
Zeb2 DNA-Binding Sites in Neuroprogenitor Cells Reveal Autoregulation and Affirm Neurodevelopmental Defects, Including in Mowat-Wilson Syndrome
 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. ESC Culture Conditions and Differentiation
2.2. Western Blots
2.3. RNA Extraction and RT-qPCR Analysis
2.4. Tag-Zeb2 Mouse ESCs
2.5. CRISPR/Cas9-Mediated Deletion of the Zeb2 Binding Site Located at chr2:45109746-45110421
2.6. Chromatin Immunoprecipitation (ChIP)
2.7. ChIP-Sequencing
2.8. ChIP-Sequencing Data Analysis
2.9. Transcription Factors Motif Enrichment Analysis
2.10. RNA-Sequencing
2.11. Meta-Analysis, Pathways Enrichment, Gene Ontology, Function Analysis, and Gene to Disease Association
2.12. Zeb2 Short Hairpin (sh) RNA-Mediated Knock-Down
3. Results
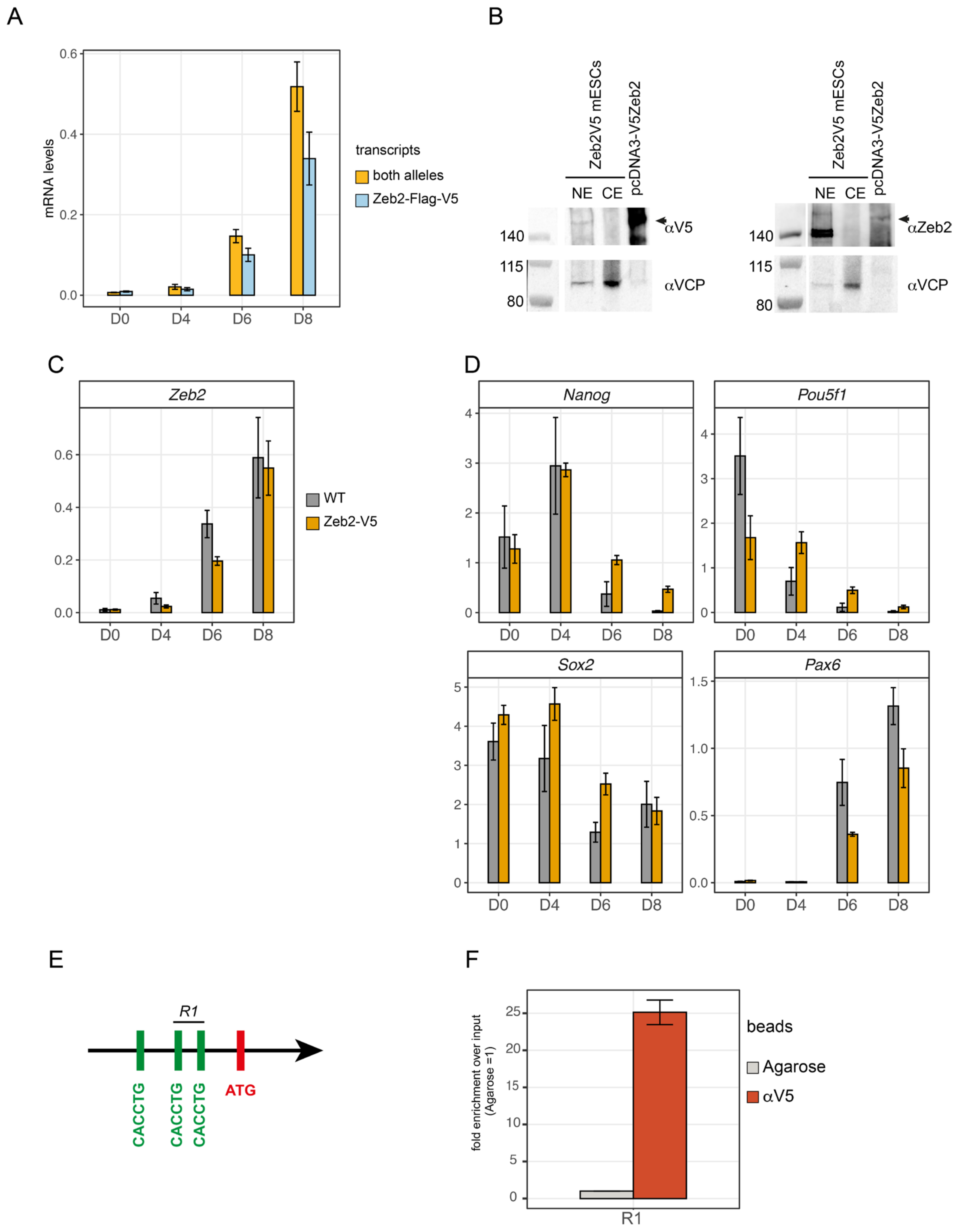
3.1. Heterozygous Zeb2-V5 ESCs Differentiate as Wild-Type Cells
3.2. One-Third of 2432 Zeb2 DNA-Binding Sites Map Close to the Transcription Start Site of System-Relevant Expressed and Protein-Encoding Genes, Including the Zeb2 Gene Itself
3.3. Zeb2 Peaks Overlap with Active Enhancers and Promoters
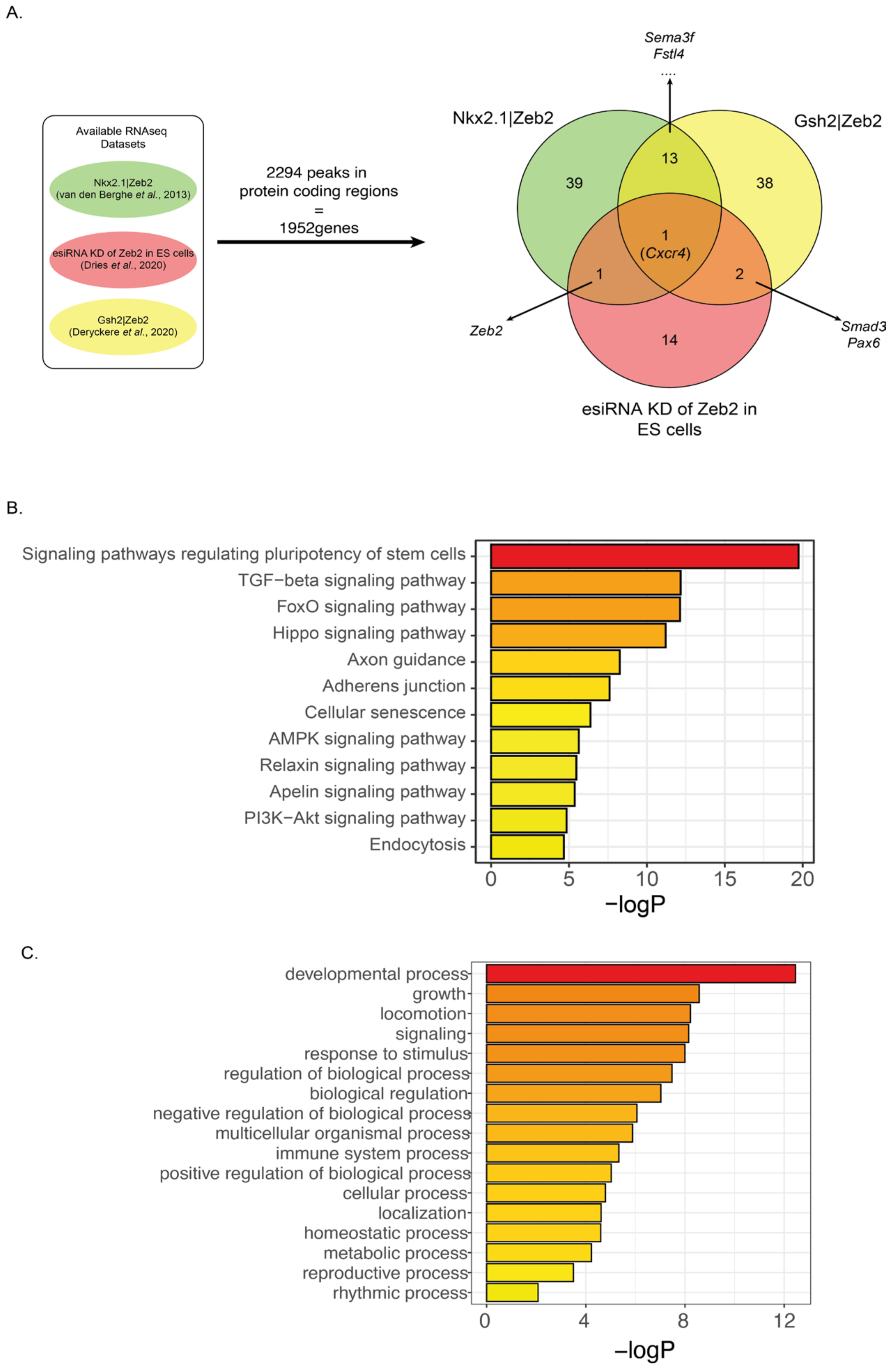
3.4. Meta-Analysis of Identified Binding Sites and Perturbed-Zeb2 RNA-Seq Data Reveal Overlapping Zeb2 Target Genes
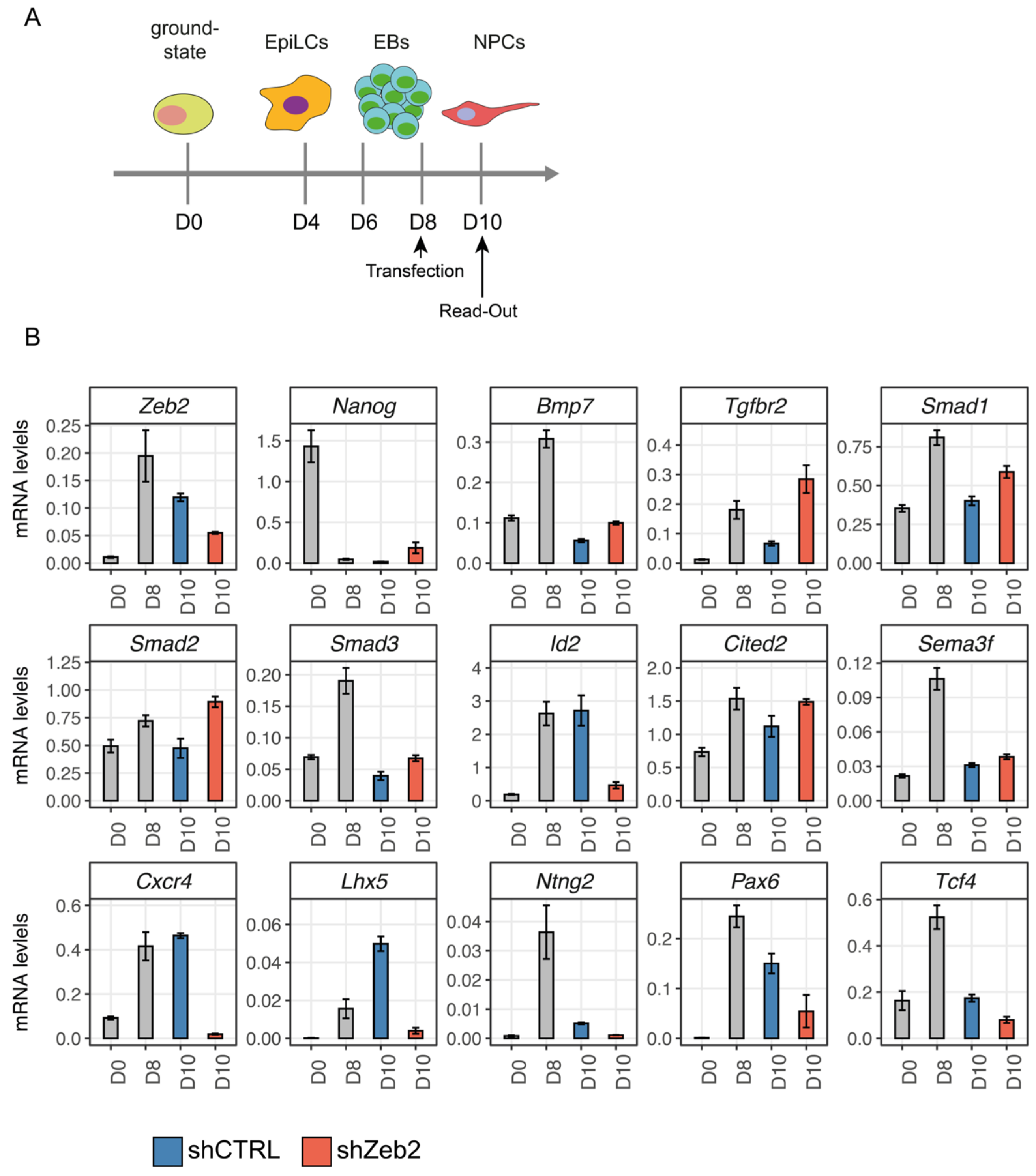
3.5. Zeb2 Directly Controls TGFβ/BMP-System Component and Neuronal Differentiation/Migration Genes
3.6. Zeb2 Potentiates Its Own Gene Expression, Which Is Crucial for Proper Control of Some of Its Direct Target Genes
3.7. Extrapolation of Zeb2 ChIP-Seq Data to Cell-Based Clinical Manifestation of MOWS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Funahashi, J.; Kamachi, Y.; Goto, K.; Kondoh, H. Identification of nuclear factor delta EF1 and its binding site essential for lens-specific activity of the delta 1-crystallin enhancer. Nucleic Acids Res. 1991, 19, 3543–3547. [Google Scholar] [CrossRef]
- Sekido, R.; Murai, K.; Funahashi, J.; Kamachi, Y.; Fujisawa-Sehara, A.; Nabeshima, Y.; Kondoh, H. The delta-crystallin enhancer-binding protein delta EF1 is a repressor of E2-box-mediated gene activation. Mol. Cell. Biol. 1994, 14, 5692–5700. [Google Scholar] [CrossRef]
- Remacle, J.E.; Kraft, H.; Lerchner, W.; Wuytens, G.; Collart, C.; Verschueren, K.; Smith, J.C.; Huylebroeck, D. New mode of DNA binding of multi-zinc finger transcription factors: DeltaEF1 family members bind with two hands to two target sites. EMBO J. 1999, 18, 5073–5084. [Google Scholar] [CrossRef] [Green Version]
- Verschueren, K.; Remacle, J.E.; Collart, C.; Kraft, H.; Baker, B.S.; Tylzanowski, P.; Nelles, L.; Wuytens, G.; Su, M.T.; Bodmer, R.; et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5’-CACCT sequences in candidate target genes. J. Biol. Chem. 1999, 274. [Google Scholar] [CrossRef] [Green Version]
- Mowat, D.R.; Wilson, M.J.; Goossens, M. Mowat-Wilson syndrome. J. Med. Genet. 2003, 40, 305–310. [Google Scholar] [CrossRef]
- Cerruti Mainardi, P.; Pastore, G.; Zweier, C.; Rauch, A. Mowat-Wilson syndrome and mutation in the zinc finger homeo box 1B gene: A well-defined clinical entity. J. Med. Genet. 2004, 41, e16. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, N.; Yamada, K.; Yamada, Y.; Miura, K.; Kato, J.; Kuwabara, N.; Hara, Y.; Kobayashi, Y.; Hoshino, K.; Nomura, Y.; et al. Clinical and molecular analysis of Mowat-Wilson syndrome associated with ZFHX1B mutations and deletions at 2q22-q24.1. J. Med. Genet. 2004, 41, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Mowat, D.R.; Croaker, G.D.; Cass, D.T.; Kerr, B.A.; Chaitow, J.; Adès, L.C.; Chia, N.L.; Wilson, M.J. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: Delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J. Med. Genet. 1998, 35, 617–623. [Google Scholar] [CrossRef]
- Cacheux, V.; Dastot-Le Moal, F.; Kääriäinen, H.; Bondurand, N.; Rintala, R.; Boissier, B.; Wilson, M.; Mowat, D.; Goossens, M. Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic Hirschsprung disease. Hum. Mol. Genet. 2001, 10, 1503–1510. [Google Scholar] [CrossRef] [Green Version]
- Wakamatsu, N.; Yamada, Y.; Yamada, K.; Ono, T.; Nomura, N.; Taniguchi, H.; Kitoh, H.; Mutoh, N.; Yamanaka, T.; Mushiake, K.; et al. Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease. Nat. Genet. 2001, 27, 369–370. [Google Scholar] [CrossRef]
- Yamada, K.; Yamada, Y.; Nomura, N.; Miura, K.; Wakako, R.; Hayakawa, C.; Matsumoto, A.; Kumagai, T.; Yoshimura, I.; Miyazaki, S.; et al. Nonsense and frameshift mutations in ZFHX1B, encoding Smad-interacting protein 1, cause a complex developmental disorder with a great variety of clinical features. Am. J. Hum. Genet. 2001, 69, 1178–1185. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.; Mowat, D.; Dastot-Le Moal, F.; Cacheux, V.; Kääriäinen, H.; Cass, D.; Donnai, D.; Clayton-Smith, J.; Townshend, S.; Curry, C.; et al. Further delineation of the phenotype associated with heterozygous mutations in ZFHX1B. Am. J. Med. Genet. A 2003, 119, 257–265. [Google Scholar] [CrossRef]
- Zweier, C.; Thiel, C.T.; Dufke, A.; Crow, Y.J.; Meinecke, P.; Suri, M.; Ala-Mello, S.; Beemer, F.; Bernasconi, S.; Bianchi, P.; et al. Clinical and mutational spectrum of Mowat-Wilson syndrome. Eur. J. Med. Genet. 2005, 48, 97–111. [Google Scholar] [CrossRef]
- Zweier, C.; Horn, D.; Kraus, C.; Rauch, A. Atypical ZFHX1B mutation associated with a mild Mowat-Wilson syndrome phenotype. Am. J. Med. Genet. A 2006, 140, 869–872. [Google Scholar] [CrossRef]
- Garavelli, L.; Zollino, M.; Mainardi, P.C.; Gurrieri, F.; Rivieri, F.; Soli, F.; Verri, R.; Albertini, E.; Favaron, E.; Zignani, M.; et al. Mowat-Wilson syndrome: Facial phenotype changing with age: Study of 19 Italian patients and review of the literature. Am. J. Med. Genet. A 2009, 149, 417–426. [Google Scholar] [CrossRef]
- Ivanovski, I.; Djuric, O.; Caraffi, S.G.; Santodirocco, D.; Pollazzon, M.; Rosato, S.; Cordelli, D.M.; Abdalla, E.; Accorsi, P.; Adam, M.P.; et al. Phenotype and genotype of 87 patients with Mowat-Wilson syndrome and recommendations for care. Genet. Med. 2018, 20, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Garavelli, L.; Ivanovski, I.; Caraffi, S.G.; Santodirocco, D.; Pollazzon, M.; Cordelli, D.M.; Abdalla, E.; Accorsi, P.; Adam, M.P.; Baldo, C.; et al. Neuroimaging findings in Mowat-Wilson syndrome: A study of 54 patients. Genet. Med. 2017, 19, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Ricci, E.; Fetta, A.; Garavelli, L.; Caraffi, S.; Ivanovski, I.; Bonanni, P.; Accorsi, P.; Giordano, L.; Pantaleoni, C.; Romeo, A.; et al. Further delineation and long-term evolution of electroclinical phenotype in Mowat Wilson Syndrome. A longitudinal study in 40 individuals. Epilepsy Behav. 2021, 124, 108315. [Google Scholar] [CrossRef]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Seuntjens, E.; Nityanandam, A.; Miquelajauregui, A.; Debruyn, J.; Stryjewska, A.; Goebbels, S.; Nave, K.A.; Huylebroeck, D.; Tarabykin, V. Sip1 regulates sequential fate decisions by feedback signalling from postmitotic neurons to progenitors. Nat. Neurosci. 2009, 12, 1373–1380. [Google Scholar] [CrossRef] [Green Version]
- Weng, Q.; Chen, Y.; Wang, H.; Xu, X.; Yang, B.; He, Q.; Shou, W.; Chen, Y.; Higashi, Y.; van den Berghe, V.; et al. Dual-mode modulation of Smad signalling by Smad-interacting protein Sip1 is required for myelination in the central nervous system. Neuron 2012, 73, 713–728. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.L.; Soen, B.; Martens, L.; Skrypek, N.; Saelens, W.; Taminau, J.; Blancke, G.; Van Isterdael, G.; Huylebroeck, D.; Haigh, J.; et al. The transcription factor Zeb2 regulates development of conventional and plasmacytoid DCs by repressing Id2. J. Exp. Med. 2016, 213, 897–911. [Google Scholar] [CrossRef] [Green Version]
- Deryckere, A.; Stappers, E.; Dries, R.; Peyre, E.; van den Berghe, V.; Conidi, A.; Zampeta, F.I.; Francis, A.; Bresseleers, M.; Stryjewska, A.; et al. Multifaceted actions of Zeb2 in postnatal neurogenesis from the ventricular-subventricular zone to the olfactory bulb. Development 2020, 147, dev184861. [Google Scholar] [CrossRef]
- Wu, L.M.; Wang, J.; Conidi, A.; Zhao, C.; Wang, H.; Ford, Z.; Zhang, L.; Zweier, C.; Ayee, B.G.; Maurel, P.; et al. Zeb2 recruits HDAC-NuRD to inhibit Notch and controls Schwann cell differentiation and remyelination. Nat. Neurosci. 2016, 19, 1060–1072. [Google Scholar] [CrossRef]
- Stryjewska, A.; Dries, R.; Pieters, T.; Verstappen, G.; Conidi, A.; Coddens, K.; Francis, A.; Umans, L.; van IJcken, W.F.; Berx, G.; et al. Zeb2 Regulates Cell Fate at the Exit from Epiblast State in Mouse Embryonic Stem Cells. Stem Cells 2017, 35, 611–625. [Google Scholar] [CrossRef]
- Menuchin-Lasowski, Y.; Dagan, B.; Conidi, A.; Cohen-Gulkar, M.; David, A.; Ehrlich, M.; Giladi, P.O.; Clark, B.S.; Blackshaw, S.; Shapira, K.; et al. Zeb2 regulates the balance between retinal interneurons and Müller glia by inhibition of BMP-Smad signalling. Dev. Biol. 2020, 468, 80–92. [Google Scholar] [CrossRef]
- Lerchner, W.; Latinkic, B.V.; Remacle, J.E.; Huylebroeck, D.; Smith, J.C. Region-specific activation of the Xenopus brachyury promoter involves active repression in ectoderm and endoderm: A study using transgenic frog embryos. Development 2000, 127, 2729–2739. [Google Scholar] [CrossRef]
- Van Grunsven, L.A.; Taelman, V.; Michiels, C.; Opdecamp, K.; Huylebroeck, D.; Bellefroid, E.J. deltaEF1 and SIP1 are differentially expressed and have overlapping activities during Xenopus embryogenesis. Dev. Dyn. 2006, 235, 1491–1500. [Google Scholar] [CrossRef]
- Conidi, A.; Cazzola, S.; Beets, K.; Coddens, K.; Collart, C.; Cornelis, F.; Cox, L.; Debruyn, J.; Dobreva, M.P.; Dries, R.; et al. Few Smad proteins and many Smad-interacting proteins yield multiple functions and action modes in TGFβ/BMP signalling in vivo. Cytokine Growth Factor Rev. 2011, 22, 287–300. [Google Scholar] [CrossRef]
- Birkhoff, J.; Huylebroeck, D.; Conidi, A. ZEB2, the Mowat-Wilson Syndrome transcription factor: Confirmations, novel functions, and continuing surprises. Genes 2021, 12, 1037. [Google Scholar] [CrossRef]
- Balcik-Ercin, P.; Cetin, M.; Yalim-Camci, I.; Odabas, G.; Tokay, N.; Sayan, A.E.; Yagci, T. Genome-wide analysis of endogenously expressed ZEB2 binding sites reveals inverse correlations between ZEB2 and GalNAc-transferase GALNT3 in human tumors. Cell. Oncol. 2018, 41, 379–393. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Toledo, E.M.; Rosmaninho, P.; Peng, C.; Uhlén, P.; Castro, D.S.; Arenas, E. A Zeb2-miR-200c loop controls midbrain dopaminergic neuron neurogenesis and migration. Commun. Biol. 2018, 1, 75. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, T.; Maruhashi, M.; Van de Putte, T.; Kondoh, H.; Huylebroeck, D.; Higashi, Y. Complementary expression pattern of Zfhx1 genes Sip1 and deltaEF1 in the mouse embryo and their genetic interaction revealed by compound mutants. Dev. Dyn. 2006, 235, 1941–1952. [Google Scholar] [CrossRef]
- Vandamme, N.; Denecker, G.; Bruneel, K.; Blancke, G.; Akay, Ö.; Taminau, J.; De Coninck, J.; De Smedt, E.; Skrypek, N.; Van Loocke, W.; et al. The EMT Transcription Factor ZEB2 Promotes Proliferation of Primary and Metastatic Melanoma While Suppressing an Invasive, Mesenchymal-Like Phenotype. Cancer Res. 2020, 80, 2983–2995. [Google Scholar] [CrossRef]
- Nelles, L.; Van de Putte, T.; van Grunsven, L.; Huylebroeck, D.; Verschueren, K. Organization of the mouse Zfhx1b gene encoding the two-handed zinc finger repressor Smad-interacting protein-1. Genomics 2003, 82, 460–469. [Google Scholar] [CrossRef]
- Van den Berghe, V.; Stappers, E.; Vandesande, B.; Dimidschstein, J.; Kroes, R.; Francis, A.; Conidi, A.; Lesage, F.; Dries, R.; Cazzola, S.; et al. Directed migration of cortical interneurons depends on the cell-autonomous action of Sip1. Neuron 2013, 77, 70–82. [Google Scholar] [CrossRef] [Green Version]
- Dries, R.; Stryjewska, A.; Coddens, K.; Okawa, S.; Notelaers, T.; Birkhoff, J.; Dekker, M.; Verfaillie, C.M.; Del Sol, A.; Mulugeta, E.; et al. Integrative and perturbation-based analysis of the transcriptional dynamics of TGFβ/BMP system components in transition from embryonic stem cells to neural progenitors. Stem Cells 2020, 38, 202–217. [Google Scholar] [CrossRef] [Green Version]
- Bibel, M.; Richter, J.; Lacroix, E.; Barde, Y.A. Generation of a defined and uniform population of CNS progenitors and neurons from mouse embryonic stem cells. Nat. Protoc. 2007, 2, 1034–1043. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Puig, R.R.; Boddie, P.; Khan, A.; Castro-Mondragon, J.A.; Mathelier, A. UniBind: Maps of high-confidence direct TF-DNA interactions across nine species. BMC Genom. 2021, 22, 482. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Pinero, J.; Ramirez-Anguita, J.M.; Sauch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [Green Version]
- Papin, C.; van Grunsven, L.A.; Verschueren, K.; Huylebroeck, D.; Smith, J.C. Dynamic regulation of Brachyury expression in the amphibian embryo by XSIP1. Mech. Dev. 2002, 111, 37–46. [Google Scholar] [CrossRef]
- Verstappen, G.; van Grunsven, L.A.; Michiels, C.; Van de Putte, T.; Souopgui, J.; Van Damme, J.; Bellefroid, E.; Vandekerckhove, J.; Huylebroeck, D. Atypical Mowat-Wilson patient confirms the importance of the novel association between ZFHX1B/SIP1 and NuRD corepressor complex. Hum. Mol. Genet. 2008, 17, 1175–1183. [Google Scholar] [CrossRef] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Sepp, M.; Pruunsild, P.; Timmusk, T. Pitt-Hopkins syndrome-associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to dominant-negative effects. Hum. Mol. Genet. 2012, 21, 2873–2888. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, J.R.; Szeto, R.A.; Carvalho, V.M.A.; Muotri, A.R.; Papes, F. Transcription factor 4 and its association with psychiatric disorders. Transl. Psychiatry 2021, 11, 19. [Google Scholar] [CrossRef]
- Corneliussen, B.; Thornell, A.; Hallberg, B.; Grundstrom, T. Helix-loop-helix transcriptional activators bind to a sequence in glucocorticoid response elements of retrovirus enhancers. J. Virol. 1991, 65, 6084–6093. [Google Scholar] [CrossRef] [Green Version]
- Forrest, M.P.; Hill, M.J.; Quantock, A.J.; Martin-Rendon, E.; Blake, D.J. The emerging roles of TCF4 in disease and development. Trends Mol. Med. 2014, 20, 322–331. [Google Scholar] [CrossRef]
- Wang, L.H.; Baker, N.E. E Proteins and ID Proteins: Helix-Loop-Helix Partners in Development and Disease. Dev. Cell 2015, 35, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.; Cai, J.; Clevers, H.; Fast, E.; Gray, S.; Greenberg, R.; Jain, M.K.; Ma, Q.; Qiu, M.; Rowitch, D.H.; et al. A genome-wide screen for spatially restricted expression patterns identifies transcription factors that regulate glial development. J. Neurosci. 2009, 29, 11399–11408. [Google Scholar] [CrossRef] [Green Version]
- Meert, L.; Birkhoff, J.C.; Conidi, A.; Poot, R.A.; Huylebroeck, D. Different E-box binding transcription factors, similar neuro-developmental defects: ZEB2 (Mowat-Wilson syndrome) and TCF4 (Pitt-Hopkins syndrome). Rare Dis. Orphan Drugs J. 2022, 1, 8. [Google Scholar] [CrossRef]
- Brockschmidt, A.; Todt, U.; Ryu, S.; Hoischen, A.; Landwehr, C.; Birnbaum, S.; Frenck, W.; Radlwimmer, B.; Lichter, P.; Engels, H.; et al. Severe mental retardation with breathing abnormalities (Pitt-Hopkins syndrome) is caused by haploinsufficiency of the neuronal bHLH transcription factor TCF4. Hum. Mol. Genet. 2007, 16, 1488–1494. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, Z.; Dong, Q.; Xiong, J.; Zhu, B. Histone H3K27 acetylation is dispensable for enhancer activity in mouse embryonic stem cells. Genome Biol. 2020, 21, 45. [Google Scholar] [CrossRef] [Green Version]
- Suh, H.; Consiglio, A.; Ray, J.; Sawai, T.; D’Amour, K.A.; Gage, F.H. In vivo fate analysis reveals the multipotent and self-renewal capacities of Sox2+ neural stem cells in the adult hippocampus. Cell Stem Cell 2007, 1, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Quintes, S.; Brinkmann, B.G.; Ebert, M.; Fröb, F.; Kungl, T.; Arlt, F.A.; Tarabykin, V.; Huylebroeck, D.; Meijer, D.; Suter, U.; et al. Zeb2 is essential for Schwann cell differentiation, myelination and nerve repair. Nat. Neurosci. 2016, 19, 1050–1059. [Google Scholar] [CrossRef] [Green Version]
- Dunn, S.J.; Martello, G.; Yordanov, B.; Emmott, S.; Smith, A.G. Defining an essential transcription factor program for naïve pluripotency. Science 2014, 344, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Rao, C.; Malaguti, M.; Mason, J.O.; Lowell, S. The transcription factor E2A drives neural differentiation in pluripotent cells. Development 2020, 147, dev184093. [Google Scholar] [CrossRef]
- El Wakil, A.; Francius, C.; Wolff, A.; Pleau-Varet, J.; Nardelli, J. The GATA2 transcription factor negatively regulates the proliferation of neuronal progenitors. Development 2006, 133, 2155–2165. [Google Scholar] [CrossRef] [Green Version]
- Beagan, J.A.; Duong, M.T.; Titus, K.R.; Zhou, L.; Cao, Z.; Ma, J.; Lachanski, C.V.; Gillis, D.R.; Phillips-Cremins, J.E. YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. Genome Res. 2017, 27, 1139–1152. [Google Scholar] [CrossRef] [Green Version]
- Hegarty, S.V.; Sullivan, A.M.; O’Keeffe, G.W. Zeb2: A multifunctional regulator of nervous system development. Prog. Neurobiol. 2015, 132, 81–95. [Google Scholar] [CrossRef]
- Stumm, R.K.; Zhou, C.; Ara, T.; Lazarini, F.; Dubois-Dalcq, M.; Nagasawa, T.; Hollt, V.; Schulz, S. CXCR4 regulates interneuron migration in the developing neocortex. J. Neurosci. 2003, 23, 5123–5130. [Google Scholar] [CrossRef] [Green Version]
- Nash, B.; Meucci, O. Functions of the chemokine receptor CXCR4 in the central nervous system and its regulation by mu-opioid receptors. Int. Rev. Neurobiol. 2014, 118, 105–128. [Google Scholar] [CrossRef] [Green Version]
- Georgala, P.A.; Carr, C.B.; Price, D.J. The role of Pax6 in forebrain development. Dev. Neurobiol. 2011, 71, 690–709. [Google Scholar] [CrossRef]
- Dias, C.M.; Punetha, J.; Zheng, C.; Mazaheri, N.; Rad, A.; Efthymiou, S.; Petersen, A.; Dehghani, M.; Pehlivan, D.; Partlow, J.N.; et al. Homozygous Missense Variants in NTNG2, Encoding a Presynaptic Netrin-G2 Adhesion Protein, Lead to a Distinct Neurodevelopmental Disorder. Am. J. Hum. Genet. 2019, 105, 1048–1056. [Google Scholar] [CrossRef] [Green Version]
- Lui, N.C.; Tam, W.Y.; Gao, C.; Huang, J.D.; Wang, C.C.; Jiang, L.; Yung, W.H.; Kwan, K.M. Lhx1/5 control dendritogenesis and spine morphogenesis of Purkinje cells via regulation of Espin. Nat. Commun. 2017, 8, 15079. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, S.F.; de Vellis, J. Id1, Id2, and Id3 gene expression in neural cells during development. Glia 1998, 24, 372–381. [Google Scholar] [CrossRef]
- Blomfield, I.M.; Rocamonde, B.; Masdeu, M.D.M.; Mulugeta, E.; Vaga, S.; van den Berg, D.L.; Huillard, E.; Guillemot, F.; Urban, N. Id4 promotes the elimination of the pro-activation factor Ascl1 to maintain quiescence of adult hippocampal stem cells. eLife 2019, 8, e48561. [Google Scholar] [CrossRef]
- Havrda, M.C.; Harris, B.T.; Mantani, A.; Ward, N.M.; Paolella, B.R.; Cuzon, V.C.; Yeh, H.H.; Israel, M.A. Id2 is required for specification of dopaminergic neurons during adult olfactory neurogenesis. J. Neurosci. 2008, 28, 14074–14086. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef] [Green Version]
- Higashi, Y.; Maruhashi, M.; Nelles, L.; Van de Putte, T.; Verschueren, K.; Miyoshi, T.; Yoshimoto, A.; Kondoh, H.; Huylebroeck, D. Generation of the floxed allele of the SIP1 (Smad-interacting protein 1) gene for Cre-mediated conditional knockout in the mouse. Genesis 2002, 32, 82–84. [Google Scholar] [CrossRef]
- Van Helden, M.J.; Goossens, S.; Daussy, C.; Mathieu, A.L.; Faure, F.; Marcais, A.; Vandamme, N.; Farla, N.; Mayol, K.; Viel, S.; et al. Terminal NK cell maturation is controlled by concerted actions of T-bet and Zeb2 and is essential for melanoma rejection. J. Exp. Med. 2015, 212, 2015–2025. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; Liu, P.; Wang, A.; Wang, B. Long noncoding RNA ZEB2AS1 affects cell proliferation and apoptosis via the miR1225p/PLK1 axis in acute myeloid leukemia. Int. J. Mol. Med. 2020, 46, 1490–1500. [Google Scholar] [CrossRef]
- Jiang, Y.; Wu, K.; Cao, W.; Xu, Q.; Wang, X.; Qin, X.; Wang, X.; Li, Y.; Zhang, J.; Chen, W. Long noncoding RNA KTN1-AS1 promotes head and neck squamous cell carcinoma cell epithelial-mesenchymal transition by targeting miR-153-3p. Epigenomics 2020, 12, 487–505. [Google Scholar] [CrossRef]
- Yao, H.; Hou, G.; Wang, Q.Y.; Xu, W.B.; Zhao, H.Q.; Xu, Y.C. LncRNA SPRY4IT1 promotes progression of osteosarcoma by regulating ZEB1 and ZEB2 expression through sponging of miR101 activity. Int. J. Oncol. 2020, 56, 85–100. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Zhao, H.; Xiao, X.; Huang, Q.; Zeng, W.; Tian, B.; Ma, T.; Lu, D.; Jin, Y.; Li, Y. Long Non-coding RNA MALAT1 Upregulates ZEB2 Expression to Promote Malignant Progression of Glioma by Attenuating miR-124. Mol. Neurobiol. 2021, 58, 1006–1016. [Google Scholar] [CrossRef]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef] [Green Version]
- Exposito-Villen, A.; Aránega, E.A.; Franco, D. Functional Role of Non-Coding RNAs during Epithelial-To-Mesenchymal Transition. Noncoding RNA 2018, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bracken, C.P.; Bert, A.G.; Goodall, G.J. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 2008, 7, 3112–3118. [Google Scholar] [CrossRef]
- Guan, H.; Liang, W.; Xie, Z.; Li, H.; Liu, J.; Liu, L.; Xiu, L.; Li, Y. Down-regulation of miR-144 promotes thyroid cancer cell invasion by targeting ZEB1 and ZEB2. Endocrine 2015, 48, 566–574. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, J.; Fu, H.; Shen, L. miR-144 functions as a tumor suppressor in breast cancer through inhibiting ZEB1/2-mediated epithelial mesenchymal transition process. Onco Targets Ther. 2016, 9, 6247–6255. [Google Scholar] [CrossRef] [Green Version]
- Nourmohammadi, B.; Tafsiri, E.; Rahimi, A.; Nourmohammadi, Z.; Daneshvar Kakhaki, A.; Cho, W.; Karimipoor, M. Expression of miR-9 and miR-200c, ZEB1, ZEB2 and E-cadherin in Non-Small Cell Lung Cancers in Iran. Asian Pac. J. Cancer Prev. 2019, 20, 1633–1639. [Google Scholar] [CrossRef] [Green Version]
- Wahab, N.A.; Othman, Z.; Nasri, N.W.M.; Mokhtar, M.H.; Ibrahim, S.F.; Hamid, A.A.; Raja Ali, R.A.; Mokhtar, N.M. Inhibition of miR-141 and miR-200a Increase DLC-1 and ZEB2 Expression, Enhance Migration and Invasion in Metastatic Serous Ovarian Cancer. Int. J. Environ. Res. Public Health 2020, 17, 2766. [Google Scholar] [CrossRef] [Green Version]
- Birkhoff, J.C.; Brouwer, R.W.W.; Kolovos, P.; Korporaal, A.L.; Bermejo-Santos, A.; Boltsis, I.; Nowosad, K.; van den Hout, M.; Grosveld, F.G.; van IJcken, W.F.J.; et al. Targeted chromatin conformation analysis identifies novel distal neural enhancers of ZEB2 in pluripotent stem cell differentiation. Hum. Mol. Genet. 2020, 29, 2535–2550. [Google Scholar] [CrossRef]
- Van Grunsven, L.A.; Papin, C.; Avalosse, B.; Opdecamp, K.; Huylebroeck, D.; Smith, J.C.; Bellefroid, E.J. XSIP1, a Xenopus zinc finger/homeodomain encoding gene highly expressed during early neural development. Mech. Dev. 2000, 94, 189–193. [Google Scholar] [CrossRef]
- Van de Putte, T.; Maruhashi, M.; Francis, A.; Nelles, L.; Kondoh, H.; Huylebroeck, D.; Higashi, Y. Mice lacking ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of Hirschsprung disease-mental retardation syndrome. Am. J. Hum. Genet. 2003, 72, 465–470. [Google Scholar] [CrossRef] [Green Version]
- Takagi, T.; Nishizaki, Y.; Matsui, F.; Wakamatsu, N.; Higashi, Y. De novo inbred heterozygous Zeb2/Sip1 mutant mice uniquely generated by germ-line conditional knockout exhibit craniofacial, callosal and behavioral defects associated with Mowat-Wilson syndrome. Hum. Mol. Genet. 2015, 24, 6390–6402. [Google Scholar] [CrossRef] [Green Version]
- Hegarty, S.V.; Wyatt, S.L.; Howard, L.; Stappers, E.; Huylebroeck, D.; Sullivan, A.M.; O’Keeffe, G.W. Zeb2 is a negative regulator of midbrain dopaminergic axon growth and target innervation. Sci. Rep. 2017, 7, 8568. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Itoh, Y.; Tabata, H.; Nakajima, K.; Akiyama, T.; Masuyama, N.; Gotoh, Y. The Wnt/β-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 2004, 131, 2791–2801. [Google Scholar] [CrossRef] [Green Version]
- Slawny, N.A.; O’Shea, K.S. Dynamic changes in Wnt signalling are required for neuronal differentiation of mouse embryonic stem cells. Mol. Cell. Neurosci. 2011, 48, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Santpere, G.; Imamura Kawasawa, Y.; Evgrafov, O.V.; Gulden, F.O.; Pochareddy, S.; Sunkin, S.M.; Li, Z.; Shin, Y.; Zhu, Y.; et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 2018, 362, eaat7615. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhu, Y.; Morozov, Y.M.; Chen, X.; Page, S.C.; Rannals, M.D.; Maher, B.J.; Rakic, P. Disruption of TCF4 regulatory networks leads to abnormal cortical development and mental disabilities. Mol. Psychiatry 2019, 24, 1235–1246. [Google Scholar] [CrossRef]
- Mesman, S.; Bakker, R.; Smidt, M.P. Tcf4 is required for correct brain development during embryogenesis. Mol. Cell. Neurosci. 2020, 106, 103502. [Google Scholar] [CrossRef]
- BabuRajendran, N.; Palasingam, P.; Narasimhan, K.; Sun, W.; Prabhakar, S.; Jauch, R.; Kolatkar, P.R. Structure of Smad1 MH1/DNA complex reveals distinctive rearrangements of BMP and TGF-β effectors. Nucleic Acids Res. 2010, 38, 3477–3488. [Google Scholar] [CrossRef]
- Morikawa, M.; Koinuma, D.; Tsutsumi, S.; Vasilaki, E.; Kanki, Y.; Heldin, C.-H.; Aburatani, H.; Miyazono, K. ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Res. 2011, 39, 8712–8727. [Google Scholar] [CrossRef]
- Liu, S.; Long, J.; Yuan, B.; Zheng, M.; Xiao, M.; Xu, J.; Lin, X.; Feng, X.H. SUMO Modification Reverses Inhibitory Effects of Smad Nuclear Interacting Protein-1 in TGF-β Responses. J. Biol. Chem. 2016, 291, 24418–24430. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Lin, X.; Feng, X.H. Posttranslational Regulation of Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022087. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Kang, M.J.; Kim, E.; Kweon, T.H.; Park, Y.S.; Ji, S.; Yang, W.H.; Yi, E.C.; Cho, J.W. O-GlcNAc stabilizes SMAD4 by inhibiting GSK-3beta-mediated proteasomal degradation. Sci. Rep. 2020, 10, 19908. [Google Scholar] [CrossRef]
- Lin, X.; Wang, Y.; Jiang, Y.; Xu, M.; Pang, Q.; Sun, J.; Yu, Y.; Shen, Z.; Lei, R.; Xu, J. Sumoylation enhances the activity of the TGF-β/SMAD and HIF-1 signalling pathways in keloids. Life Sci. 2020, 255, 117859. [Google Scholar] [CrossRef]
- McKinsey, G.L.; Lindtner, S.; Trzcinski, B.; Visel, A.; Pennacchio, L.A.; Huylebroeck, D.; Higashi, Y.; Rubenstein, J.L. Dlx1&2-dependent expression of Zfhx1b (Sip1, Zeb2) regulates the fate switch between cortical and striatal interneurons. Neuron 2013, 77, 83–98. [Google Scholar] [CrossRef] [Green Version]
- De Haan, W.; Dheedene, W.; Apelt, K.; Décombas-Deschamps, S.; Vinckier, S.; Verhulst, S.; Conidi, A.; Deffieux, T.; Staring, M.W.; Vandervoort, P.; et al. Endothelial Zeb2 preserves the hepatic angioarchitecture and protects against liver fibrosis. Cardiovasc. Res. 2022, 118, 1262–1275. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sense/Antisense | Sequence (5′ → 3′) | Application |
|---|---|---|---|
| FlV5mZeb2Ex9_Fwd | Sense | GGCTTACCTGCAGAGCATCA | genotyping |
| FlV5mZeb2Ex9_Rev | Antisense | CTCCATCTAACTCTGTCTTGGC | genotyping |
| FlV5_Fwd | Sense | CTACTCGCAGCACATGAATC | genotyping |
| FlV5_Rev | Antisense | GAGAGGGTTAGGGATAGGC | genotyping |
| ΔZP_P1_Fwd | Sense | GTCAGTCCGTCCCCAGGTTT | genotyping |
| ΔZP_P2_Rev | Antisense | GGCATGCTAGCTGGGCTGGT | genotyping |
| LN249_Fwd | Sense | GGAGCAAACTGAACAAAACCTCGCC | genotyping |
| LN249_Rev | Antisense | GGCGAGGTTTTGTTCAGTTTGCTCC | genotyping |
| LN209_Fwd | Sense | AGCGGATCAGATGGCAGTTCGCATG | genotyping |
| LN209_Rev | Antisense | CATGCGAACTGCCATCTGATCCGCT | genotyping |
| Zeb2_Fwd | Sense | CAATGCAGCACTTAGGTGTA | qPCR |
| Zeb2_Rev | Antisense | TTGCCTAGAAACCGTATTGT | qPCR |
| Zeb2V5_Fwd | Sense | GAAACGATACGGGATGAGGA | qPCR |
| Zeb2V5_Rev | Antisense | AGGAGAGGGTTAGGGATAGG | qPCR |
| Nanog_Fwd | Sense | TCTTCCTGGTCCCCACAGTTT | qPCR |
| Nanog_Rev | Antisense | GCAAGAATAGTTCTCGGGATGAA | qPCR |
| Pou5f1_Fwd | Sense | AGAGGATCACCTTGGGGTACA | qPCR |
| Pou5f1_Rev | Antisense | CGAAGCGACAGATGGTGG TC | qPCR |
| Sox2_Fwd | Sense | GCGGAGTGGAAACTTTTGTCC | qPCR |
| Sox2_Rev | Antisense | CGGGAAGCGTGTACTTATCCTT | qPCR |
| Pax6_Fwd | Sense | ACATCTTTTACCCAAGAGCA | qPCR |
| Pax6_Rev | Antisense | GGCAAACACATCTGGATAAT | qPCR |
| Acrv1b_Fwd | Sense | CTGCCTACAGACCAACTACACC | qPCR |
| Acrv1b_Rev | Antisense | CCACGCCATCCAGGTTAAAGA | qPCR |
| Lhx5_Fwd | Sense | AGAACCGAAGGTCCAAAGAA | qPCR |
| Lhx5_Rev | Antisense | TCACTTTGGTAGTCTCCGTA | qPCR |
| Ntng2_Fwd | Sense | CAAGGACTCTACGCTTTTCG | qPCR |
| Ntng2_Rev | Antisense | AGCACTCGCAGTCTTGAAAT | qPCR |
| Sema3f_Fwd | Sense | CTACACAGCATCCTCCAAGA | qPCR |
| Sema3f_Rev | Antisense | ACGGCATTCTTGTTTGCATT | qPCR |
| Smad1_Fwd | Sense | TACTATGAGCTCAACAACCG | qPCR |
| Smad1_Rev | Antisense | GAAGCGGTTCTTATTGTTGG | qPCR |
| Smad3_Fwd | Sense | CACGCAGAACGTGAACACC | qPCR |
| Smad3_Rev | Antisense | GGCAGTAGATAACGTGAGGGA | qPCR |
| Sox13_Fwd | Sense | CTTACAGGAGGTTGTGCCA | qPCR |
| Sox13_Rev | Antisense | TCCTTAGCTTCCACATTGCT | qPCR |
| Stat3_Fwd | Sense | CAATACCATTGACCTGCCGAT | qPCR |
| Stat3_Rev | Antisense | GAGCGACTCAAACTGCCCT | qPCR |
| Tcf4_Fwd | Sense | TTGAAGATGTTTTCGCCTCC | qPCR |
| Tcf4_Rev | Antisense | CCTGCTAGTCATGTGGTCAT | qPCR |
| Tgfbr2_Fwd | Sense | GAAGGAAAAGAAAAGGGCGG | qPCR |
| Tgfbr2_Rev | Antisense | TGCTGGTGGTGTATTCTTCC | qPCR |
| Amylase_Fwd | Sense | GGCTGAGTGTTCTGGGAT | ChIP-qPCR |
| Amylase_Rev | Antisense | CACGGTGCTCTGGTAGAT | ChIP-qPCR |
| Cdh1_R1_Fwd | Sense | GCTAGGCTAGGATTCGAACGAC | ChIP-qPCR |
| Cdh1_R1_Rev | Antisense | TGCAGGGCCCTCAACTT | ChIP-qPCR |
| Name | Sequence | CRISPR/Cas9 |
|---|---|---|
| Flag-V5 donor template 1 | aaaatggaaaccaaatcagaccacgaagaagacaatatggaagatggcatcgaaGACTACAAAGACGATGACGACAAGgatatcGGTAAGCCTATCCCTAACCCTCTCCTCGGTCTCGATTCTACGTAAactactgcattttaagcttcctattttttttttccagtagtattgtt | in-frame knock-in of Flag-V5 tag |
| gRNA_ex9_1 | GGAAACCAAATCAGACCACGAGG | |
| gRNA_ΔZP1 | CCCGCGCGCGTTTCAATGGGCGC | Zeb2 ΔP deletion |
| gRNA_ΔZP2 | CCCTCGCGAGTGCAACACACCAA | |
| gRNA_ΔZP3 | GGGCTCGGAGCGCTGCCGATCGG | |
| gRNA_ΔZP4 | CCGCTGGACCGGGGGGGAGTTGA |
| Name | Sequence |
|---|---|
| shZeb2_1 | CCGGCCGAATGAGAAACAATATCAACTCGAGTTGATATTGTTTCTCATTCGGTTTTTG |
| shZeb2_2 | CCGGCCTCAGGAATTTGTGAAGGAACTCGAGTTCCTTCACAAATTCCTGAGGTTTTTG |
| shZeb2_3 | CCGGCCAGTGTCAGATTTGTAAGAACTCGAGTTCTTACAAATCTGACACTGGTTTTTG |
| shZeb2_4 | CCGGCCCATTTAGTGCCAAGCCTTTCTCGAGAAAGGCTTGGCACTAAATGGGTTTTTG |
| shCTRL | CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birkhoff, J.C.; Korporaal, A.L.; Brouwer, R.W.W.; Nowosad, K.; Milazzo, C.; Mouratidou, L.; van den Hout, M.C.G.N.; van IJcken, W.F.J.; Huylebroeck, D.; Conidi, A. Zeb2 DNA-Binding Sites in Neuroprogenitor Cells Reveal Autoregulation and Affirm Neurodevelopmental Defects, Including in Mowat-Wilson Syndrome. Genes 2023, 14, 629. https://doi.org/10.3390/genes14030629
Birkhoff JC, Korporaal AL, Brouwer RWW, Nowosad K, Milazzo C, Mouratidou L, van den Hout MCGN, van IJcken WFJ, Huylebroeck D, Conidi A. Zeb2 DNA-Binding Sites in Neuroprogenitor Cells Reveal Autoregulation and Affirm Neurodevelopmental Defects, Including in Mowat-Wilson Syndrome. Genes. 2023; 14(3):629. https://doi.org/10.3390/genes14030629
Chicago/Turabian StyleBirkhoff, Judith C., Anne L. Korporaal, Rutger W. W. Brouwer, Karol Nowosad, Claudia Milazzo, Lidia Mouratidou, Mirjam C. G. N. van den Hout, Wilfred F. J. van IJcken, Danny Huylebroeck, and Andrea Conidi. 2023. "Zeb2 DNA-Binding Sites in Neuroprogenitor Cells Reveal Autoregulation and Affirm Neurodevelopmental Defects, Including in Mowat-Wilson Syndrome" Genes 14, no. 3: 629. https://doi.org/10.3390/genes14030629
APA StyleBirkhoff, J. C., Korporaal, A. L., Brouwer, R. W. W., Nowosad, K., Milazzo, C., Mouratidou, L., van den Hout, M. C. G. N., van IJcken, W. F. J., Huylebroeck, D., & Conidi, A. (2023). Zeb2 DNA-Binding Sites in Neuroprogenitor Cells Reveal Autoregulation and Affirm Neurodevelopmental Defects, Including in Mowat-Wilson Syndrome. Genes, 14(3), 629. https://doi.org/10.3390/genes14030629