
Chromosome-Level Genome Assembly Provides Insights into the Evolution of the Special Morphology and Behaviour of Lepturacanthus savala
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA and RNA Extraction for Library Construction and Sequencing
2.3. Evaluation of Genome Size, Heterozygosity, and Contamination
2.4. Genome Assembly and Integrity Assessment
2.5. Chromosome Assembly by Hi-C
2.6. Genome Repetition, Structure, Function, and Noncoding RNA Annotation
2.7. Genome Evolution, Gene Family Dynamics, and Positive Selection Analyses
3. Results
3.1. Genome Size Estimation and Initial Characterization of the Genome
3.2. Genome Assembly and Evaluation
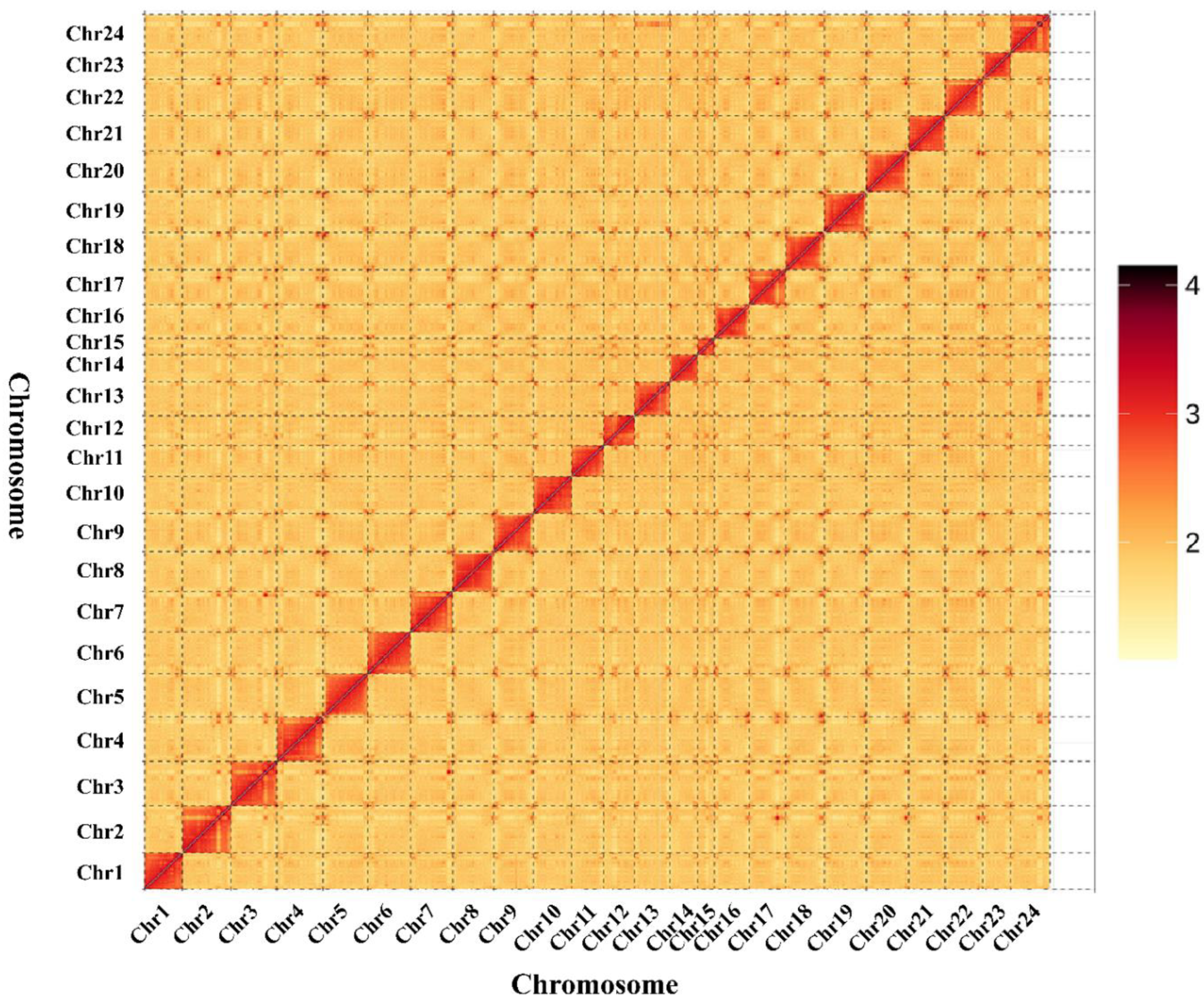
3.3. Chromosome Assembly by Hi-C Data
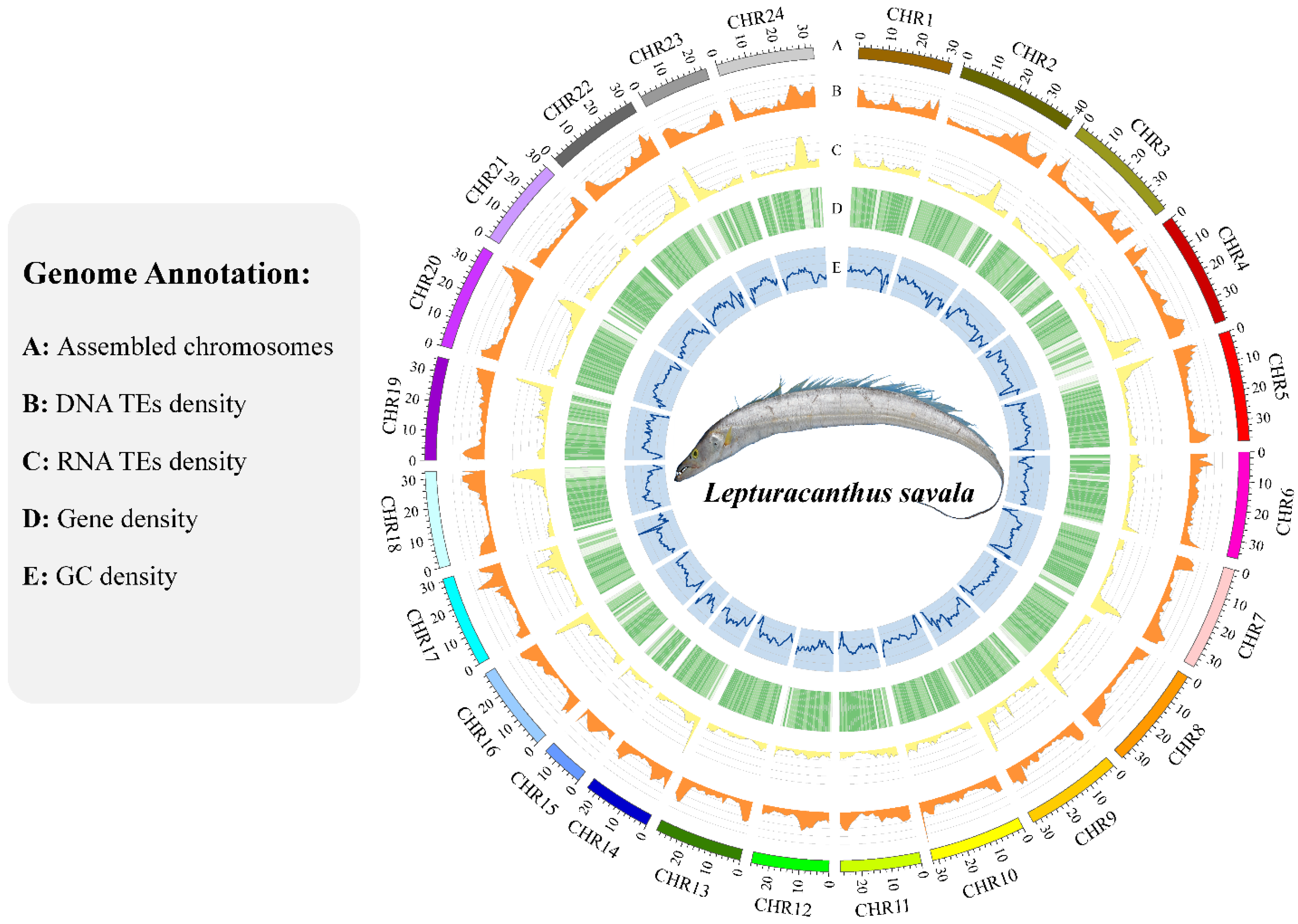
3.4. Genome Annotation
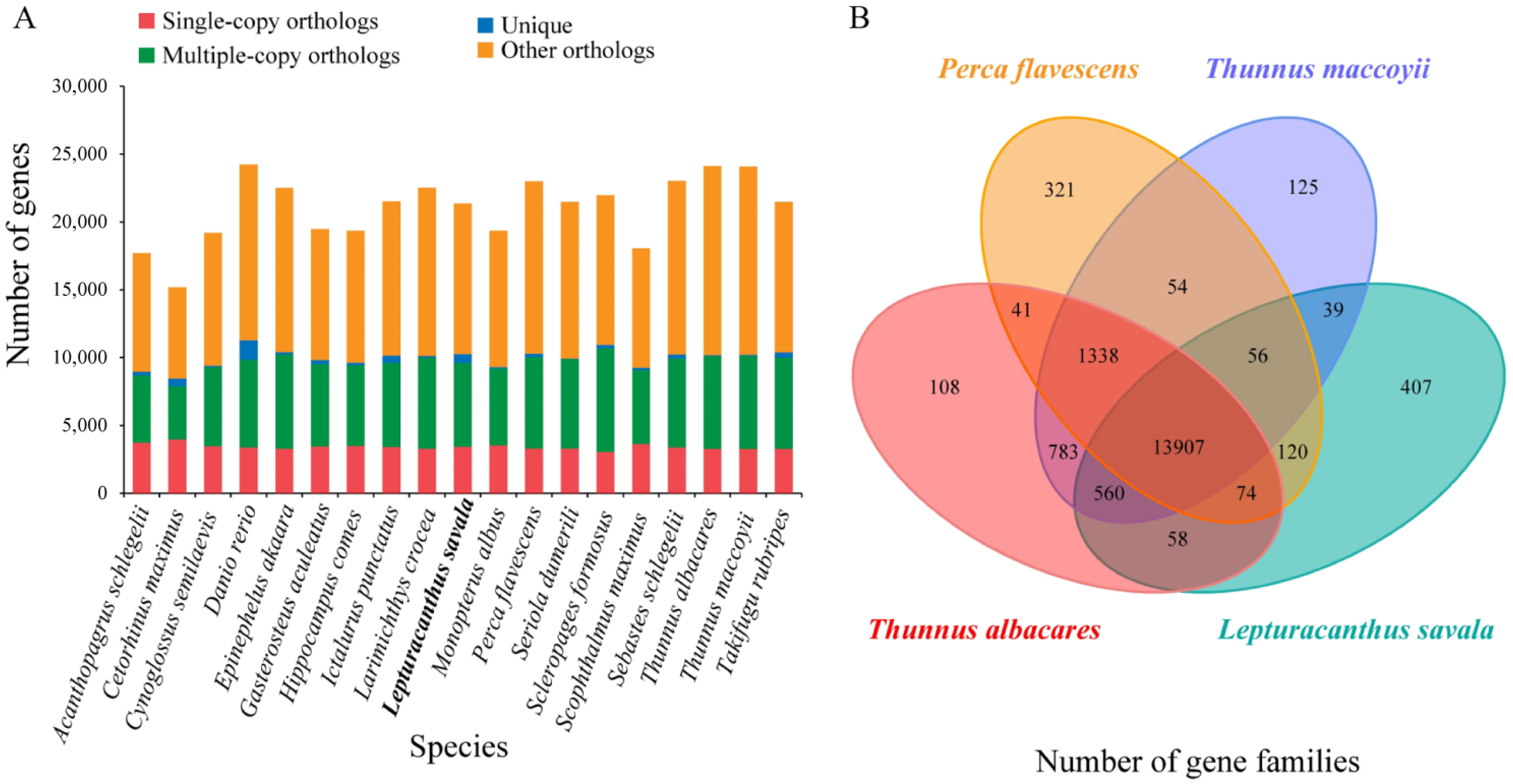
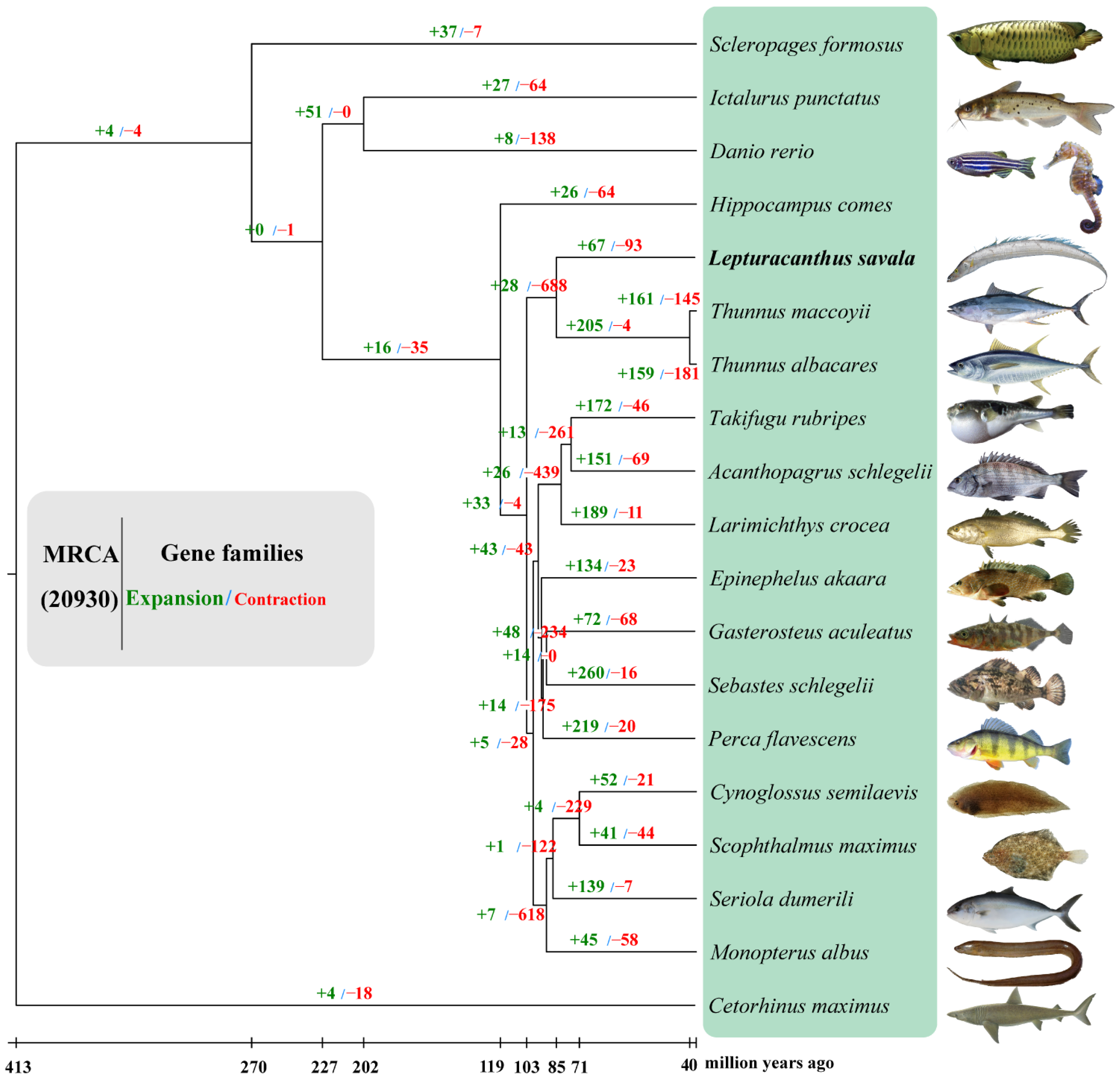
3.5. Gene Family Clustering, Expansion and Contraction, and Phylogenetic Analyses
3.6. Positive Selection Analysis
4. Discussion
4.1. Quality Evaluation of the L. savala Genome
4.2. Genes Associated with the Specific Morphology of L. savala
4.3. Movement and Immunity in L. savala
4.4. Contribution of DNA Repair Mechanisms to the Maintenance of Genomic Stability in L. savala
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nelson, J.S.; Grande, T.C.; Wilson, M.V.H. Fishes of the World, 5th ed.; John Wiley & Sons: New York, NY, USA, 2016; pp. 1–12. [Google Scholar]
- Shao, K.T. The Fish Database of Taiwan. Available online: http://fishdb.sinica.edu.tw (accessed on 28 March 2023).
- Ravi, V.; Venkatesh, B. Rapidly evolving fish genomes and teleost diversity. Curr. Opin. Genet. Dev. 2008, 18, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.S.; Park, J.C.; Kim, M.S.; Han, J.; Kim, D.H.; Hagiwara, A.; Sakakura, Y.; Hwang, U.K.; Lee, B.Y.; Lee, J.S. The reference genome of the selfing fish Kryptolebias hermaphroditus: Identification of phases I and II detoxification genes. Comp. Biochem. Physiol. Part D Genom. Proteom. 2020, 35, 100684. [Google Scholar] [CrossRef]
- Leder, E.H.; Andre, C.; Le Moan, A.L.; Topel, M.; Blomberg, A.; Havenhand, J.N.; Lindstrom, K.; Volckaert, F.A.M.; Kvarnemo, C.; Johannesson, K.; et al. Post-glacial establishment of locally adapted fish populations over a steep salinity gradient. J. Evol. Biol. 2021, 34, 138–156. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, S.; Chapman, J.; Stupka, E.; Putnam, N.; Chia, J.M.; Dehal, P.; Christoffels, A.; Rash, S.; Hoon, S.; Smit, A.; et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 2002, 297, 1301–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Gao, D.; Sims, Y.; Fang, W.; Collins, J.; Torrance, J.; Lin, G.; Xie, J.; Liu, J.; Howe, K. Chromosome-level Genome Assembly of Acanthopagrus latus Provides Insights into Salinity Stress Adaptation of Sparidae. Mar. Biotechnol. 2022, 24, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Gao, H.; Zhang, Y.; Fan, G.; Xu, H.; Zhai, J.; Xu, W.; Chen, Z.; Zhang, H.; Liu, S.; et al. A chromosome-level genome assembly of the giant grouper (Epinephelus lanceolatus) provides insights into its innate immunity and rapid growth. Mol. Ecol. Resour. 2019, 19, 1322–1332. [Google Scholar] [CrossRef]
- Chen, H.Y.; Chen, Y.Y.; Li, R.; Xiao, H.; Chen, S.Y. Research advances in whole-genome sequencing of representative fish species. J. Biol. 2017, 34, 73–77. [Google Scholar]
- Ahmad, S.F.; Jehangir, M.; Srikulnath, K.; Martins, C. Fish genomics and its impact on fundamental and applied research of vertebrate biology. Rev. Fish Biol. Fish. 2022, 32, 357–385. [Google Scholar] [CrossRef]
- Cossins, A.R.; Crawford, D.L. Fish as models for environmental genomics. Nat. Rev. Genet. 2005, 6, 324–333. [Google Scholar] [CrossRef]
- Ahti, P.A.; Kuparinen, A.; Uusi-Heikkilä, S. Size does matter—the eco-evolutionary effects of changing body size in fish. Environ. Rev. 2020, 28, 311–324. [Google Scholar] [CrossRef]
- Giammona, F.F. Form and Function of the Caudal Fin Throughout the Phylogeny of Fishes. Integr. Comp. Biol. 2021, 61, 550–572. [Google Scholar] [CrossRef]
- Bradic, M.; Beerli, P.; García-de León, F.J.; Esquivel-Bobadilla, S.; Borowsky, R.L. Gene flow and population structure in the Mexican blind cavefish complex (Astyanax mexicanus). BMC Evol. Biol. 2012, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Protas, M.; Conrad, M.; Gross, J.B.; Tabin, C.; Borowsky, R. Regressive Evolution in the Mexican Cave Tetra, Astyanax mexicanus. Curr. Biol. 2007, 17, 452–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGaugh, S.E.; Gross, J.B.; Aken, B.; Blin, M.; Borowsky, R.; Chalopin, D.; Hinaux, H.; Jeffery, W.R.; Keene, A.; Ma, L.; et al. The cavefish genome reveals candidate genes for eye loss. Nat. Commun. 2014, 5, 5307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, B.; Lee, A.P.; Ravi, V.; Maurya, A.K.; Lian, M.M.; Swann, J.B.; Ohta, Y.; Flajnik, M.F.; Sutoh, Y.; Kasahara, M.; et al. Elephant shark genome provides unique insights into gnathostome evolution. Nature 2014, 505, 174–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Fan, S.; Zhang, Y.; Xu, M.; Zhang, H.; Yang, Y.; Lee, A.P.; Woltering, J.M.; Ravi, V.; Gunter, H.M.; et al. The seahorse genome and the evolution of its specialized morphology. Nature 2016, 540, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Yu, H.; Ravi, V.; Li, C.; Lee, A.P.; Lian, M.M.; Tay, B.H.; Brenner, S.; Wang, J.; Yang, H.; et al. The genome of the largest bony fish, ocean sunfish (Mola mola), provides insights into its fast growth rate. Gigascience 2016, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Fan, G.; Chan, J.; Ma, K.; Yang, B.; Zhang, H.; Yang, X.; Shi, C.; Law, H.C.H.; Ren, Z.; Xu, Q.; et al. Chromosome-level reference genome of the Siamese fighting fish Betta splendens, a model species for the study of aggression. Gigascience 2018, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Huang, Y.; Bian, C.; You, X.; Zhang, X.; Chen, J.; Wang, M.; Hu, C.; Xu, Y.; Xu, J.; et al. Whole genome sequencing of the fast-swimming Southern bluefin tuna (Thunnus maccoyii). Front. Genet. 2022, 13, 1020017. [Google Scholar] [CrossRef]
- Froese, R.; Pauly, D.; FishBase. World Wide Web Electronic Publication. Version (02/2023). Available online: http://www.fishbase.org (accessed on 28 March 2023).
- James, P. MBAI Memoir No. 1: The Ribbon-Fishes of the Family Trichiuridae of India; Western Printers & Printers, Bombay-13: Madras State, India, 1967; pp. 15–28. [Google Scholar]
- Nakamura, I.; Parin, N.V. An annotated and illustrated catalogue of the Snake Mackerels, Snoeks, Escolars, Gemfishes, Sackfishes, Domine, Oilfish, Cutlassfishes, Scabbardfishes, Hairtails and Frostfishes known to date. FAO Fish. Synopis 1993, 125, 100–101. [Google Scholar]
- Liu, J.; Wu, R.X.; Kang, B.; Ma, L. Fishes of Beibu Gulf; Science Press: Beijing, China, 2016; p. 340. [Google Scholar]
- Chakravarty, M.S.; Pavani, B.; Ganesh, P.R.C. Gonado-somatic index and fecundity studies in two species of ribbon fishes, Trichiurus lepturus (Linnaeus, 1758) and Lepturacanthus savala (Cuvier, 1829) off Visakhapatnam, east coast of India. Indian J. Fish. 2013, 60, 163–165. [Google Scholar]
- Memon, K.H.; Liu, Q.; Kalhoro, M.A.; Chang, M.S.; Baochao, L.; Memon, A.M.; Hyder, S.; Tabassum, S. Growth and mortality parameters of hairtail Lepturacanthus savala from Pakistan waters. Pak. J. Zool. 2016, 48, 829–837. [Google Scholar]
- Fischer, W.; Bianchi, G. FAO Species Identification Sheets for Fishery Purposes: Western Indian Ocean (Fishing Area 51); Food and Agriculture Organization of the United Nations: Rome, Italy, 1984; Volume IV, Trichiuridae Lept 2. [Google Scholar]
- Yi, M.R. Based on Skeletal Comparison and COI Sequence Analysis for 6 Species of Cutlassfishes Trichiuridae Systematic Classification in Chinese Costal Water. Master’s Thesis, Guangdong Ocean University, Zhanjiang, China, 2019. [Google Scholar]
- Wang, Y.M.; Tang, W.Q. A Comparative Study of the Number of Vertebrae in Chinese Teleost Fishes. In Proceedings of the 2012 Symposium of Ichthyology Branch of Chinese Marine Lake and Marsh Society and Ichthyology Branch of Chinese Zoological Society, Lanzhou City, China, 1 September 2012; Chinese Zoological Society Press: Beijing, China, 2012; p. 35. [Google Scholar]
- Pakhmode, P.K.; Mohite, S.A.; Mohite, A.S. Morphological characters and morphometric relationship of ribbonfish, Lepturacanthus savala (Cuvier, 1929) off Ratnagiri coast, Maharashtra. Species 2013, 5, 18–22. [Google Scholar]
- Zhang, B. Preliminary Studies on Marine Food Web and Trophodynamics in China Coastal Seas. PhD Thesis, Ocean University of China, Qingdao, China, 2005. [Google Scholar]
- Kudale, S.; Rathod, J. Sex Ratio of Ribbonfish, Lepturacanthus Savala (Cuvier, 1829) From Karwar Waters, Karnataka. IOSR J. Environ. Sci. Toxicol. Food Technol. 2014, 8, 07–10. [Google Scholar] [CrossRef]
- Pakhmode, P.K.; Mohite, S.A. Study of gonad development using ova diameter analysis in ribbonfish, Lepturacanthus savala (Cuvier, 1829). IQSR J. Agri. Vet. Sci. 2016, 9, 01–05. [Google Scholar]
- Ahmed, Q.; Benzer, S.; Ali, Q.M. Heavy Metal Concentration in Largehead Hairtail (Trichiurus lepturus Linneaus, 1758) and Savalai Hairtail (Lepturacanthus savala (Cuvier, 1829)) Obtained from Karachi Fish Harbour, Pakistan. Bull. Environ. Contam. Toxicol. 2018, 101, 467–472. [Google Scholar] [CrossRef]
- Pakhmode, P.K.; Mohite, S.A. Feeding biology of ribbonfish, Lepturacanthus savala (Cuvier, 1929) off Ratnagiri coast, Maharashtra. Int. J. Fish. Aquat. Stud. 2014, 1, 123–129. [Google Scholar]
- Cai, C.; Song, N.; Zhao, L.; Gao, T. The complete mitogenome of the Lepturacanthus savala (Perciformes: Trichiuridae) from the Yellow Sea. Mitochondrial DNA B Resour. 2020, 5, 2815–2816. [Google Scholar] [CrossRef]
- Zhang, H.R.; Liang, Z.B.; Wu, R.X.; Niu, S.F.; Liang, Y.; Wang, Q.; Wei, H.; Xiao, Y.; Sun, B. Microsatellite Loci Isolation in the Savalai hairtail (Lepturacanthus savala) Based on SLAF-seq Technology and Generality in the Related Species. Genom. Appl. Biol. 2018, 37, 3331–3338. [Google Scholar]
- Sambrook, J.; Russell, D.W. The Inoue Method for Preparation and Transformation of Competent E. Coli: “Ultra-Competent” Cells. Cold Spring Harb. Protoc. 2006, 1, pdb.prot3944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread Polycistronic Transcripts in Fungi Revealed by Single-Molecule mRNA Sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. Quant. Biol. 2012, arXiv:1207.3907. [Google Scholar]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingett, S.; Ewels, P.; Furlan-Magaril, M.; Nagano, T.; Schoenfelder, S.; Fraser, P.; Andrews, S. HiCUP: Pipeline for mapping and processing Hi-C data [version 1; referees: 2 approved, 1 approved with reservations]. F1000Res. 2015, 4, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, S.; Zhao, Q.; Ming, R.; Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 2019, 5, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Stanke, M. Predicting Genes in Single Genomes with AUGUSTUS. Curr. Protoc. Bioinf. 2019, 65, e57. [Google Scholar] [CrossRef] [Green Version]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef] [Green Version]
- Alioto, T.; Blanco, E.; Parra, G.; Guigó, R. Using geneid to Identify Genes. Curr. Protoc. Bioinf. 2018, 64, e56. [Google Scholar] [CrossRef]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic. Acids. Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic. Acids. Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic. Acids. Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinforma. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic. Acids. Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic. Acids. Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. CABIOS, Comput. Appl. Biosci. 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Song, N.; Zhao, X.; Cai, C.; Gao, T. Profile of the genomic characteristics and comparative studies of five Trichiuridae species by genome survey sequencing. Front. Mar. Sci. 2022, 9, 962307. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, T.; Chen, J.; Wu, L.; Wu, X.; Zhang, W.; Luo, J.; Xia, J.; Meng, Z.; Liu, X. Whole-genome sequencing of brown-marbled grouper (Epinephelus fuscoguttatus) provides insights into adaptive evolution and growth differences. Mol. Ecol. Resour. 2021, 22, 711–723. [Google Scholar] [CrossRef]
- Earl, D.; Bradnam, K.; John, J.S.; Darling, A.; Lin, D.; Fass, J.; Yu, H.O.K.; Buffalo, V.; Zerbino, D.R.; Diekhans, M.; et al. Assemblathon 1: A competitive assessment of de novo short read assembly methods. Genome Res. 2011, 21, 2224–2241. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Kim, J.H.; Jo, E.; Lee, S.J.; Jung, J.; Kim, B.M.; Lee, J.H.; Oh, T.J.; Yum, S.; Rhee, J.S.; et al. 2020. Chromosomal-level assembly of Takifugu obscurus (Abe, 1949) genome using third-generation DNA sequencing and Hi-C analysis. Mol. Ecol. Resour. 2019, 20, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, J.; Dong, J.; Niu, Y.; Hu, J.; Lian, J.; Li, W.; Li, J.; Tian, Y.; Shi, Q.; et al. Chromosome-level genome assembly for the largemouth bass Micropterus salmoides provides insights into adaptation to fresh and brackish water. Mol. Ecol. Resour. 2021, 21, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Xiao, Z.; Ma, D.; Liu, J.; Li, J. Genome sequence of the barred knifejaw Oplegnathus fasciatus (Temminck & Schlegel, 1844): The first chromosome-level draft genome in the family Oplegnathidae. Gigascience 2019, 8, giz013. [Google Scholar] [PubMed] [Green Version]
- Coutts, A.S.; MacKenzie, E.; Griffith, E.; Black, D.M. TES is a novel focal adhesion protein with a role in cell spreading. J. Cell. Sci. 2003, 116, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Garvalov, B.K.; Higgins, T.E.; Sutherland, J.D.; Zettl, M.; Scaplehorn, N.; Kocher, T.; Piddini, E.; Griffiths, G.; Way, M. The conformational state of Tes regulates its zyxin-dependent recruitment to focal adhesions. J. Cell. Biol. 2003, 161, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, E.; Coutts, A.S.; Black, D.M. RNAi Knockdown of the Focal Adhesion Protein TES Reveals Its Role in Actin Stress Fibre Organisation. Cell Motil. Cytoskelet. 2005, 60, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Dingwell, K.S.; Smith, J.C. Tes regulates neural crest migration and axial elongation in Xenopus. Dev. Biol. 2006, 293, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Coleman, N.; Subbiah, V.; Pant, S.; Patel, K.; Roy-Chowdhuri, S.; Yedururi, S.; Johnson, A.; Yap, T.A.; Rodon, J.; Shaw, K.; et al. Emergence of mTOR mutation as an acquired resistance mechanism to AKT inhibition, and subsequent response to mTORC1/2 inhibition. NPJ. Precis. Oncol. 2021, 5, 99. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Villa, E.; Sahu, U.; O’Hara, B.P.; Ali, E.S.; Helmin, K.A.; Asara, J.M.; Gao, P.; Singer, B.D.; Ben-Sahra, I. mTORC1 stimulates cell growth through SAM synthesis and m6A mRNA-dependent control of protein synthesis. Mol. Cell 2021, 81, 2076–2093.e9. [Google Scholar] [CrossRef]
- Chen, J.; Long, F. mTORC1 signaling controls mammalian skeletal growth through stimulation of protein synthesis. Development 2014, 141, 2848–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitter, S.; Matthews, M.P.; Martin, S.K.; Xie, J.; Ooi, S.S.; Walkley, C.R.; Codrington, J.D.; Ruegg, M.A.; Hall, M.N.; Proud, C.G.; et al. mTORC1 Plays an Important Role in Skeletal Development by Controlling Preosteoblast Differentiation. Mol. Cell. Biol. 2017, 37, e00668-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, E.; Berna-Erro, A.; Lopez, J.J.; Granados, M.P.; Bermejo, N.; Brull, J.M.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. Role of mTOR1 and mTOR2 complexes in MEG-01 cell physiology. Thromb. Haemost. 2015, 114, 969–981. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Holguin, N.; Shi, Y.; Silva, M.J.; Long, F. mTORC2 Signaling Promotes Skeletal Growth and Bone Formation in Mice. J. Bone. Miner. Res. 2015, 30, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Matoba, K.; Noda, N.N. Structural catalog of core Atg proteins opens new era of autophagy research. J. Biochem. 2021, 169, 517–525. [Google Scholar] [CrossRef]
- Fang, D.; Xie, H.; Hu, T.; Shan, H.; Li, M. Binding Features and Functions of ATG3. Front. Cell. Dev. Biol. 2021, 9, 685625. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell. Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Oral, O.; Oz-Arslan, D.; Itah, Z.; Naghavi, A.; Deveci, R.; Karacali, S.; Gozuacik, D. Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis 2012, 17, 810–820. [Google Scholar] [CrossRef]
- Li, M.; Hou, Y.; Wang, J.; Chen, X.; Shao, Z.; Yin, X. Kinetics Comparisons of Mammalian Atg4 Homologues Indicate Selective Preferences toward Diverse Atg8 Substrates. J. Biol. Chem. 2011, 286, 7327–7338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.N.; Padman, B.S.; Zellner, S.; Khuu, G.; Uoselis, L.; Lam, W.K.; Skulsuppaisarn, M.; Lindblom, R.S.J.; Watts, E.M.; Behrends, C.; et al. ATG4 family proteins drive phagophore growth independently of the LC3/GABARAP lipidation system. Mol. Cell 2021, 81, 2013–2030. [Google Scholar]
- Bartolomeo, S.D.; Nazio, F.; Cecconi, F. The role of autophagy during development in higher eukaryotes. Traffic 2010, 11, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, F.; Levine, B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Dev. Cell. 2008, 15, 344–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luciani, M.F.; Giusti, C.; Harms, B.; Oshima, Y.; Kikuchi, H.; Kubohara, Y.; Golstein, P. Atg1 allows second-signaled autophagic cell death in Dictyostelium. Autophagy 2011, 7, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Wei, W.; Ablimit, M.; Ma, Y.; Fu, T.; Liu, K.; Peng, J.; Li, Y.; Hong, H. Responses of two insect cell lines to starvation: Autophagy prevents them from undergoing apoptosis and necrosis, respectively. J. Insect. Physiol. 2011, 57, 723–734. [Google Scholar] [CrossRef]
- Malagoli, D.; Abdalla, F.C.; Cao, Y.; Feng, Q.; Fujisaki, K.; Gregorc, A.; Matsuo, T.; Nezis, I.P.; Papassideri, I.S.; Sass, M.; et al. Autophagy and its physiological relevance in arthropods: Current knowledge and perspectives. Autophagy 2010, 6, 575–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanelli, D.; Casati, B.; Franzetti, E.; Tettamanti, G. A Molecular View of Autophagy in Lepidoptera. Biomed. Res. Int. 2014, 2014, 902315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzetti, E.; Huang, Z.J.; Shi, Y.X.; Xie, K.; Deng, X.J.; Li, J.P.; Li, Q.R.; Yang, W.Y.; Zeng, W.N.; Casartelli, M.; et al. Autophagy precedes apoptosis during the remodeling of silkworm larval midgut. Apoptosis 2012, 17, 305–324. [Google Scholar] [CrossRef] [PubMed]
- Varga, M.; Sass, M.; Papp, D.; Takacs-Vellai, K.; Kobolak, J.; Dinnyes, A.; Klionsky, D.J.; Vellai, T. Autophagy is required for zebrafish caudal fin regeneration. Cell Death Differ. 2014, 21, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bircher, J.E.; Koleske, A.J. Trio family proteins as regulators of cell migration and morphogenesis in development and disease-mechanisms and cellular contexts. J. Cell. Sci. 2021, 134, jcs248393. [Google Scholar] [CrossRef]
- Chen, H.; Guo, S.; Xia, Y.; Yuan, L.; Lu, M.; Zhou, M.; Fang, M.; Meng, L.; Xiao, Z.; Ma, J. The role of Rho-GEF Trio in regulating tooth root development through the p38 MAPK pathway. Exp. Cell. Res. 2018, 372, 158–167. [Google Scholar] [CrossRef]
- Lv, W.; Ma, A.; Chi, X.; Li, Q.; Pang, Y.; Su, P. A novel complement factor I involving in the complement system immune response from Lampetra morii. Fish Shellfish. Immunol. 2020, 98, 988–994. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Overview of Complement Activation and Regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimiyan, H.; Mostafaei, S.; Aslani, S.; Faezi, S.T.; Farhadi, E.; Jamshidi, A.; Mahmoudi, M. Association between complement gene polymorphisms and systemic lupus erythematosus: A systematic review and meta-analysis. Clin. Exp. Med. 2022, 22, 427–438. [Google Scholar] [CrossRef]
- Nayak, A.; Pednekar, L.; Reid, K.B.; Kishore, U. Complement and non-complement activating functions of C1q: A prototypical innate immune molecule. Innate Immun. 2012, 18, 350–363. [Google Scholar] [CrossRef]
- Eggleton, P.; Tenner, A.J.; Reid, K.B. C1q receptors. Clin. Exp. Immunol. 2020, 120, 406–412. [Google Scholar] [CrossRef]
- Oikonomopoulou, K.; Ricklin, D.; Ward, P.A.; Lambris, J.D. Interactions between coagulation and complement—Their role in inflammation. Semin. Immunopathol. 2012, 34, 151–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajic, G.; Degn, S.E.; Thiel, S.; Andersen, G.R. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J. 2015, 34, 2735–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavia, L.; Santiesteban-Lores, L.E.; Carneiro, M.C.; Prodocimo, M.M. Advances in the complement system of a teleost fish, Oreochromisniloticus. Fish Shellfish. Immunol. 2022, 123, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Su, Y.C.; Riesbeck, K. Vitronectin in bacterial pathogenesis: A host protein used in complement escape and cellular invasion. Mol. Microbiol. 2010, 78, 545–560. [Google Scholar] [CrossRef]
- Li, S.; Hao, G.; Peng, W.; Geng, X.; Sun, J. Expression and functional characterization of vitronectin gene from Japanese flounder (Paralichthys olivaceus). Fish Shellfish. Immunol. 2017, 65, 9–16. [Google Scholar] [CrossRef]
- Hebenstreit, D.; Wirnsberger, G.; Horejs-Hoeck, J.; Duschl, A. Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev. 2006, 17, 173–188. [Google Scholar] [CrossRef]
- Chen, H.; Sun, H.; You, F.; Sun, W.; Zhou, X.; Chen, L.; Yang, J.; Wang, Y.; Tang, H.; Guan, Y.; et al. Activation of STAT6 by STING Is Critical for Antiviral Innate Immunity. Cell 2011, 147, 436–446. [Google Scholar] [CrossRef] [Green Version]
- Secombes, C.J.; Wang, T.; Bird, S. The interleukins of fish. Dev. Comp. Immunol. 2011, 35, 1336–1345. [Google Scholar] [CrossRef]
- El Kasmi, K.C.; Smith, A.M.; Williams, L.; Neale, G.; Panopoulos, A.D.; Watowich, S.S.; Hacker, H.; Foxwell, B.M.; Murray, P.J. Cutting Edge: A Transcriptional Repressor and Corepressor Induced by the STAT3-Regulated Anti-Inflammatory Signaling Pathway. J. Immunol. 2007, 179, 7215–7219. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.I.; Logsdon, N.J.; Sheikh, F.; Donnelly, R.P.; Walter, M.R. Conformational Changes Mediate Interleukin-10 Receptor 2 (IL-10R2) Binding to IL-10 and Assembly of the Signaling Complex. J. Biol. Chem. 2006, 281, 35088–35096. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.P.; Turner, J.E. Debunking the Myth of Exercise-Induced Immune Suppression: Redefining the Impact of Exercise on Immunological Health Across the Lifespan. Front. Immunol. 2018, 9, 648. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, J.G.; Matson, K.D. Ecological Immunology through the Lens of Exercise Immunology: New Perspective on the Links between Physical Activity and Immune Function and Disease Susceptibility in Wild Animals. Integr. Comp. Biol. 2016, 56, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, V.; Grisdale-Helland, B.; Jørgensen, S.M.; Helgerud, J.; Claireaux, G.; Farrell, A.P.; Krasnov, A.; Helland, S.J.; Takle, H. Disease resistance is related to inherent swimming performance in Atlantic salmon. BMC Physiol. 2013, 13, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, N.A.; Timmerhaus, G.; Baranski, M.; Andersen, O.; Takle, H.; Krasnov, A. Training the salmon’s genes: Influence of aerobic exercise, swimming performance and selection on gene expression in Atlantic salmon. BMC Genom. 2017, 18, 971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Q.; Fu, S.; Huang, T.; Li, X.; Shi, X. Effects of Aerobic Exercise Training on the Growth, Swimming Performance, Antipredation Ability and Immune Parameters of Juvenile Rock Carp (Procypris rabaudi). Animals 2022, 12, 257. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Song, B.; Lin, X.; Xu, Z. Effects of water-current speed on hematological, biochemical and immune parameters in juvenile tinfoil barb, Barbonymus schwanenfeldii (Bleeker, 1854). Chin. J. Oceanol. Limnol. 2015, 34, 118–124. [Google Scholar] [CrossRef]
- Areal, L.B.; Pereira, L.P.; Ribeiro, F.M.; Olmo, I.G.; Muniz, M.R.; Rodrigues, M.D.C.; Costa, P.F.; Martins-Silva, C.; Ferguson, S.S.G.; Guimarães, D.A.M.; et al. Role of Dynein Axonemal Heavy Chain 6 Gene Expression as a Possible Biomarker for Huntington’s Disease: A Translational Study. J. Mol. Neurosci. 2017, 63, 342–348. [Google Scholar] [CrossRef]
- Whitfield, M.; Thomas, L.; Bequignon, E.; Schmitt, A.; Stouvenel, L.; Montantin, G.; Tissier, S.; Duquesnoy, P.; Copin, B.; Chantot, S.; et al. Mutations in DNAH17, Encoding a Sperm-Specific Axonemal Outer Dynein Arm Heavy Chain, Cause Isolated Male Infertility Due to Asthenozoospermia. Am. J. Hum. Genet. 2019, 105, 198–212. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yagi, H.; Onuoha, E.O.; Damerla, R.R.; Francis, R.; Furutani, Y.; Tariq, M.; King, S.M.; Hendricks, G.; Cui, C.; et al. DNAH6 and Its Interactions with PCD Genes in Heterotaxy and Primary Ciliary Dyskinesia. PLoS Genet. 2016, 12, e1005821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Miyata, H.; Gao, Y.; Sha, Y.; Tang, S.; Xu, Z.; Whitfield, M.; Patrat, C.; Wu, H.; Dulioust, E.; et al. Bi-allelic DNAH8 Variants Lead to Multiple Morphological Abnormalities of the Sperm Flagella and Primary Male Infertility. Am. J. Hum. Genet. 2020, 107, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Xu, K.; Zhou, Y.; Wu, C.; Wang, S.; Xiao, J.; Wen, M.; Zhao, R.; Luo, K.; Tao, M.; et al. Different expression patterns of sperm motility-related genes in testis of diploid and tetraploid cyprinid fish. Biol. Reprod. 2017, 96, 907–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgers, P.M.; Koonin, E.V.; Bruford, E.; Blanco, L.; Burtis, K.C.; Christman, M.F.; Copeland, W.C.; Friedberg, E.C.; Hanaoka, F.; Hinkle, D.C.; et al. Eukaryotic DNA polymerases: Proposal for a revised nomenclature. J. Biol. Chem. 2001, 276, 43487–43490. [Google Scholar] [CrossRef] [Green Version]
- Nick McElhinny, S.A.; Ramsden, D.A. Polymerase Mu Is a DNA-Directed DNA/RNA Polymerase. Mol. Cell Biol. 2003, 23, 2309–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladdy, R.A.; Nutter, L.M.; Kunath, T.; Danska, J.S.; Guidos, C.J. p53-Independent Apoptosis Disrupts Early Organogenesis in Embryos Lacking Both Ataxia-Telangiectasia Mutated and Prkdc. Mol. Cancer Res. 2006, 4, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.-C.W.; Hwang, L.-Y.; Bowcock, A.M.; Baer, R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996, 14, 430–440. [Google Scholar] [CrossRef]
- Laufer, M.; Nandula, S.V.; Modi, A.P.; Wang, S.; Jasin, M.; Murty, V.V.; Ludwig, T.; Baer, R. Structural Requirements for the BARD1 Tumor Suppressor in Chromosomal Stability and Homology-directed DNA Repair. J. Biol. Chem. 2007, 282, 34325–34333. [Google Scholar] [CrossRef] [Green Version]
- Westermark, U.K.; Reyngold, M.; Olshen, A.B.; Baer, R.; Jasin, M.; Moynahan, M.E. BARD1 Participates with BRCA1 in Homology-Directed Repair of Chromosome Breaks. Mol. Cell. Biol. 2003, 23, 7926–7936. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.R.; Solomon, E. BRCA1: BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum. Mol. Genet. 2004, 13, 807–817. [Google Scholar] [CrossRef] [Green Version]
- Wu-Baer, F.; Ludwig, T.; Baer, R. The UBXN1 Protein Associates with Autoubiquitinated Forms of the BRCA1 Tumor Suppressor and Inhibits Its Enzymatic Function. Mol. Cell. Biol. 2010, 30, 2787–2798. [Google Scholar] [CrossRef] [Green Version]
- Stracker, T.H. Chaperoning the DNA damage response. FEBS J. 2017, 284, 2375–2377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuhlke, K.A.; Johnson, A.M.; Okoth, L.A.; Stoffel, E.M.; Robbins, C.M.; Tembe, W.A.; Salinas, C.A.; Zheng, S.L.; Xu, J.F.; Carpten, J.D.; et al. Identification of a novel NBN truncating mutation in a family with hereditary prostate cancer. Fam. Cancer 2012, 11, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Lamerdin, J.E.; Tebbs, R.S.; Schild, D.; Tucker, J.D.; Shen, M.R.; Brookman, K.W.; Siciliano, M.J.; Walter, C.A.; Fan, W.F.; et al. XRCC2 and XRCC3, New Human Rad51-Family Members, Promote Chromosome Stability and Protect against DNA Cross-Links and Other Damages. Mol. Cell 1998, 1, 783–793. [Google Scholar] [CrossRef]
- Cui, X.; Brenneman, M.; Meyne, J.; Oshimura, M.; Goodwin, E.H.; Chen, D.J. The XRCC2 and XRCC3 repair genes are required for chromosome stability in mammalian cells. Mutat. Res. DNA Repair. 1999, 434, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Ling, C.; Coulthard, R.; Yan, Z.; Xue, Y.; Meetei, A.R.; Laghmani, E.H.; Joenje, H.; McDonald, N.; de Winter, J.P.; et al. Identification of FAAP24, a Fanconi Anemia Core Complex Protein that Interacts with FANCM. Mol. Cell. 2007, 25, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Ishiai, M.; Ali, A.M.; Medhurst, A.L.; Neveling, K.; Kalb, R.; Yan, Z.; Xue, Y.; Oostra, A.B.; Auerbach, A.D.; et al. FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO J. 2007, 26, 2104–2114. [Google Scholar] [CrossRef]
- Kristensen, A.S.; Andersen, J.; Jorgensen, T.N.; Sorensen, L.; Eriksen, J.; Loland, C.J.; Stromgaard, K.; Gether, U. SLC6 Neurotransmitter Transporters: Structure, Function, and Regulation. Pharmacol. Rev. 2011, 63, 585–640. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, K.; Das, A.K.; Luethi, D.; Szöllősi, D.; Schütz, G.J.; Reith, M.E.A.; Sitte, H.H.; Stockner, T. SLC6 transporter oligomerization. J. Neurochem. 2020, 157, 919–929. [Google Scholar] [CrossRef]
- Meuwissen, M.E.; Halley, D.J.; Smit, L.S.; Lequin, M.H.; Cobben, J.M.; de Coo, R.; van Harssel, J.; Sallevelt, S.; Woldringh, G.; van der Knaap, M.S.; et al. The expanding phenotype of COL4A1 and COL4A2 mutations: Clinical data on 13 newly identified families and a review of the literature. Genet. Med. 2015, 17, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Billhaq, D.H.; Lee, S.H.; Lee, S. The potential function of endometrial-secreted factors for endometrium remodeling during the estrous cycle. Anim. Sci. J. 2020, 91, e13333. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Wang, C.; Umesaki, N. Autocrine/paracrine regulation of human endometrial stromal remodeling by laminin and type IV collagen. Int. J. Mol. Med. 2008, 22, 581–587. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
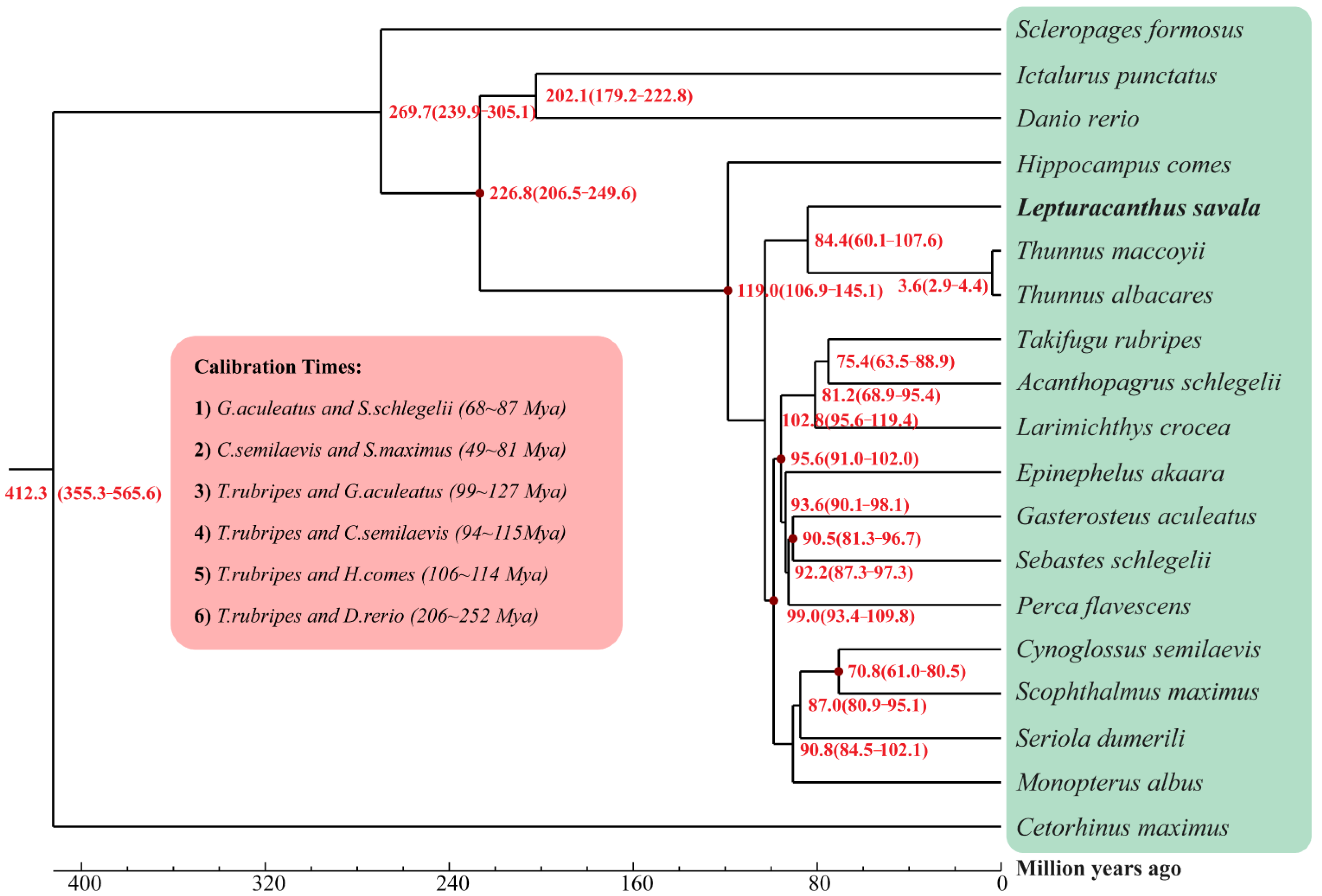{kind=link}
| Methods | Gene Set | Number | Average Transcript Length (bp) | Average CDS Length (bp) | Average Exons per Gene | Average Exon Length (bp) | Average Intron Length (bp) |
|---|---|---|---|---|---|---|---|
| De novo | Augustus | 33,486 | 8827.52 | 1201.39 | 6.87 | 174.85 | 1298.98 |
| GlimmerHMM | 76,383 | 8606.22 | 686.71 | 4.6 | 149.37 | 2201.43 | |
| SNAP | 64,956 | 11,484.45 | 799.65 | 5.76 | 138.71 | 2242.37 | |
| Geneid | 34,350 | 14,311.26 | 1231.61 | 6.09 | 202.2 | 2569.11 | |
| GenScan | 33,084 | 16,121.64 | 1490.15 | 8.22 | 181.34 | 2027.19 | |
| Homolog | Danio rerio | 23,256 | 10,252.69 | 1494.80 | 7.53 | 198.53 | 1341.28 |
| Etheostoma spectabile | 22,973 | 11,643.93 | 1604.77 | 8.3 | 193.26 | 1374.52 | |
| Gasterosteus aculeatus | 27,165 | 8780.31 | 1235.59 | 6.74 | 183.24 | 1313.76 | |
| Homo sapiens | 18,148 | 11,303.80 | 1465.64 | 7.95 | 184.38 | 1415.76 | |
| Oryzias latipes | 23,446 | 11,206.10 | 1630.43 | 8.19 | 199.16 | 1332.48 | |
| Perca flavescens | 26,554 | 10,560.30 | 1481.67 | 7.73 | 191.65 | 1348.74 | |
| Perca fluviatilis | 25,661 | 10,889.74 | 1529.66 | 7.87 | 194.37 | 1362.46 | |
| Sander lucioperca | 25,313 | 11,255.57 | 1578.82 | 8.21 | 192.27 | 1341.86 | |
| Thunnus albacares | 25,361 | 11,411.78 | 1589.75 | 8.25 | 192.62 | 1354.14 | |
| Thunnus maccoyii | 24,446 | 11,716.77 | 1631.43 | 8.48 | 192.5 | 1349.21 | |
| Takifugu rubripes | 22,038 | 11,995.55 | 1634.46 | 8.63 | 189.42 | 1358.19 | |
| RNA-Seq | PASA | 43,445 | 11,533.68 | 1469.51 | 9.05 | 162.38 | 1250.24 |
| Cufflinks | 37,916 | 13,980.13 | 2755.81 | 8.75 | 315.05 | 1448.83 | |
| EVM (EVidenceModeler) | 31,876 | 10,753.39 | 1307.47 | 7.49 | 174.54 | 1455.28 | |
| PASA-update * | 31,434 | 11,153.72 | 1339.72 | 7.67 | 174.73 | 1471.95 | |
| Final set ** | 23,625 | 13,717.34 | 1620.82 | 9.38 | 172.74 | 1442.96 | |
| 1. Contraction (93 Gene Families, 13 KEGG Pathways) | ||
| KEGG pathways | p-value | Genes |
| Synaptic vesicle cycle | 1.61 × 10−6 | SLC6A13, SLC6A1, SLC6A11 |
| GABAergic synapse | 5.39 × 10−6 | SLC6A13, SLC6A1, SLC6A11 |
| Choline metabolism in cancer | 7.27 × 10−5 | SLC22A5, SLC5A7 |
| NOD-like receptor signaling pathway | 0.0028841 | NLRC3, GVIN1, URGCP |
| Small cell lung cancer | 0.0040207 | COL4A1, COL4A2, COL4A6 |
| Protein digestion and absorption | 0.0044306 | COL4A1, COL4A2, COL6A3, SLC6A19, COL4A6, SLC6A19 |
| Pathogenic Escherichia coli infection | 0.0060438 | TUBB1, COL6A3 |
| Necroptosis | 0.0111952 | NLRC3, COL6A3, CAPN2, ALOX5 |
| Mineral absorption | 0.0208705 | SLC6A19 |
| Gap junction | 0.0305233 | TUBB1, COL6A3 |
| Arachidonic acid metabolism | 0.0311748 | ALOX5 |
| ECM–receptor interaction | 0.0359224 | COL4A1, COL4A2, COL6A3, COL6A6 |
| Focal adhesion | 0.0466035 | COL4A1, COL4A2, COL6A3, COL6A6 |
| 2. Expansion (67 gene families, 18 KEGG pathways) | ||
| KEGG pathways | p-value | Genes |
| Focal adhesion | 0.00 | TRIO, TES |
| ECM–receptor interaction | 0.00 | TRIO, TES |
| Platelet activation | 0.00 | TRIO, TES |
| Relaxin signaling pathway | 0.00 | TRIO, TES |
| AGE-RAGE signaling pathway in diabetic complications | 0.00 | TRIO, TES |
| Protein digestion and absorption | 0.00 | TRIO, TES |
| Amoebiasis | 0.00 | TRIO, IGHM, GPR119, TES |
| Human papillomavirus infection | 1.36 × 10−262 | TRIO, F5, EIF3A, TES |
| PI3K-Akt signaling pathway | 6.51 × 10−251 | TRIO, IGHM, TES |
| Olfactory transduction | 4.99 × 10−40 | NONE |
| Lysine degradation | 2.09 × 10−6 | KMT5AA, KMT5A, SET-1 |
| Huntington disease | 4.89 × 10−5 | DNAH7, DNAH11, DNAH9, NES, DNAH3, DNAH5, DNAH8, DNAH2, DHC10, KLF18, SGS4, DNAH1, DNAH6, QRICH2, DNAH10 |
| Staphylococcus aureus infection | 0.0079023 | IGLV1-51, IGHM, SFTPD, MBL, MBL2, IFITM3 |
| Cortisol synthesis and secretion | 0.0119558 | CACNA1G, CACNA1H, CACNA1I, CACNA1H |
| PPAR signaling pathway | 0.0169356 | SAMD3 |
| Bacterial secretion system | 0.017091 | SECA3, SECA |
| Allograft rejection | 0.0267611 | IGLV1-51, IGHM, PRF1 |
| Glycosphingolipid biosynthesis | 0.0462553 | ST3GAL1 |
| Group 1 (Genes: 903; GO Terms: 62; KEGG Pathways: 17) | ||
| A: L. savala; B. A. schlegelii, L. crocea, P. flavescens | ||
| GO terms | KEGG Pathways | Genes screened |
| DNA metabolic process | JAK-STAT signaling pathway | HIRA, IL15RA, PRLR, etc. |
| Nuclear chromosome | Novobiocin biosynthesis | TAT |
| DNA repair | Fanconi anemia pathway | EME2, FAAP100, BRCA1, etc. |
| Nucleic acid binding | Cytokine–cytokine receptor interaction | INHBA, HIRA, TNFRSF26, etc. |
| Helicase activity | Sulfur relay system | SYNPR, MOCS2, NFS1 |
| Nuclease activity | Ether lipid metabolism | TPT1, SH3BGRL3, PLA2G3, etc. |
| Checkpoint clamp complex | Arginine biosynthesis | NOS1, ASL, NAGS, GLS2 |
| Spindle | RNA transport | RANBP2, EIF2B3, RPP30, etc. |
| Hyaluronic acid binding | Homologous recombination | BARD1, EME2, BRCA1, etc. |
| Chromatin binding | Phenylalanine, tyrosine, and tryptophan biosynthesis | TAT |
| Ino80 complex | Tropane, piperidine, and pyridine alkaloid biosynthesis | TAT |
| Protein homodimerization activity | Alanine, aspartate, and glutamate metabolism | ASNS, ASL, ABAT, etc. |
| 7S RNA binding | Ubiquinone and other terpenoid-quinone biosynthesis | TAT, COQ6 |
| Signal recognition particle | Thiamine metabolism | AK5, CFAP61, NFS1 |
| ATPase activity | Ribosome biogenesis in eukaryotes | UTP14A, RIOK1, HEATR1, etc. |
| Isomerase activity | Nonhomologous end-joining | PRKDC, POLM |
| DNA damage checkpoint | Complement and coagulation cascades | F5, PLAU, CPB2, etc. |
| Group 2 (Genes: 922; GO terms: 70; KEGG Pathways: 18) | ||
| A. L. savala, M. albus; B. A. schlegelii, L. crocea, P. flavescens | ||
| GO terms | KEGG Pathways | Genes screened |
| Methyltransferase activity | Cytokine–cytokine receptor interaction | TNFRSF13B, OSMR, PRLR, etc. |
| Aminomethyltransferase activity | JAK-STAT signaling pathway | OSMR, PRLR, IL15RA, etc. |
| Nucleic acid binding | RNA transport | UPF3A, EIF3F, EIF3C, etc. |
| Neurotransmitter metabolic process | Thyroid cancer | ANKDD1A, RET, CCDC6, etc. |
| Catabolic process | Ribosome biogenesis in eukaryotes | HEATR1, REXO1, VSTM2A, etc. |
| Organic substance catabolic process | Autophagy—other | ATG3, TRIM14, MTOR, etc. |
| Glycine catabolic process | Pancreatic cancer | E2F3, ANKDD1A, VEGFAA, etc. |
| RNA cap binding complex | Intestinal immune network for IgA production | TNFRSF13B, IL15RA, CD28, etc. |
| RNA binding | Autophagy—yeast | ATG3, TRIM14, MTOR, etc. |
| Organonitrogen compound catabolic process | Chronic myeloid leukemia | E2F3, ANKDD1A, GRAP, etc. |
| LUBAC complex | EGFR tyrosine kinase inhibitor resistance | VEGFAA, MTOR, GRAP, etc. |
| N-methyltransferase activity | Fanconi anemia pathway | BRCA1, ANKDD1A, RMI1, etc. |
| Phospholipase A2 activity | Phenazine biosynthesis | PBLD |
| Drug catabolic process | Ubiquinone and other terpenoid-quinone biosynthesis | COQ2, TAT, COQ6 |
| Threonine-type endopeptidase activity | Glycine, serine, and threonine metabolism | AMT, CHDH, DMGDH, etc. |
| Proteasome core complex | Nonhomologous end-joining | XRCC6, DCLRE1C, DNTT |
| Kinetochore | Prostate cancer | E2F3, MTOR, GRAP, BAD, etc. |
| Protein homodimerization activity | Acute myeloid leukemia | MTOR, GRAP, BAD, etc. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, R.-X.; Miao, B.-B.; Han, F.-Y.; Niu, S.-F.; Liang, Y.-S.; Liang, Z.-B.; Wang, Q.-H. Chromosome-Level Genome Assembly Provides Insights into the Evolution of the Special Morphology and Behaviour of Lepturacanthus savala. Genes 2023, 14, 1268. https://doi.org/10.3390/genes14061268
Wu R-X, Miao B-B, Han F-Y, Niu S-F, Liang Y-S, Liang Z-B, Wang Q-H. Chromosome-Level Genome Assembly Provides Insights into the Evolution of the Special Morphology and Behaviour of Lepturacanthus savala. Genes. 2023; 14(6):1268. https://doi.org/10.3390/genes14061268
Chicago/Turabian StyleWu, Ren-Xie, Ben-Ben Miao, Fang-Yuan Han, Su-Fang Niu, Yan-Shan Liang, Zhen-Bang Liang, and Qing-Hua Wang. 2023. "Chromosome-Level Genome Assembly Provides Insights into the Evolution of the Special Morphology and Behaviour of Lepturacanthus savala" Genes 14, no. 6: 1268. https://doi.org/10.3390/genes14061268
APA StyleWu, R. -X., Miao, B. -B., Han, F. -Y., Niu, S. -F., Liang, Y. -S., Liang, Z. -B., & Wang, Q. -H. (2023). Chromosome-Level Genome Assembly Provides Insights into the Evolution of the Special Morphology and Behaviour of Lepturacanthus savala. Genes, 14(6), 1268. https://doi.org/10.3390/genes14061268