CrossRef]
- Rousseau, F.; Bonaventure, J.; Legeai-Mallet, L.; Pelet, A.; Rozet, J.M.; Maroteaux, P.; Le Merrer, M.; Munnich, A. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature 1994, 371, 252–254. [Google Scholar] [CrossRef]
- Shiang, R.; Thompson, L.M.; Zhu, Y.Z.; Church, D.M.; Fielder, T.J.; Bocian, M.; Winokur, S.T.; Wasmuth, J.J. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 1994, 78, 335–342. [Google Scholar] [CrossRef]
- Horton, W.A.; Hall, J.G.; Hecht, J.T. Achondroplasia. Lancet 2007, 370, 162–172. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Marie, P.J. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 2015, 29, 1463–1486. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| mcEDS-CHST14 | mcEDS- DSE | Chst14-/- Mouse | Dse-/- Mouse | |
|---|---|---|---|---|
| Perinatal lethality | ? | ? | + * [43,44,45] | + * [34,35] |
| Reduced body weight | + [51] | ± [13] | − #, + $ [43,44] | + [34] |
| Reduced Body length | + [51] | − [13] | + [43,44] | + [34] |
| Skin fragility | + (90%) [14] | + (29%) [14] | + [42,44,46] | + [34] |
| Dermal collagen fibril deformity | + [11,13,46,52] | + [11,13,46,52] | + [46] | + [34] |
| Large subcutaneous hematoma | + (81%) [14] | + (67%) [14] | − † [43] | ? |
| Spinal deformities | + (87%) [14] | + (57%) [14] | + § [42,44] | ? § [34,35] |
| Talipes deformities | + (98%) [14] | + (100%) [14] | − [42] | ? |
| Hypotonia | + (86%) [14] | + (75%) [14] | + [42] | ? |
| Motor developmental delay | + (87%) [14] | + (75%) [14] | + [42] | ? |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshizawa, T.; Kosho, T. Mouse Models of Musculocontractural Ehlers-Danlos Syndrome. Genes 2023, 14, 436. https://doi.org/10.3390/genes14020436
Yoshizawa T, Kosho T. Mouse Models of Musculocontractural Ehlers-Danlos Syndrome. Genes. 2023; 14(2):436. https://doi.org/10.3390/genes14020436
Chicago/Turabian StyleYoshizawa, Takahiro, and Tomoki Kosho. 2023. "Mouse Models of Musculocontractural Ehlers-Danlos Syndrome" Genes 14, no. 2: 436. https://doi.org/10.3390/genes14020436
APA StyleYoshizawa, T., & Kosho, T. (2023). Mouse Models of Musculocontractural Ehlers-Danlos Syndrome. Genes, 14(2), 436. https://doi.org/10.3390/genes14020436