
Mitogenomes of Eight Nymphalidae Butterfly Species and Reconstructed Phylogeny of Nymphalidae (Nymphalidae: Lepidoptera)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, Identification, Sequencing, and Mitogenome Assembly
2.2. Mitogenome Annotation and Characteristics Analysis
2.3. Phylogenetic Analysis
3. Results
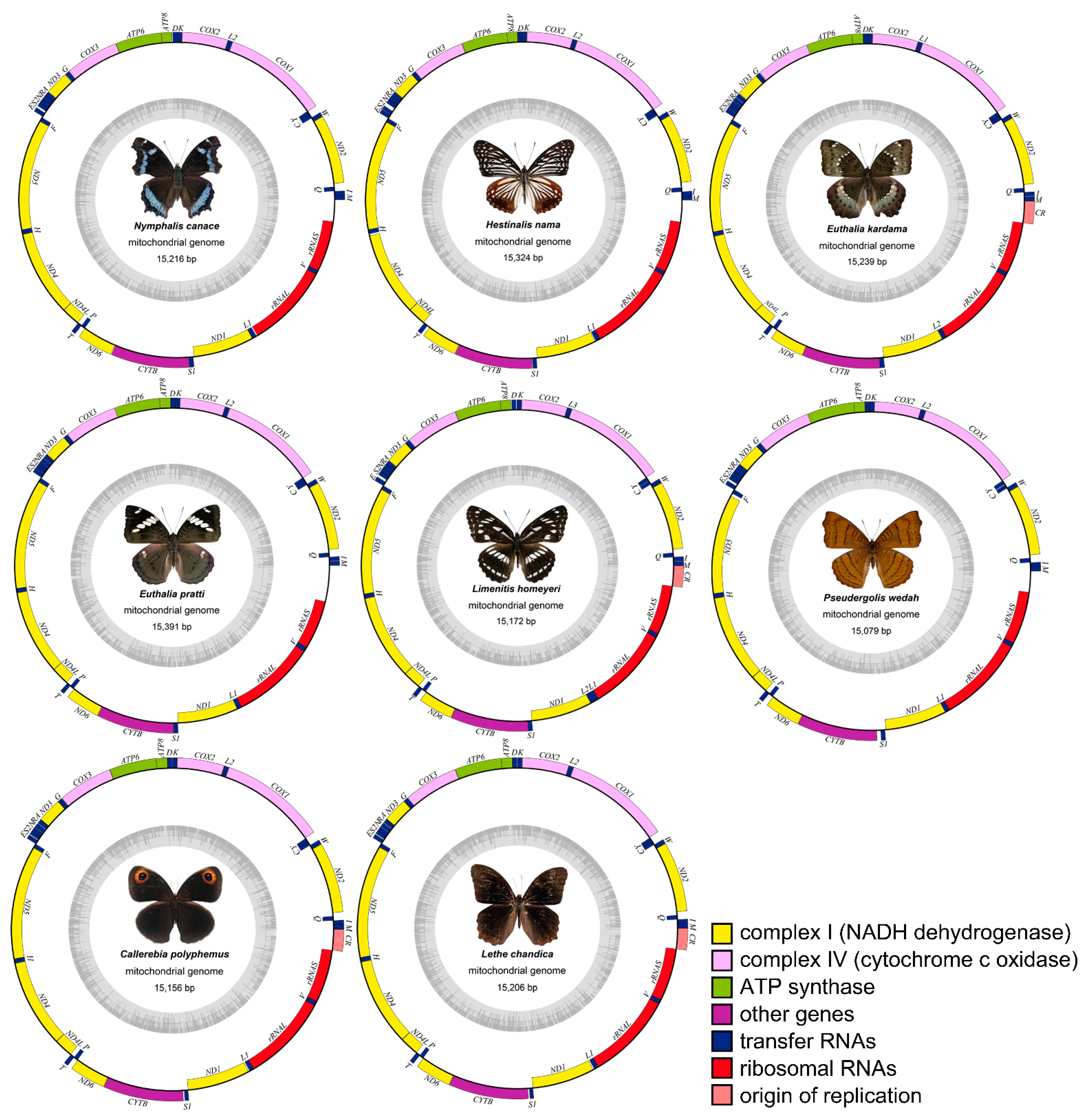
3.1. Genome Organization and Nucleotide Composition
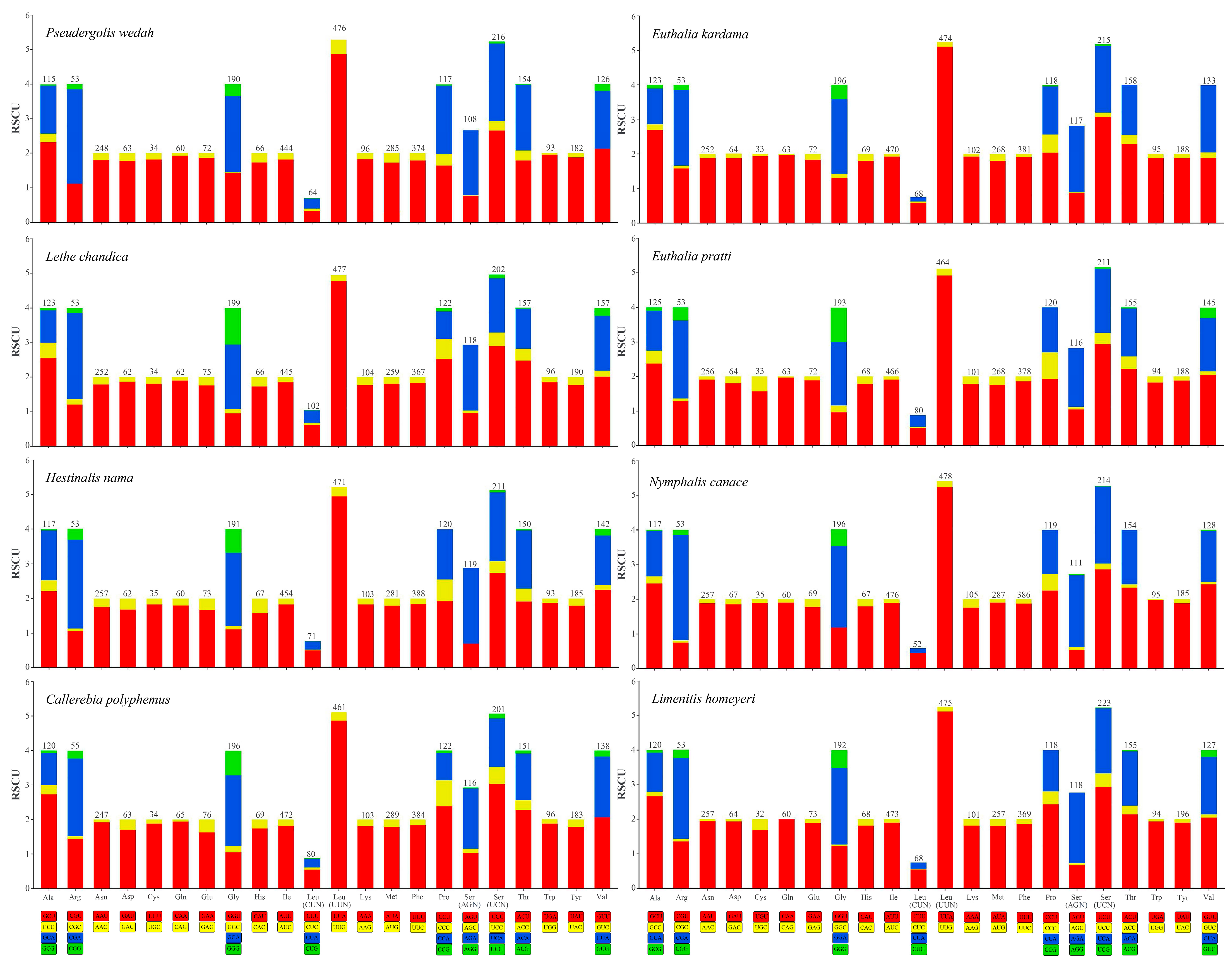
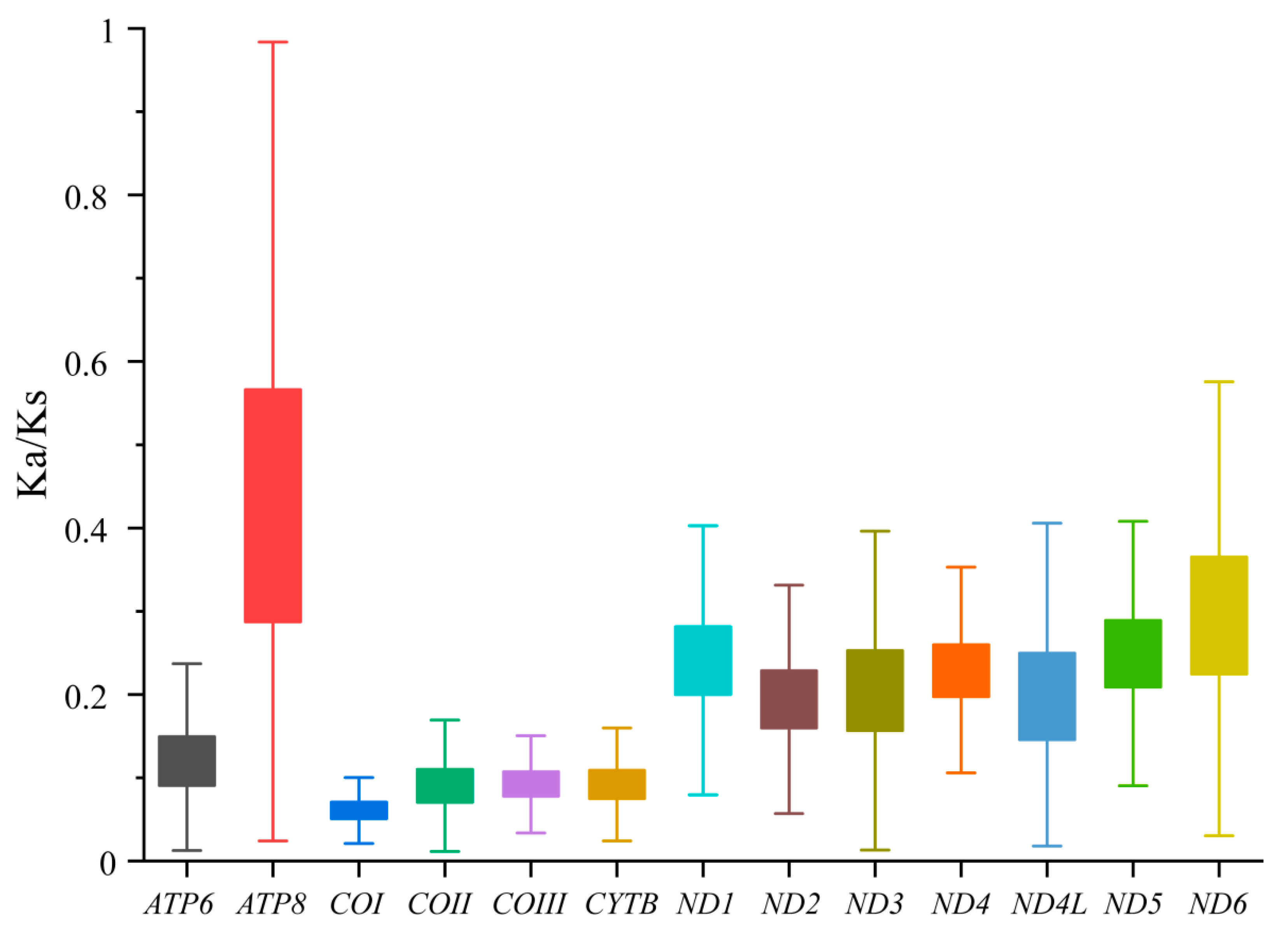
3.2. Characteristics of Mitogenomes Genes
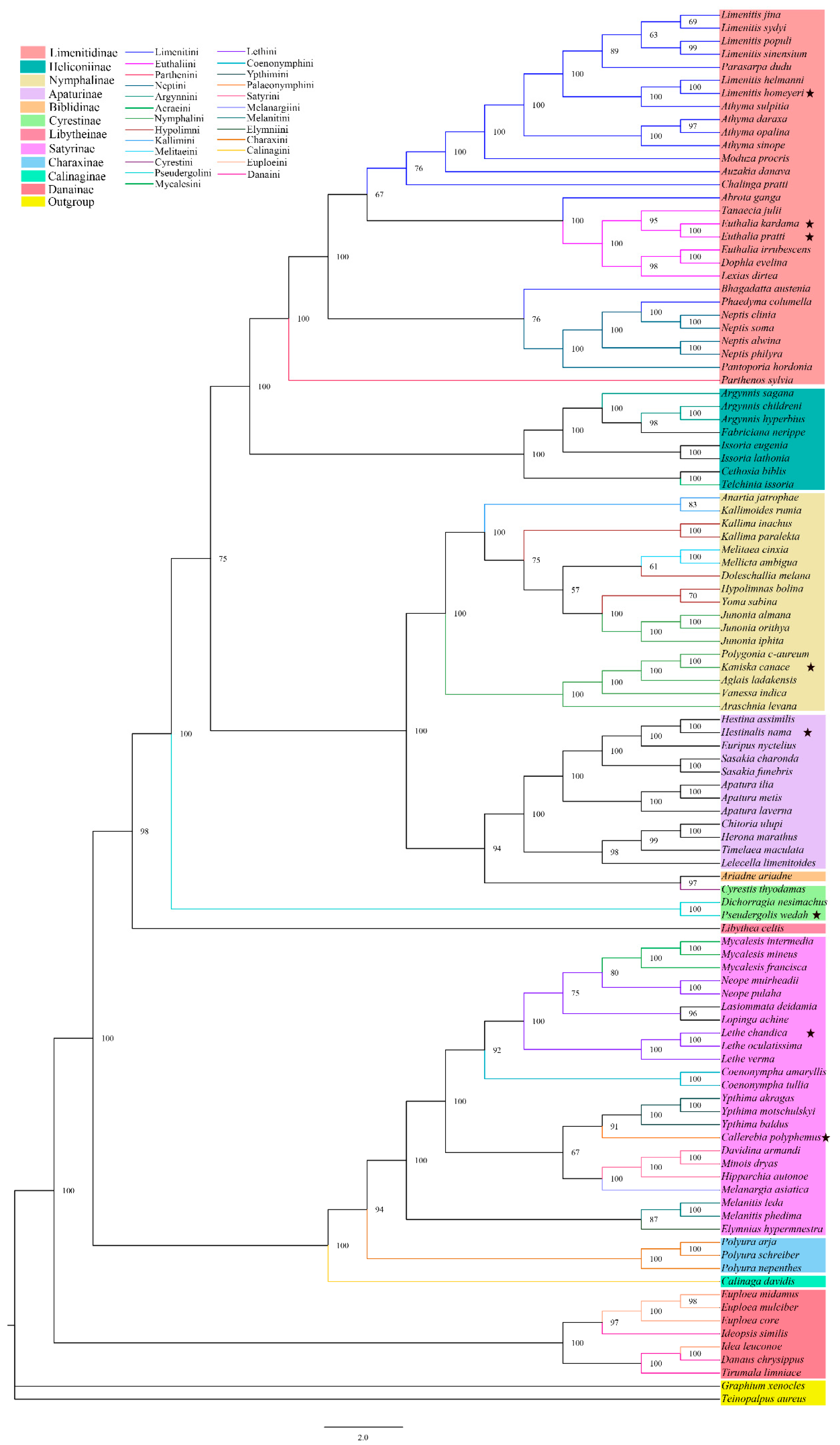
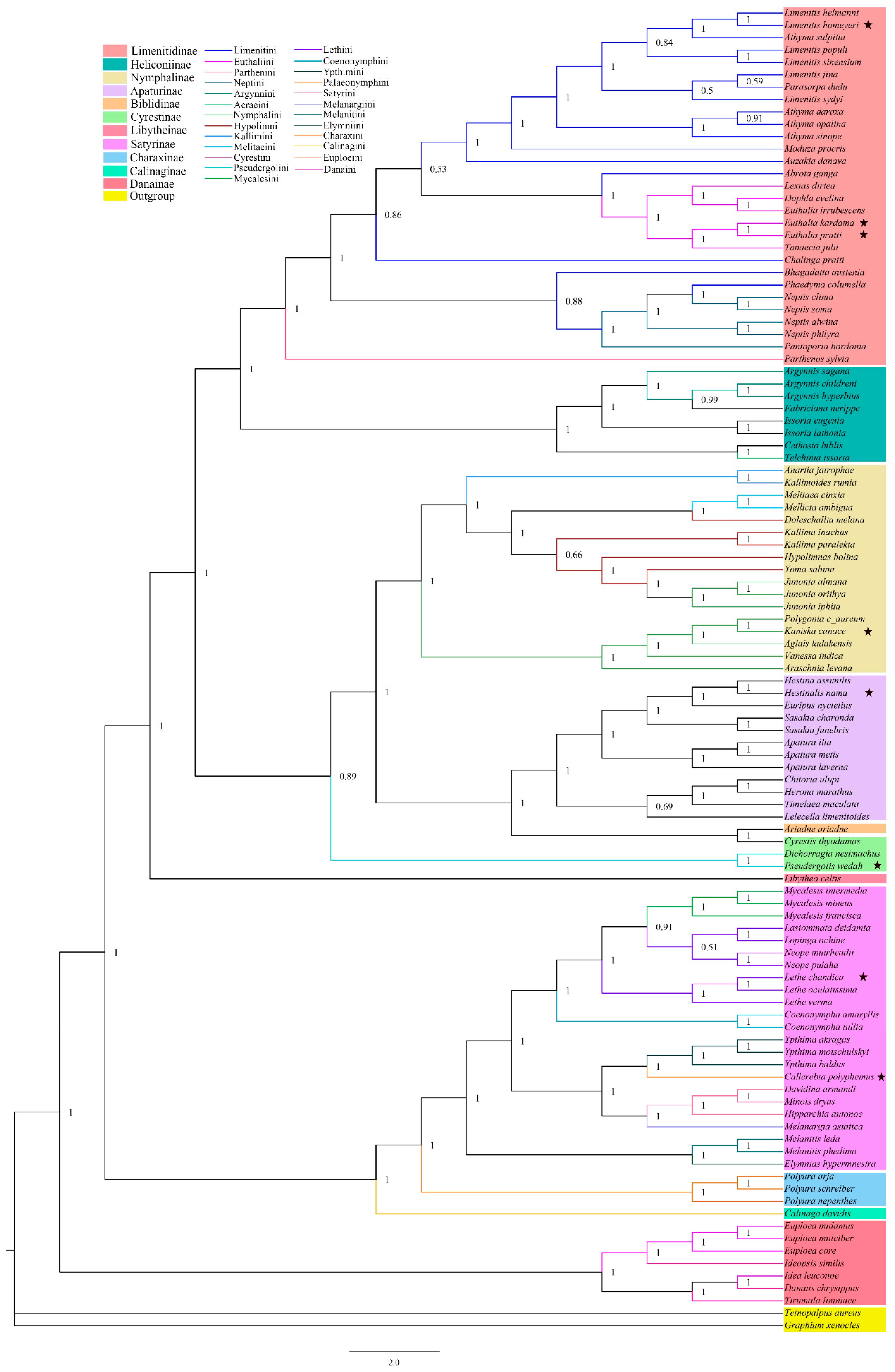
3.3. Phylogenetic Relationships
4. Discussion
4.1. General Characteristics
4.2. Subfamily-Level Phylogenetic Relationships in Nymphalidae
4.3. Tribal-Level Phylogenetic Relationships in Nymphalidae
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lang, S.Y. The Nymphalidae of China (Lepidoptera, Rhopalocera). Part I. Libytheinae, Danainae, Calinaginae, Morphinae, Heliconninae, Nymphalinae, Charaxinae, Apaturinae, Cyrestinae, Biblidinae, Limenitinae; Tshikolovets Publications: Pardubice, Czech Republic, 2012; Volume 456, pp. 4–41. [Google Scholar]
- Zhou, Y. Monograph of Chinese Butterflies; Henan Scientific and Technological Publishing House: Zhengzhou, China, 2000; Volume 1. [Google Scholar]
- Carroll, S.B.; Gates, J.; Keys, D.N.; Paddock, S.W.; Panganiban, G.E.F.; Selegue, J.E.; Williams, J.A. Pattern formation and eyespot determination in butterfly wings. Science 1994, 265, 109–114. [Google Scholar] [CrossRef]
- Gilbert, L.E. Biodiversity of a Central American Heliconius Community: Pattern, process, and problems. In Plant-Animal Interactions: Evolutionary Ecology in Tropical and Temperate Region; Wiley: New York, NY, USA, 1991; pp. 403–427. [Google Scholar]
- Monteriro, A.; Brakefield, P.M.; French, V. Butterfly eyespots: The genetics and development of the color rings. Evolution 1997, 51, 1207–1216. [Google Scholar] [CrossRef]
- Wahlberg, N.; Moilanen, A.; Hanski, I. Predicting the occurrence of endangered species in fragmented landscapes. Science 1996, 273, 1536–1538. [Google Scholar] [CrossRef]
- Brower, A.V.Z. Phylogenetic relationships among the Nymphalidae (Lepidoptera) inferrd from partial sequnces of the wingless gene. Proc. Biol. Sci. 2000, 267, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Boggs, C.L.; Watt, W.B.; Ehrlich, P.R. Butterflies: Evolution and Ecology Taking Flight; The University of Chicago Press: Chicago, IL, USA, 2003; 736p. [Google Scholar]
- Ackery, P.R.; de Jong, R.; Vane-Wright, R.I. The butterflies: Hedyloidea, Hesperioidea and Papilionoidea. Evol. Syst. Biogeogr. 1999, 35, 263–300. [Google Scholar]
- Zhou, Y. Classification and Identification of Chinese Butterflies; Science and Technology of Henan Press: Zhengzhou, China, 1998. [Google Scholar]
- Wahlberg, N.; Braby, M.F.; Brower, A.V.Z.; de Jong, R.; Lee, M.-M.; Nylin, S.; Pierce, N.E.; Sperling, F.A.H.; Vila, R.; Warren, A.D.; et al. Synergistic effects of combining morphological and molecular data in resolving the phylogeny of butterflies and skippers. Proc. Biol. Sci. 2005, 272, 1577–1586. [Google Scholar] [CrossRef]
- Weller, S.J.; Pashley, D.P.; Martin, J.A. Reassessment of butterfly family relationships using independent genes and morphology. Ann. Entomol. Soc. Am. 1996, 89, 184–192. [Google Scholar] [CrossRef]
- Chai, H.N.; Du, Y.Z.; Zhai, B.P. Characterization of the complete mitochondrial genomes of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae). Int. J. Biol. Sci. 2012, 8, 561–579. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Gabaldón, T.; Huynen, M. Reconstraction of the Proto-Mitochondrial Metabolism. Science 2003, 301, 609. [Google Scholar] [CrossRef]
- De Salle, R.; Williams, A.; George, M. Isolation and manipulation of animal mitochondrial DNA. Mothods Enzymol. 1993, 224, 176–204. [Google Scholar]
- Galtier, N.; Nabholz, B.; Glémin, S.; Hurst, G.D.D. Mitochondrial DNA as a marker of molecular diversity: A reappraisal. Mol. Ecol. 2009, 18, 4541–4550. [Google Scholar] [CrossRef]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- He, H.Y.; Yu, W.D.; Jiang, W.B. Research progress in mitochondrial genomics of butterflies. Chin. Bull. Life Sci. 2016, 28, 978–985. [Google Scholar]
- Shi, H. Molecular Phylogenetic Analysis of Papilionidea Based on 16S rDNA, Cytb and COI Sequence; Nanjing Normal University: Nanjing, China, 2006; (In Chinese with English abstract). [Google Scholar]
- Gray, M.W. Origin and evolution of mitochondrial DNA. Annu. Rev. Cell Biol. 1989, 5, 25–50. [Google Scholar] [CrossRef]
- Yuan, F.; Yuan, X.Q. Advanced in Molecular Systematics of Butterflies. ACTA Agric. Boreali-Occident. Sin. 2013, 22, 1–14. [Google Scholar]
- Qin, X.M. The Study on Mitochondrial Genomes and Molecular Phylogenetics of Butterflies; Beijing Science Technology Publishing House: Beijing, China, 2017; 236p. [Google Scholar]
- Wilson, A.C.; Cann, R.L.; Carr, S.M.; George, M.; Gyllensten, U.B.; Helm-Bychowski, K.M.; Higuchi, R.G.; Palumbi, S.R.; Prager, E.M.; Sage, R.D.; et al. Mitochondrial DNA and two Perspectives on the evolutionary genetics. Biol. J. Linn. Soc. 1985, 26, 375–400. [Google Scholar] [CrossRef]
- Wu, C.S.; Xu, Y.F. Illustrated Handbook of Chinese Butterflies; Straits Publishing House: Fuzhou, China, 2017. [Google Scholar]
- Lin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; Depamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 1–31. [Google Scholar]
- Meng, G.L.; Li, Y.Y.; Yang, C.T.; Liu, S.L. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, 63–64. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothad, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef]
- Koichiro, T.; Glen, S.; Daniel, P.; Alan, F.; Sudhir, K. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- OriginPro, Version 2019b; OriginLab Corporation: Northampton, MA, USA, 2019.
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. Partitionfinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Enrico, N.; Massimiliano, B.; Tomaso, P. The mitochondrial genome of the ascalaphid owlfly Libelloides macaronious and comparative evolutionary mitochondriomics of neuropterid insects. BMC Genom. 2011, 12, 1–26. [Google Scholar]
- Xia, X.H.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Xia, X.; Lemey, P. Assessing substitution saturation with DAMBE. In The Phylogenetic Handbook: A Practical Approach to DNA and Protein Phylogeny, 2nd ed.; Lemey, P., Salemi, M., Vandamme, A.-M., Eds.; Cambridge University Press: Cambridge, UK, 2009; pp. 615–630. [Google Scholar]
- Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta (lnsecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 2008, 408, 112–123. [Google Scholar] [CrossRef]
- Gong, Y.-J.; Shi, B.-C.; Kang, Z.-J.; Zhang, F.; Wei, S.-J. The complete mitochondrial genome of the oriental fruit moth Grapholita molesta (Busck) (Lepidoptera: Tortricidae). Mol. Biol. Rep. 2012, 39, 2893–2900. [Google Scholar] [CrossRef]
- Cao, Y.Q.; Ma, C.; Chen, J.Y.; Yang, D.R. The complete mitochondrial genomes of two ghost moths, Thitarodes renzhiensis and Thitarodes yunnanensis: The ancestral gene arrangement in Lepidoptera. BMC Genom. 2012, 13, 276. [Google Scholar] [CrossRef]
- Peng, G.Z.; Chen, L.L.; Tian, D.C. Progress in the Study of Gene Duplication. Hereditas 2006, 7, 886–892. [Google Scholar]
- Wahlberg, N.; Weingartner, E.; Nylin, S. Towards a better understanding of the higher systematics of Nymphalidae (Lepidoptera: Papilionoidea). Mol. Phylogenet. Evol. 2003, 28, 473–484. [Google Scholar] [CrossRef]
- Wahlberg, N.; Leneveu, J.; Kodandaramaiah, U.; Peña, C.; Nylin, S.; Freitas, A.V.; Brower, A.V. Nymphalid butterflies diversify following near demise at the Cretaceous/Tertiary boundary. Proc. R. Soc. B 2009, 276, 4295–4302. [Google Scholar] [CrossRef]
- Wu, L.W.; Lin, L.H.; Lee, D.C.; Hsu, Y.F. Mitogenomic sequences effectively recover relationships within brush-footed butterflies, Lepidoptera: Nymphalidae. BMC Genom. 2014, 15, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.H.; Sun, X.Y.; Wang, Y.L.; Hao, J.S.; Yang, Q. Morphological characters are compatible with mitogenomic data in resolving the phylogeny of nymphalid butterflies (Lepidoptera: Papilionoidea: Nymphalidae). PLoS ONE 2015, 10, e0124349. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Li, N.; Yang, P.; Sun, C.; Fang, J.; Wang, S. The complete mitochondrial genome of Damora sagana and phylogenetic analyses of the family Nymphalidae. Genes Genom. 2018, 40, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.S.; Sun, M.; Shi, Q.H.; Sun, X.Y.; Shao, L.; Yang, Q. Complete Mitogenomes of Euploea mulciber (Nymphalidae: Danainae) and Libythea celtis (Nymphalidae: Libytheinae) and Their Phylogenetic Implications; International Scholarly Research Notices: New York, NY, USA, 2013; pp. 1–14. [Google Scholar]
- Wu, D.X. Molecular Phylogenetic Analysis of Some Species of Limenitinae Based on Mitochondrial Cytb and COI Sequences; Anhui Normal University: Wuhu, China, 2007. [Google Scholar]
- Wu, D.-x.; Zhu, G.-p.; Chen, N.; Su, C.-y.; Hao, J.s. Molecular Phylogenetic Analysis of Limenitinae Based on Mitochondrial COI Gene Sequences. Life Sci. Res. 2007, 11, 64–71. [Google Scholar]
- Chen, Z.H. Systematic Study of the Butterfly Tribe Neptini from China (Lepidoptera: Nymphalidae: Limenitinae); Northwest A&F University: Xianyang, China, 2015. [Google Scholar]
- Wang, J.P.; Cao, T.W.; Zhang, Y.; Fan, R.J.; Zhang, M.; Shi, B.M.; Peng, F.C. Sequencing and Analusis of the Complete miyochondrial Genome of Limenitis helmanni (Lepidoptera: Nymphalidae). Acta Entomol. Sin. 2017, 60, 950–961. [Google Scholar]
- Peña, C.; Wahlberg, N.; Weingartner, E.; Kodandaramaiah, U.; Nylin, S.; Freitas, A.V.; Brower, A.V. Higher level phylogeny of Satyrinae butterflies (Lepidoptera: Nymphalidae) based on DNA sequence data. Mol. Phylogenet. Evol. 2006, 40, 29–49. [Google Scholar] [CrossRef]
- Penz, C.M. Higher Level Phylogeny for the Passion-Vine Butterflies (Nymphalidae, Heliconiinae) Based on early Stage and Adult Morphology. Zool. J. Linn. Soc. 1999, 127, 277–344. [Google Scholar] [CrossRef]
- Win, N.Z.; Choi, E.Y.; Park, J.; Park, J.K. Molecular phylogenetic relationship of the subfamily Nymphalinae (Lepidoptera: Nymphalidae) in Myanmar, inferred from mitochondrial gene sequences. J. Asia-Pac. Biodivers. 2017, 10, 86–90. [Google Scholar] [CrossRef]
- Liu, N.; Fang, L.J.; Zhang, Y. The Complete Mitochondrial Genomes of Four Species in the Subfamily Limenitidinae (Lepidoptera, Nymphalidae) and a Phylogenetic Analysis. Insects 2021, 13, 16. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Subfamily | Species | Accession No. | Collected Location |
|---|---|---|---|---|
| 1 | Nymphalinae | Kaniska canace Linnaeus, 1763 | MZ481931 | Zhaomu Mountain Forest Park, Chongqing (106.514° E, 29.633° N) |
| 2 | Apaturinae | Hestina nama Douleday, 1844 | MZ501810 | Love ladder, Jiangjin District, Chongqing (106.335° E, 28.728° N) |
| 3 | Limenitinae | Euthalia kardama Moore, 1859 | MZ501803 | Jinfo Mountain, Chongqing (107.134° E, 29.054° N) |
| 4 | Euthalia pratti Leech, 1891 | MZ501809 | Jinfo Mountain, Chongqing (107.134° E, 29.054° N) | |
| 5 | Limenitis homeyeri Tancre,1881 | MZ501806 | Wangxiangtai, Simian Mountain, Jiangjin District, Chongqing (106.45° E, 28.679° N) | |
| 6 | Cyrestinae | Pseudergolis wedah Collar, 1844 | MZ501808 | Nanshan Botanical Garden, Chongqing (106.635° E, 29.561° N) |
| 7 | Satyrinae | Callerebia polyphemus Oberthür, 1877 | MZ491831 | Jinfo Mountain, Chongqing (107.134° E, 29.054° N) |
| 8 | Lethe chandica Moore, 1858 | MZ501804 | Nanshan Botanical Garden, Chongqing (106.635° E, 29.561° N) |
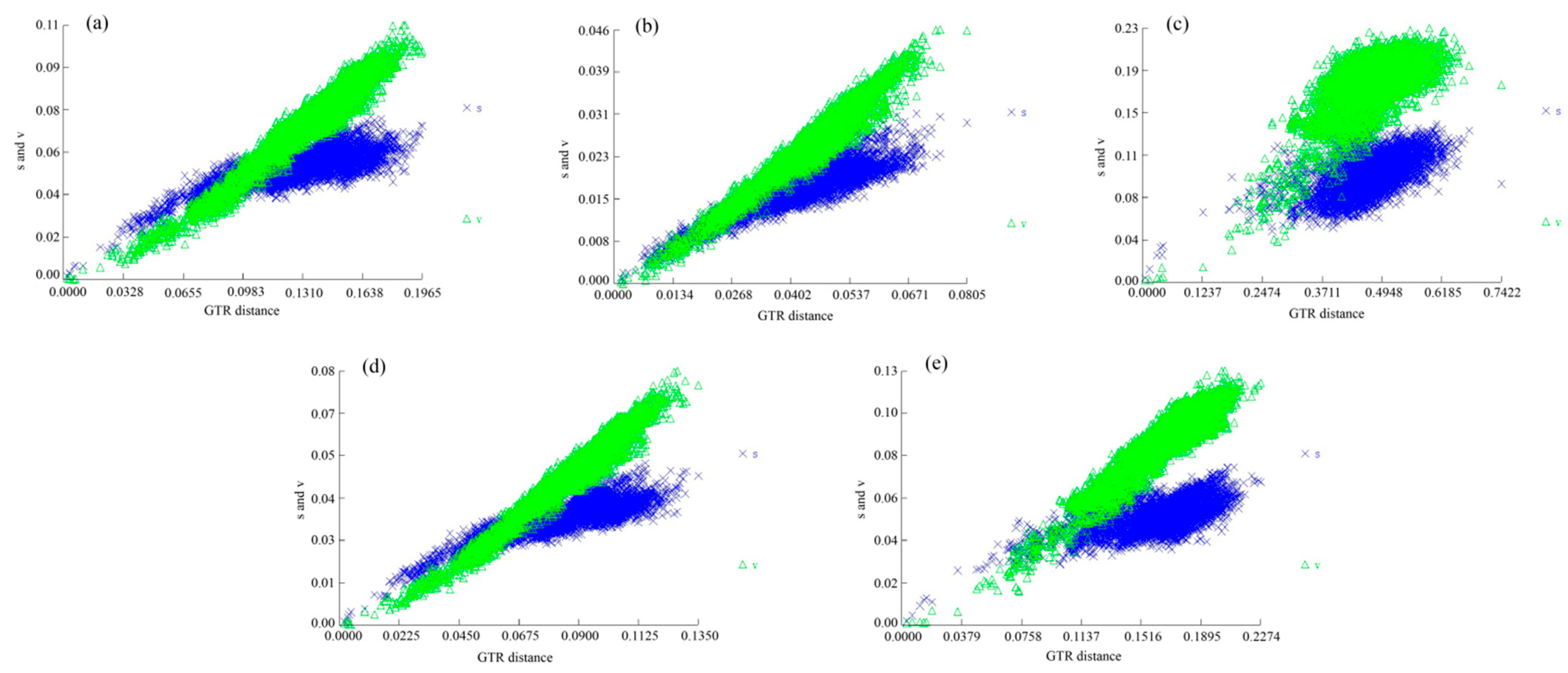
| Data Partition | Iss | Iss.cSym † | Psym ‡ | Iss.cAsym § | Pasym ¶ |
|---|---|---|---|---|---|
| PCG123 | 0.224 | 0.857 | 0 | 0.846 | 0 |
| PCG12 | 0.126 | 0.853 | 0 | 0.845 | 0 |
| PCG1 | 0.172 | 0.849 | 0 | 0.836 | 0 |
| PCG2 | 0.077 | 0.849 | 0 | 0.836 | 0 |
| PCG3 | 0.489 | 0.849 | 0 | 0.836 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Z.-T.; Fan, Z.-H.; He, S.-L.; Wang, X.-Q.; Chen, B.; Luo, S.-T. Mitogenomes of Eight Nymphalidae Butterfly Species and Reconstructed Phylogeny of Nymphalidae (Nymphalidae: Lepidoptera). Genes 2023, 14, 1018. https://doi.org/10.3390/genes14051018
Yan Z-T, Fan Z-H, He S-L, Wang X-Q, Chen B, Luo S-T. Mitogenomes of Eight Nymphalidae Butterfly Species and Reconstructed Phylogeny of Nymphalidae (Nymphalidae: Lepidoptera). Genes. 2023; 14(5):1018. https://doi.org/10.3390/genes14051018
Chicago/Turabian StyleYan, Zhen-Tian, Zhen-Huai Fan, Shu-Lin He, Xue-Qian Wang, Bin Chen, and Si-Te Luo. 2023. "Mitogenomes of Eight Nymphalidae Butterfly Species and Reconstructed Phylogeny of Nymphalidae (Nymphalidae: Lepidoptera)" Genes 14, no. 5: 1018. https://doi.org/10.3390/genes14051018
APA StyleYan, Z. -T., Fan, Z. -H., He, S. -L., Wang, X. -Q., Chen, B., & Luo, S. -T. (2023). Mitogenomes of Eight Nymphalidae Butterfly Species and Reconstructed Phylogeny of Nymphalidae (Nymphalidae: Lepidoptera). Genes, 14(5), 1018. https://doi.org/10.3390/genes14051018