
mRNA Signatures in Peripheral White Blood Cells Predict Reproductive Potential in Beef Heifers at Weaning

 ,
,  ,
, 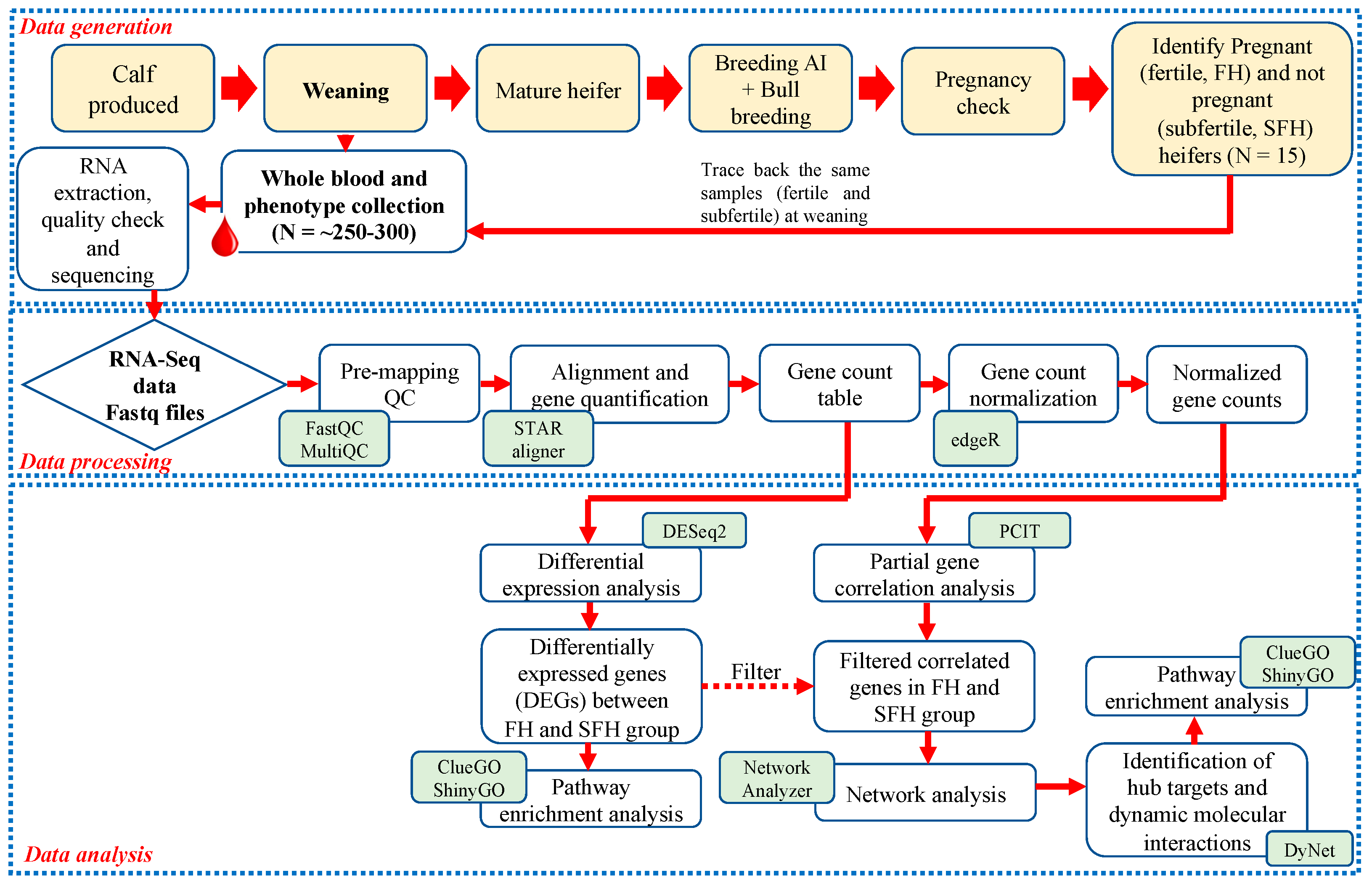{kind=link}
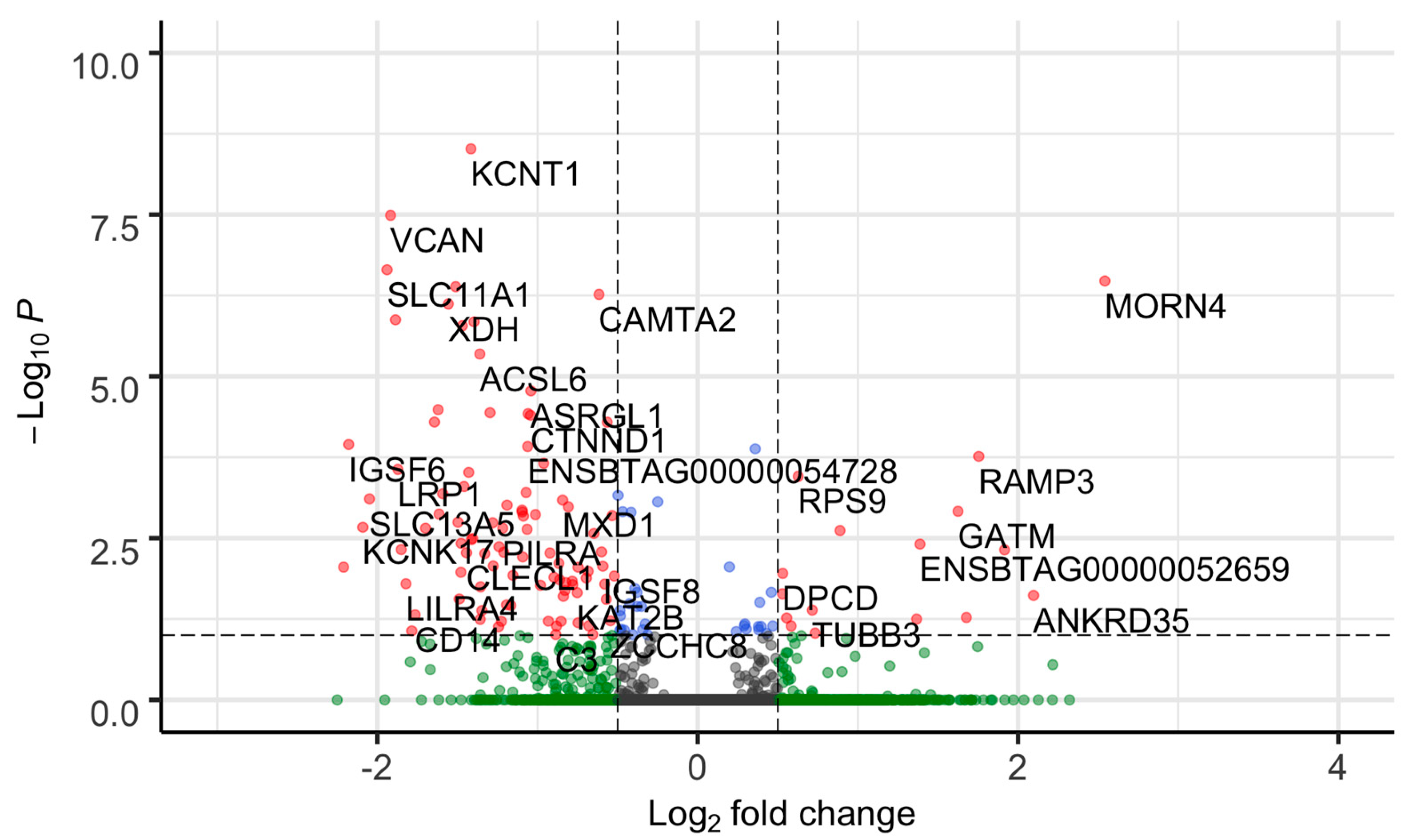{kind=link}
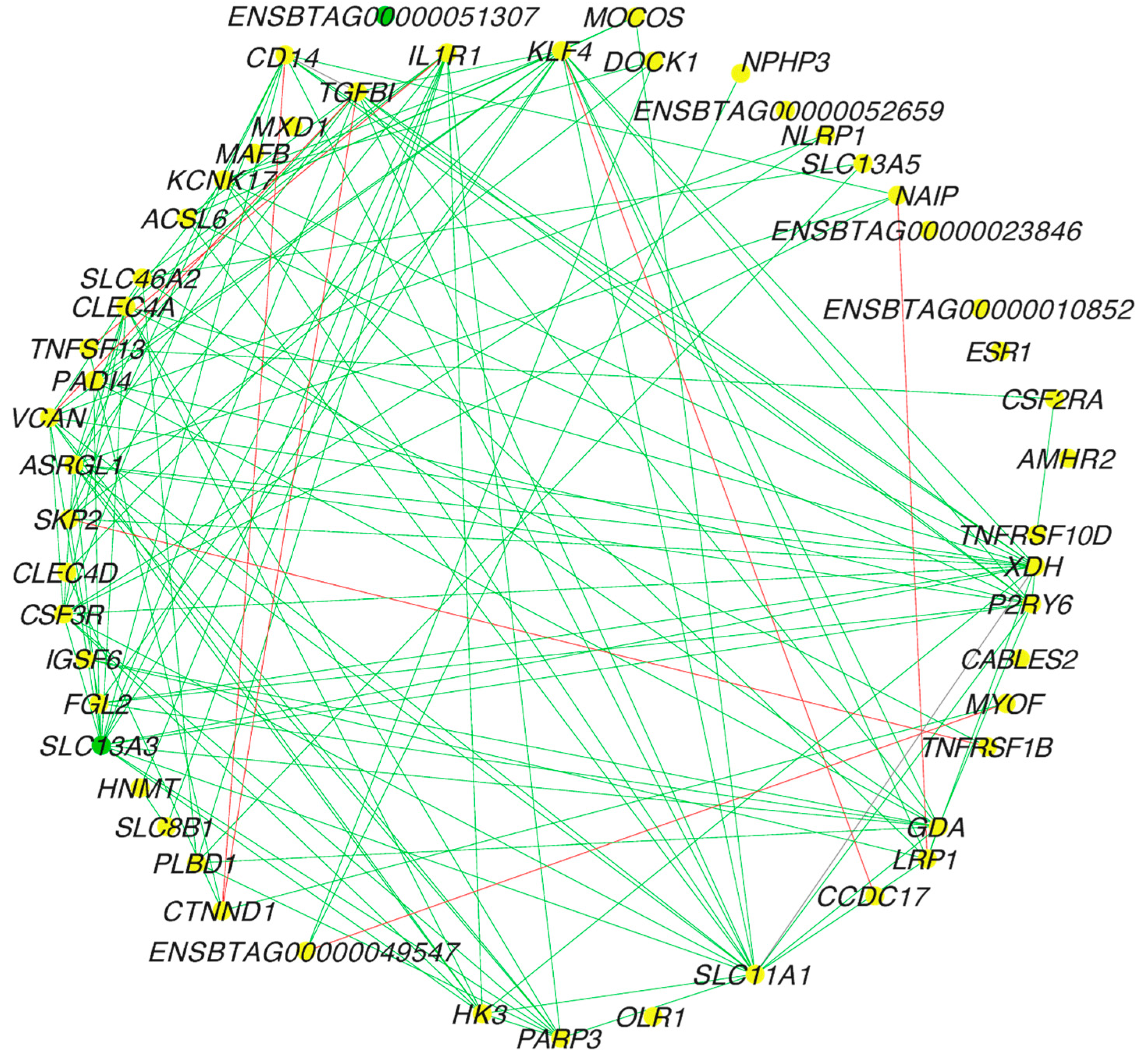{kind=link}
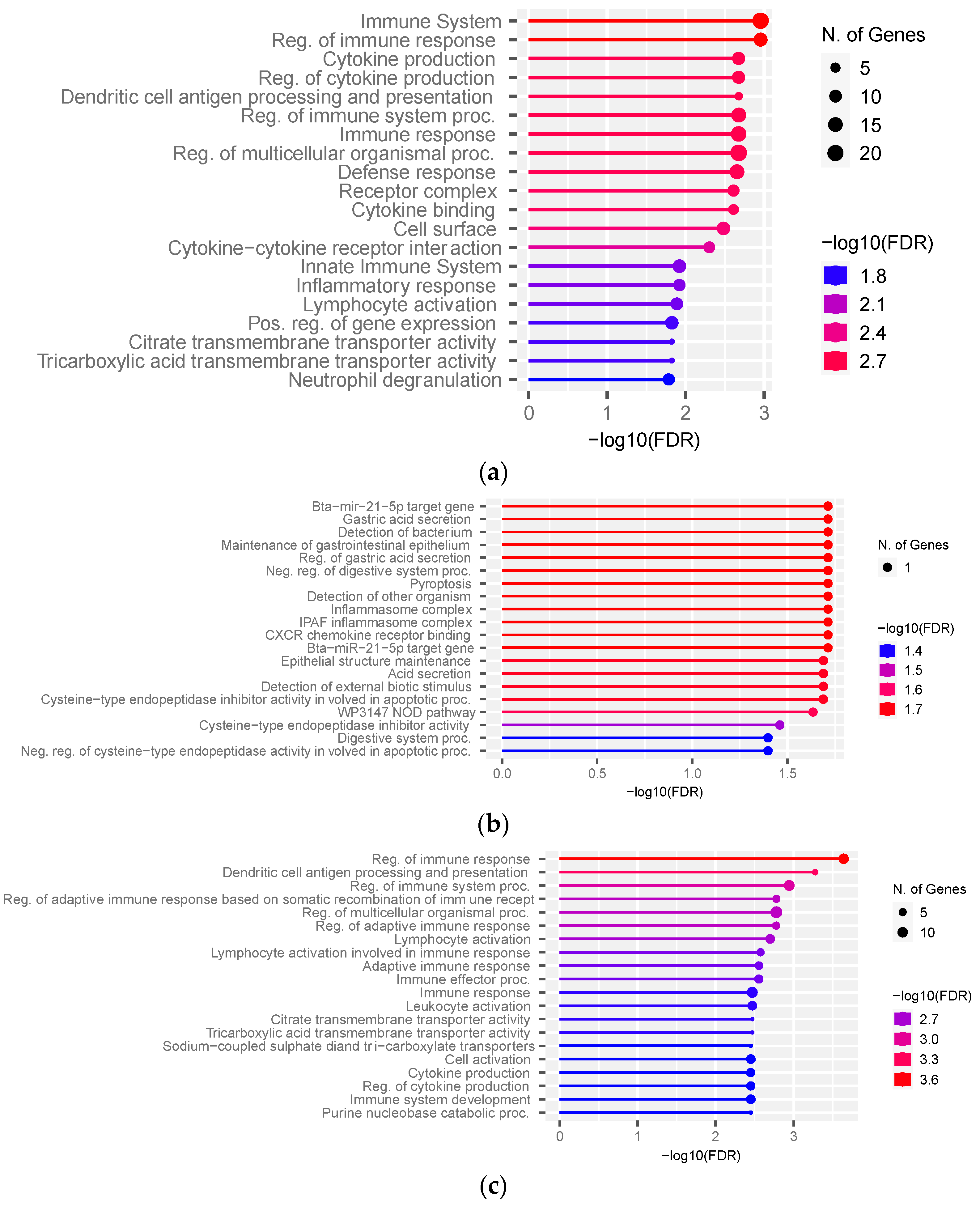{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Handling and Phenotype Collection
2.2. RNA Extraction, Library Preparation, and Sequencing
2.3. Data Processing and Differential Expression
2.4. Co-Expression Profile, DEGs Filtering, and Gene Networks
2.5. Pathway Analysis
2.6. mRNA Expression of Top Targets at Weaning with RT-qPCR
2.7. Statistical Analysis
3. Results
3.1. Transcriptome Profiling from PWBCs and Differential Expression Analysis
3.2. PCIT and Network Analysis
3.3. Functional Over-Representation Analysis
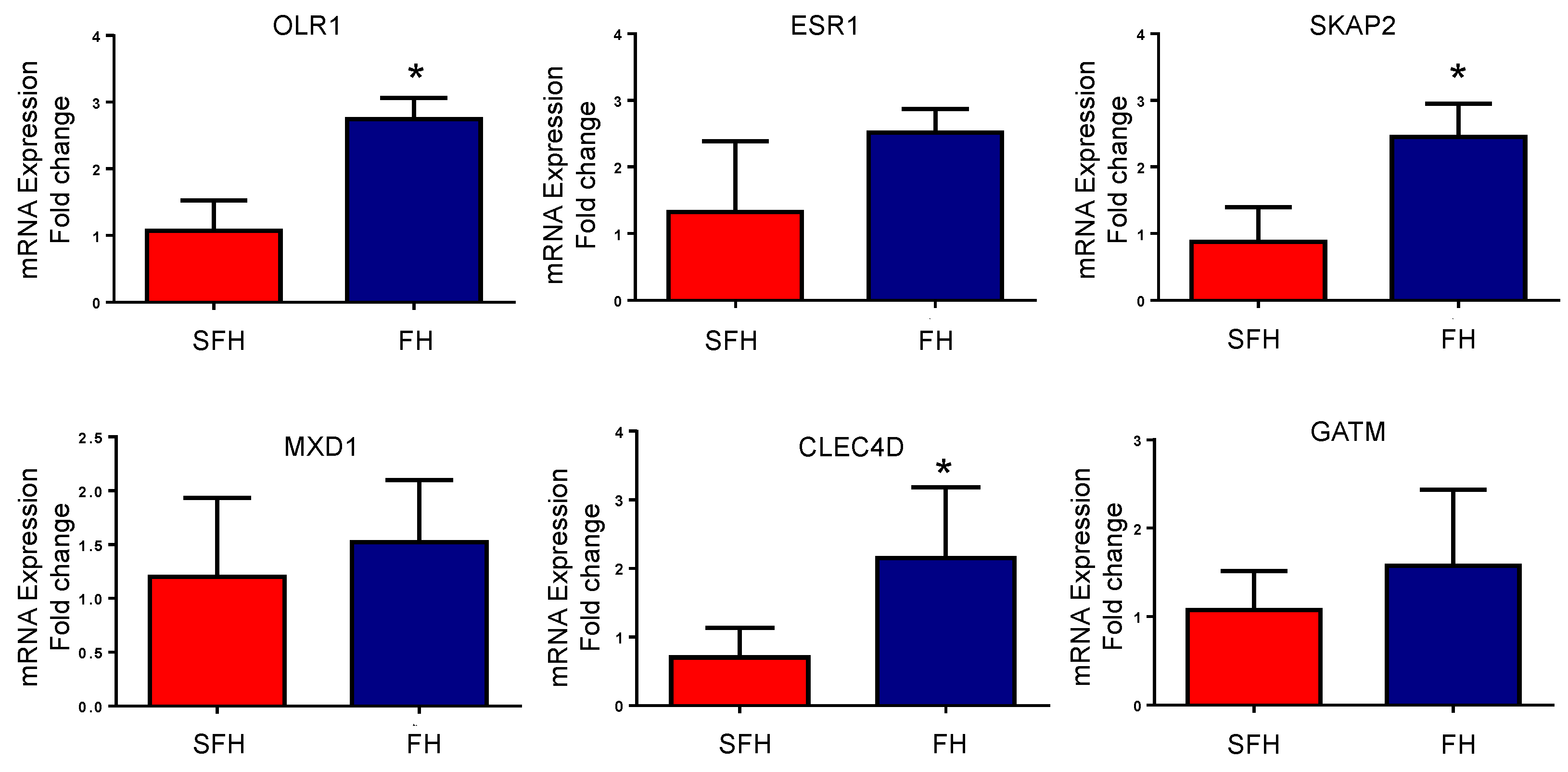
3.4. mRNA Expression of the Identified DEGs and Hub Targets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moorey, S.E.; Biase, F.H. Beef Heifer Fertility: Importance of Management Practices and Technological Advancements. J. Anim. Sci. Biotechnol. 2020, 11, 97. [Google Scholar] [CrossRef]
- Wathes, D.C.; Brickell, J.S.; Bourne, N.E.; Swali, A.; Cheng, Z. Factors Influencing Heifer Survival and Fertility on Commercial Dairy Farms. Animal 2008, 2, 1135–1143. [Google Scholar] [CrossRef] [Green Version]
- Sprott, D.B.H.; Sprott, L.R. Body Condition, Nutrition and Reproduction of Beef Cows. AgriLife Ext. Tex. 1998, B-1526, 1–11. [Google Scholar]
- Markusfeld, O.; Galon, N.; Ezra, E. Body Condition Score, Health, Yield and Fertility in Dairy Cows. Vet. Rec. 1997, 141, 67–72. [Google Scholar] [CrossRef]
- Anderson, K.J.; LeFever, D.G.; Brinks, J.S.; Odde, K.G. The Use of Reproductive Tract Scoring in Beef Heifers. Agri-Practice 1991, 12, 19–26. [Google Scholar]
- Neville, W.E.; Mullinix, B.G.; Smith, J.B.; McCormick, W.C. Growth Patterns for Pelvic Dimensions and Other Body Measurements of Beef Females. J. Anim. Sci. 1978, 47, 1080–1088. [Google Scholar] [CrossRef] [Green Version]
- Veerkamp, R.F.; Beerda, B. Genetics and Genomics to Improve Fertility in High Producing Dairy Cows. Theriogenology 2007, 68, S266–S273. [Google Scholar] [CrossRef]
- Kim, K.; Zakharkin, S.O.; Allison, D.B. Expectations, Validity, and Reality in Gene Expression Profiling. J. Clin. Epidemiol. 2010, 63, 950–959. [Google Scholar] [CrossRef] [Green Version]
- Graf, A.; Krebs, S.; Zakhartchenko, V.; Schwalb, B.; Blum, H.; Wolf, E. Fine Mapping of Genome Activation in Bovine Embryos by RNA Sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 4139–4144. [Google Scholar] [CrossRef] [Green Version]
- Chitwood, J.L.; Rincon, G.; Kaiser, G.G.; Medrano, J.F.; Ross, P.J. RNA-Seq Analysis of Single Bovine Blastocysts. BMC Genom. 2013, 14, 350. [Google Scholar] [CrossRef] [Green Version]
- Forde, N.; Carter, F.; Fair, T.; Crowe, M.A.; Evans, A.C.O.; Spencer, T.E.; Bazer, F.W.; McBride, R.; Boland, M.P.; O’Gaora, P.; et al. Progesterone-Regulated Changes in Endometrial Gene Expression Contribute to Advanced Conceptus Development in Cattle. Biol. Reprod. 2009, 81, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Mazzoni, G.; Pedersen, H.S.; Rabaglino, M.B.; Hyttel, P.; Callesen, H.; Kadarmideen, H.N. Characterization of the Endometrial Transcriptome in Early Diestrus Influencing Pregnancy Status in Dairy Cattle after Transfer of in Vitro-Produced Embryos. Physiol. Genom. 2020, 52, 269–279. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Ramos, R.S.; Pugliesi, G.; Andrade, S.C.S.; Van Hoeck, V.; Langbeen, A.; Oliveira, M.L.; Gonella-Diaza, A.M.; Gasparin, G.; Fukumasu, H.; et al. Endometrial Transcriptional Profiling of a Bovine Fertility Model by Next-Generation Sequencing. Genom. Data 2016, 7, 26–28. [Google Scholar] [CrossRef] [Green Version]
- Diniz, W.J.S.; Banerjee, P.; Rodning, S.P.; Dyce, P.W. Machine Learning-Based Co-Expression Network Analysis Unravels Potential Fertility-Related Genes in Beef Cows. Animals 2022, 12, 2715. [Google Scholar] [CrossRef]
- Binelli, M.; Scolari, S.C.; Pugliesi, G.; Van Hoeck, V.; Gonella-Diaza, A.M.; Andrade, S.C.S.; Gasparin, G.R.; Coutinho, L.L. The Transcriptome Signature of the Receptive Bovine Uterus Determined at Early Gestation. PLoS ONE 2015, 10, e0122874. [Google Scholar] [CrossRef] [Green Version]
- Moorey, S.E.; Walker, B.N.; Elmore, M.F.; Elmore, J.B.; Rodning, S.P.; Biase, F.H. Rewiring of Gene Expression in Circulating White Blood Cells Is Associated with Pregnancy Outcome in Heifers (Bos Taurus). Sci. Rep. 2020, 10, 16786. [Google Scholar] [CrossRef]
- Dickinson, S.E.; Griffin, B.A.; Elmore, M.F.; Kriese-Anderson, L.; Elmore, J.B.; Dyce, P.W.; Rodning, S.P.; Biase, F.H. Transcriptome Profiles in Peripheral White Blood Cells at the Time of Artificial Insemination Discriminate Beef Heifers with Different Fertility Potential. BMC Genom. 2018, 19, 129. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Rodning, S.P.; Diniz, W.J.S.; Dyce, P.W. Co-Expression Network and Integrative Analysis of Metabolome and Transcriptome Uncovers Biological Pathways for Fertility in Beef Heifers. Metabolites 2022, 12, 708. [Google Scholar] [CrossRef]
- Hudson, N.J.; Dalrymple, B.P.; Reverter, A. Beyond Differential Expression: The Quest for Causal Mutations and Effector Molecules. BMC Genom. 2012, 13, 356. [Google Scholar] [CrossRef] [Green Version]
- Basu, M.; Wang, K.; Ruppin, E.; Hannenhalli, S. Predicting Tissue-Specific Gene Expression from Whole Blood Transcriptome. Sci. Adv. 2021, 7, eabd6991. [Google Scholar] [CrossRef]
- Koh, W.; Pan, W.; Gawad, C.; Fan, H.C.; Kerchner, G.A.; Wyss-Coray, T.; Blumenfeld, Y.J.; El-Sayed, Y.Y.; Quake, S.R. Noninvasive in Vivo Monitoring of Tissue-Specific Global Gene Expression in Humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7361–7366. [Google Scholar] [CrossRef] [Green Version]
- Michou, V.I.; Kanavaros, P.; Athanassiou, V.; Chronis, G.B.; Stabamas, S.; Tsilivakos, V. Fraction of the Peripheral Blood Concentration of CD56+/CD16−/CD3− Cells in Total Natural Killer Cells as an Indication of Fertility and Infertility. Fertil. Steril. 2003, 80, 691–697. [Google Scholar] [CrossRef]
- Thum, M.Y.; Bhaskaran, S.; Abdalla, H.I.; Ford, B.; Sumar, N.; Shehata, H.; Bansal, A.S. An Increase in the Absolute Count of CD56dimCD16+CD69+ NK Cells in the Peripheral Blood Is Associated with a Poorer IVF Treatment and Pregnancy Outcome. Hum. Reprod. 2004, 19, 2395–2400. [Google Scholar] [CrossRef] [Green Version]
- van Dam, S.; Võsa, U.; van der Graaf, A.; Franke, L.; de Magalhães, J.P. Gene Co-Expression Analysis for Functional Classification and Gene–Disease Predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef]
- Gaiteri, C.; Ding, Y.; French, B.; Tseng, G.C.; Sibille, E. Beyond Modules and Hubs: The Potential of Gene Coexpression Networks for Investigating Molecular Mechanisms of Complex Brain Disorders. Genes Brain Behav. 2014, 13, 13–24. [Google Scholar] [CrossRef] [Green Version]
- de la Fuente, A. From ‘Differential Expression’ to ‘Differential Networking’—Identification of Dysfunctional Regulatory Networks in Diseases. Trends Genet. 2010, 26, 326–333. [Google Scholar] [CrossRef]
- Gustafsson, M.; Nestor, C.E.; Zhang, H.; Barabási, A.-L.; Baranzini, S.; Brunak, S.; Chung, K.F.; Federoff, H.J.; Gavin, A.-C.; Meehan, R.R.; et al. Modules, Networks and Systems Medicine for Understanding Disease and Aiding Diagnosis. Genome Med. 2014, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Zhang, B.; Hoffman, E.P.; Clarke, R.; Zhang, Z.; Shih, I.-M.; Xuan, J.; Herrington, D.M.; Wang, Y. Knowledge-Fused Differential Dependency Network Models for Detecting Significant Rewiring in Biological Networks. BMC Syst. Biol. 2014, 8, 87. [Google Scholar] [CrossRef] [Green Version]
- Andrew, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 16 August 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2018. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 24 August 2022).
- Reverter, A.; Chan, E.K.F. Combining Partial Correlation and an Information Theory Approach to the Reversed Engineering of Gene Co-Expression Networks. Bioinformatics 2008, 24, 2491–2497. [Google Scholar] [CrossRef] [Green Version]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.-E.; Lengauer, T.; Albrecht, M. Computing Topological Parameters of Biological Networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Fuller, T.F.; Ghazalpour, A.; Aten, J.E.; Drake, T.A.; Lusis, A.J.; Horvath, S. Weighted Gene Coexpression Network Analysis Strategies Applied to Mouse Weight. Mamm. Genome 2007, 18, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A Graphical Gene-Set Enrichment Tool for Animals and Plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Pohler, K.G.; Franco, G.A.; Reese, S.T.; Smith, M.F. Physiology and Pregnancy of Beef Cattle. Animal Agriculture 2020, 37–55. [Google Scholar] [CrossRef]
- Gutierrez, K.; Kasimanickam, R.; Tibary, A.; Gay, J.M.; Kastelic, J.P.; Hall, J.B.; Whittier, W.D. Effect of Reproductive Tract Scoring on Reproductive Efficiency in Beef Heifers Bred by Timed Insemination and Natural Service versus Only Natural Service. Theriogenology 2014, 81, 918–924. [Google Scholar] [CrossRef]
- Bruno, V.; Rizzacasa, B.; Pietropolli, A.; Capogna, M.V.; Massoud, R.; Ticconi, C.; Piccione, E.; Cortese, C.; Novelli, G.; Amati, F. OLR1 and Loxin Expression in PBMCs of Women with a History of Unexplained Recurrent Miscarriage: A Pilot Study. Genet. Test. Mol. Biomark. 2017, 21, 363–372. [Google Scholar] [CrossRef]
- Mohammed, M.M.; Al-Thuwaini, T.M.; Al-Shuhaib, M.B.S. A Novel p.K116Q SNP in the OLR1 Gene and Its Relation to Fecundity in Awassi Ewes. Theriogenology 2022, 184, 185–190. [Google Scholar] [CrossRef]
- Cao, C.; Pressman, E.K.; Cooper, E.M.; Guillet, R.; Westerman, M.; O’Brien, K.O. Placental Heme Receptor LRP1 Correlates with the Heme Exporter FLVCR1 and Neonatal Iron Status. Reproduction 2014, 148, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Bretherick, K.L.; Hanna, C.W.; Currie, L.M.; Fluker, M.R.; Hammond, G.L.; Robinson, W.P. Estrogen Receptor α Gene Polymorphisms Are Associated with Idiopathic Premature Ovarian Failure. Fertil. Steril. 2008, 89, 318–324. [Google Scholar] [CrossRef]
- Bahia, W.; Soltani, I.; Haddad, A.; Soua, A.; Radhouani, A.; Mahdhi, A.; Ferchichi, S. Association of Genetic Variants in Estrogen Receptor (ESR)1 and ESR2 with Susceptibility to Recurrent Pregnancy Loss in Tunisian Women: A Case Control Study. Gene 2020, 736, 144406. [Google Scholar] [CrossRef]
- Jin, X.; Chen, Y.; Sheng, Z.; Wang, X.; Zhang, Z.; Huang, J.; Zhou, J.; Lin, F. SKAP2 Is Downregulated in the Villous Tissues of Patients with Missed Abortion and Regulates Growth and Migration in Trophoblasts through the WAVE2-ARP2/3 Signaling Pathway. Placenta 2022, 128, 100–111. [Google Scholar] [CrossRef]
- Moufarrej, M.N.; Vorperian, S.K.; Wong, R.J.; Campos, A.A.; Quaintance, C.C.; Sit, R.V.; Tan, M.; Detweiler, A.M.; Mekonen, H.; Neff, N.F.; et al. Early Prediction of Preeclampsia in Pregnancy with Cell-Free RNA. Nature 2022, 602, 689–694. [Google Scholar] [CrossRef]
- Ellery, S.J.; Murthi, P.; Della Gatta, P.A.; May, A.K.; Davies-Tuck, M.L.; Kowalski, G.M.; Callahan, D.L.; Bruce, C.R.; Wallace, E.M.; Walker, D.W.; et al. The Effects of Early-Onset Pre-Eclampsia on Placental Creatine Metabolism in the Third Trimester. Int. J. Mol. Sci. 2020, 21, 806. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.G.A.; Pereira, L.A.A.C.; Viana, J.H.M.; Russo, R.C.; Campos-Junior, P.H.A. The CC-Chemokine Receptor 2 Is Involved in the Control of Ovarian Folliculogenesis and Fertility Lifespan in Mice. J. Reprod. Immunol. 2020, 141, 103174. [Google Scholar] [CrossRef]
- Kong, F.; Su, Z.; Zhou, C.; Sun, C.; Liu, Y.; Zheng, D.; Yuan, H.; Yin, J.; Fang, J.; Wang, S.; et al. Role of Positive Selection in Functional Divergence of Mammalian Neuronal Apoptosis Inhibitor Proteins during Evolution. J. Biomed. Biotechnol. 2011, 2011, 809765. [Google Scholar] [CrossRef] [Green Version]
- Morón-Calvente, V.; Romero-Pinedo, S.; Toribio-Castelló, S.; Plaza-Díaz, J.; Abadía-Molina, A.C.; Rojas-Barros, D.I.; Beug, S.T.; LaCasse, E.C.; MacKenzie, A.; Korneluk, R.; et al. Inhibitor of Apoptosis Proteins, NAIP, CIAP1 and CIAP2 Expression during Macrophage Differentiation and M1/M2 Polarization. PLoS ONE 2018, 13, e0193643. [Google Scholar] [CrossRef]
- Yockey, L.J.; Iwasaki, A. Role of Interferons and Proinflammatory Cytokines in Pregnancy and Fetal Development. Immunity 2018, 49, 397–412. [Google Scholar] [CrossRef] [Green Version]
- Perry, I.D.; Krishnan, L.; Murphy, S.P. SLC11A1 Is Expressed in the Human Placenta across Multiple Gestational Ages. Placenta 2019, 75, 23–26. [Google Scholar] [CrossRef]
- Evans, P.M.; Liu, C. Role of Kruppel-like Factor 4 in Normal Homeostasis, Cancer and Stem Cells. Acta Biochim. Biophys. Sin. 2008, 40, 554–564. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, H.N.; Sales, K.J.; Catalano, R.D.; Norman, J.E. Inflammatory Pathways in Female Reproductive Health and Disease. Reproduction 2009, 138, 903–919. [Google Scholar] [CrossRef]
- Hamada, M.; Tsunakawa, Y.; Jeon, H.; Yadav, M.K.; Takahashi, S. Role of MafB in Macrophages. Exp. Anim. 2020, 69, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Beloosesky, R.; Maravi, N.; Weiner, Z.; Khativ, N.; Ross, M.G.; Itskovitz-Eldor, J. 482: Maternal Lipopolysaccharide (LPS) Induced Inflammation during Pregnancy Programs Impaired Offspring Innate Immune Responses. Am. J. Obstet. Gynecol. 2008, 199, S143. [Google Scholar] [CrossRef]
- Alharatani, R.; Ververi, A.; Beleza-Meireles, A.; Ji, W.; Mis, E.; Patterson, Q.T.; Griffin, J.N.; Bhujel, N.; Chang, C.A.; Dixit, A.; et al. Novel Truncating Mutations in CTNND1 Cause a Dominant Craniofacial and Cardiac Syndrome. Hum. Mol. Genet. 2020, 29, 1900–1921. [Google Scholar] [CrossRef] [Green Version]
- Oas, R.G.; Xiao, K.; Summers, S.; Wittich, K.B.; Chiasson, C.M.; Martin, W.D.; Grossniklaus, H.E.; Vincent, P.A.; Reynolds, A.B.; Kowalczyk, A.P. P120-Catenin Is Required for Mouse Vascular Development. Circ. Res. 2010, 106, 941–951. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, P.; Diniz, W.J.S.; Hollingsworth, R.; Rodning, S.P.; Dyce, P.W. mRNA Signatures in Peripheral White Blood Cells Predict Reproductive Potential in Beef Heifers at Weaning. Genes 2023, 14, 498. https://doi.org/10.3390/genes14020498
Banerjee P, Diniz WJS, Hollingsworth R, Rodning SP, Dyce PW. mRNA Signatures in Peripheral White Blood Cells Predict Reproductive Potential in Beef Heifers at Weaning. Genes. 2023; 14(2):498. https://doi.org/10.3390/genes14020498
Chicago/Turabian StyleBanerjee, Priyanka, Wellison J. S. Diniz, Rachel Hollingsworth, Soren P. Rodning, and Paul W. Dyce. 2023. "mRNA Signatures in Peripheral White Blood Cells Predict Reproductive Potential in Beef Heifers at Weaning" Genes 14, no. 2: 498. https://doi.org/10.3390/genes14020498
APA StyleBanerjee, P., Diniz, W. J. S., Hollingsworth, R., Rodning, S. P., & Dyce, P. W. (2023). mRNA Signatures in Peripheral White Blood Cells Predict Reproductive Potential in Beef Heifers at Weaning. Genes, 14(2), 498. https://doi.org/10.3390/genes14020498