


The State of Squamate Genomics: Past, Present, and Future of Genome Research in the Most Speciose Terrestrial Vertebrate Order

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
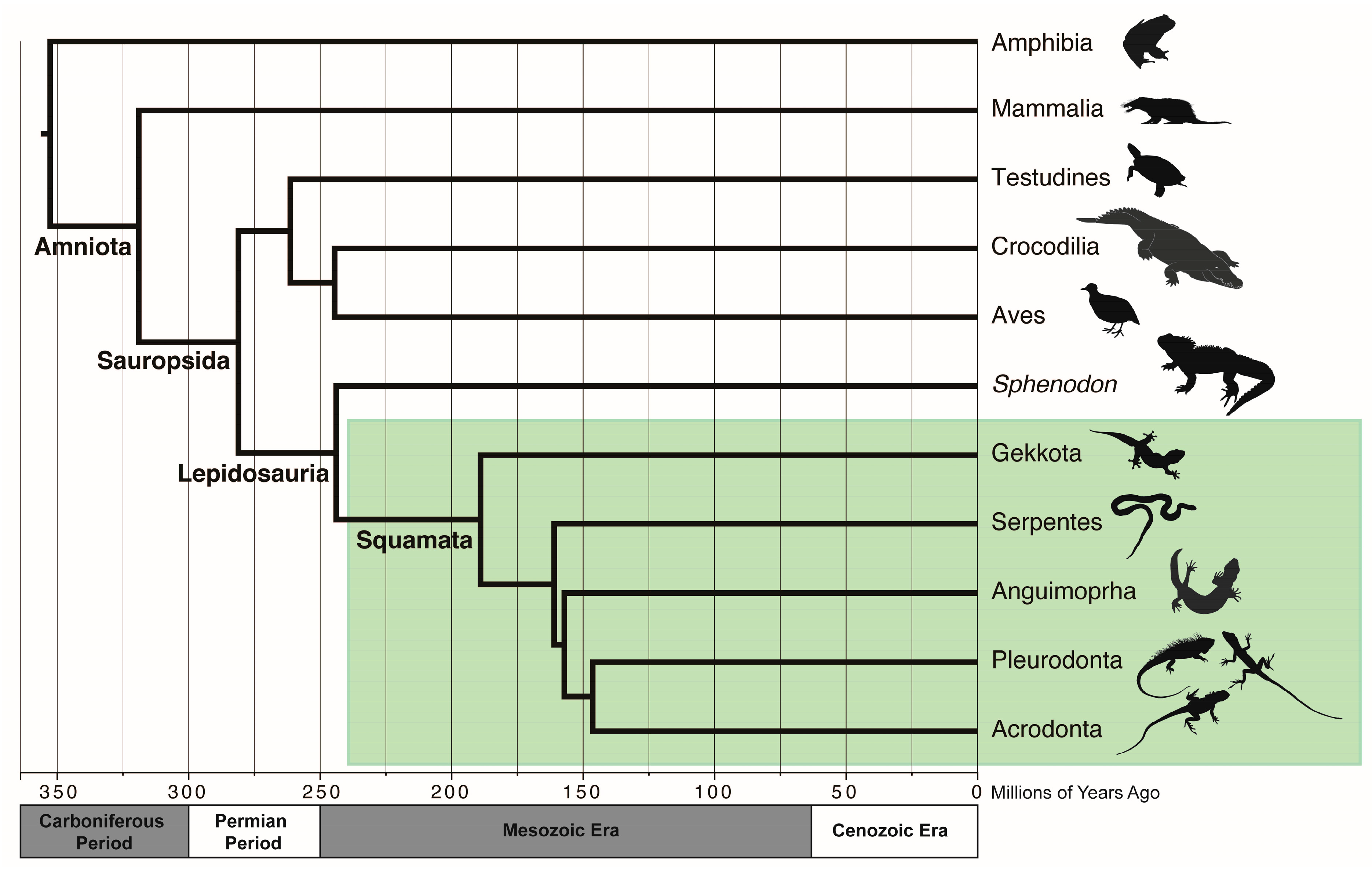
2. What Are Squamates (and “Reptiles” for That Matter)?

3. The Era of Amniote Genomes and The Long Road to Squamate Genome Representation
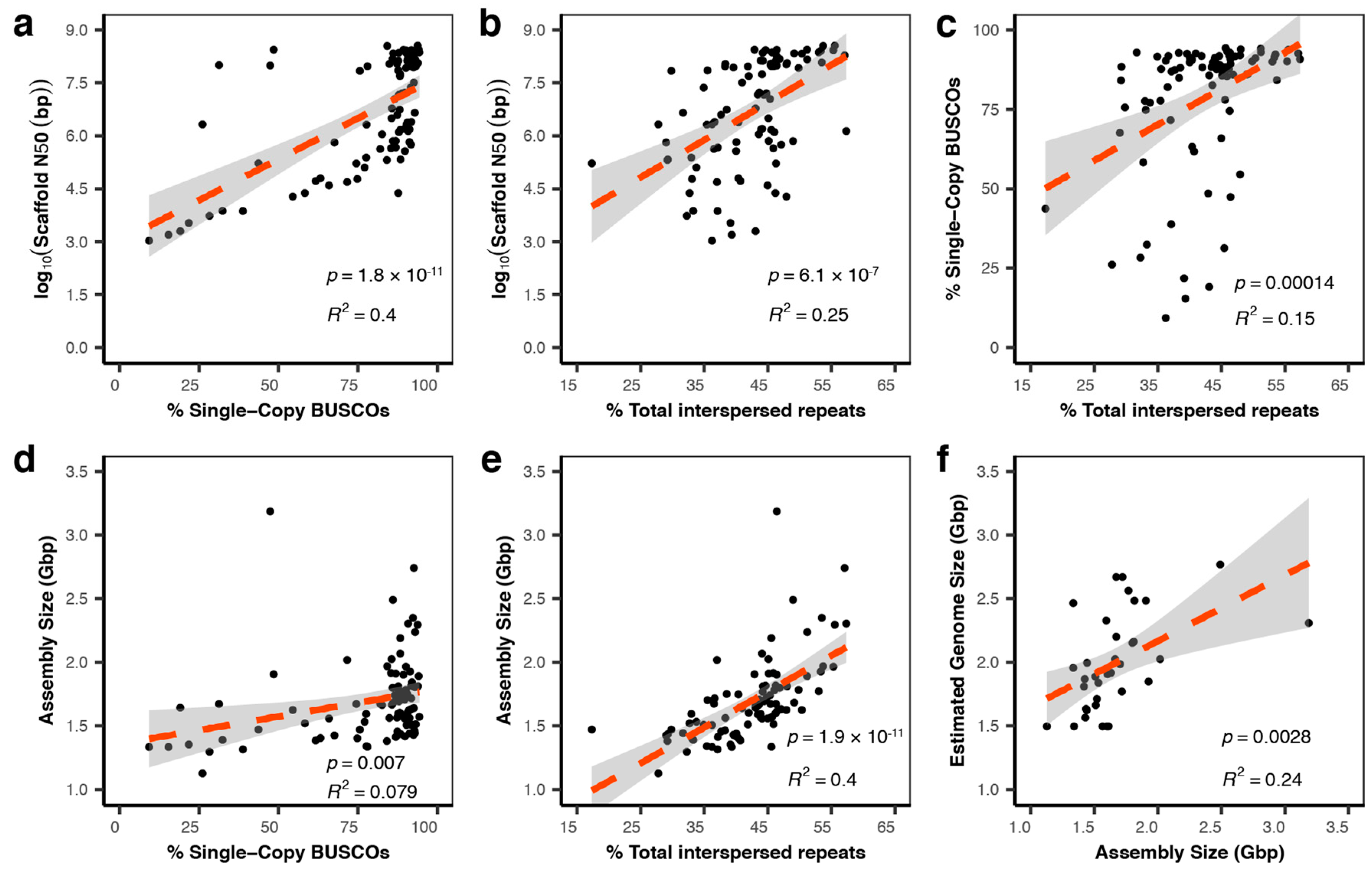
3.1. Sequencing and Assembly Quality Impact the Utility of Genomes
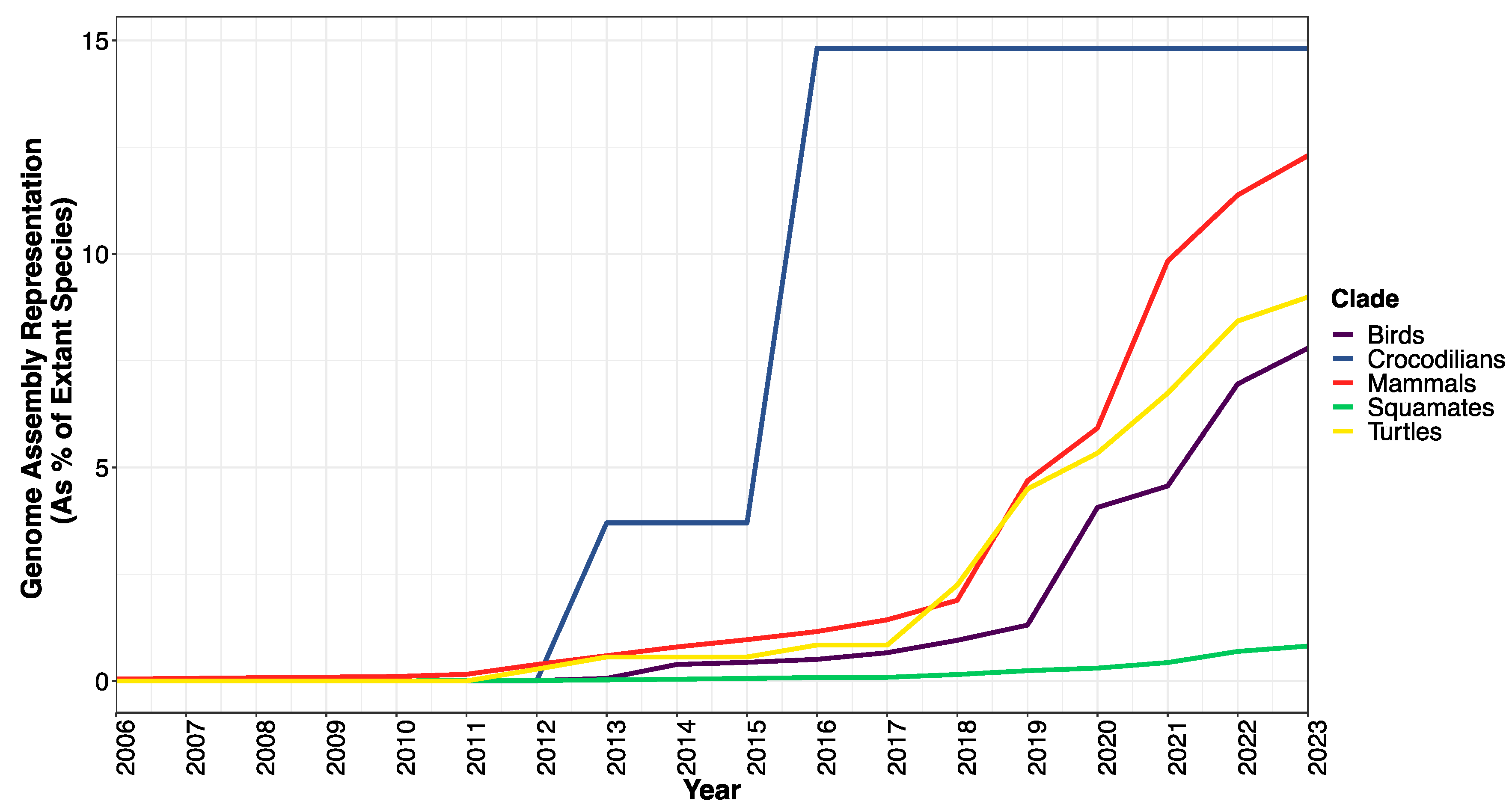
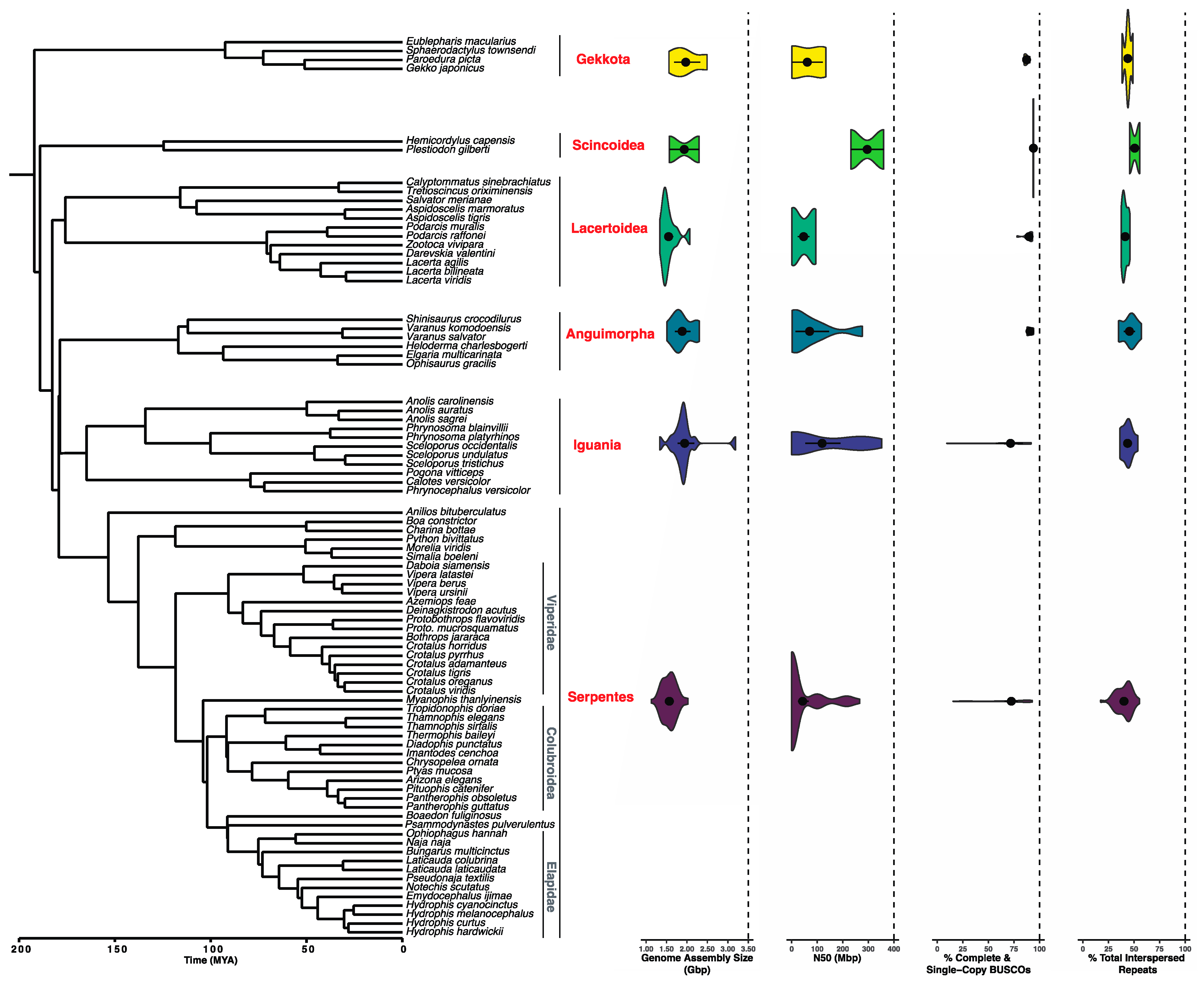
3.2. Where Does the Availability and Quality of Squamate Genomes Currently Stand?
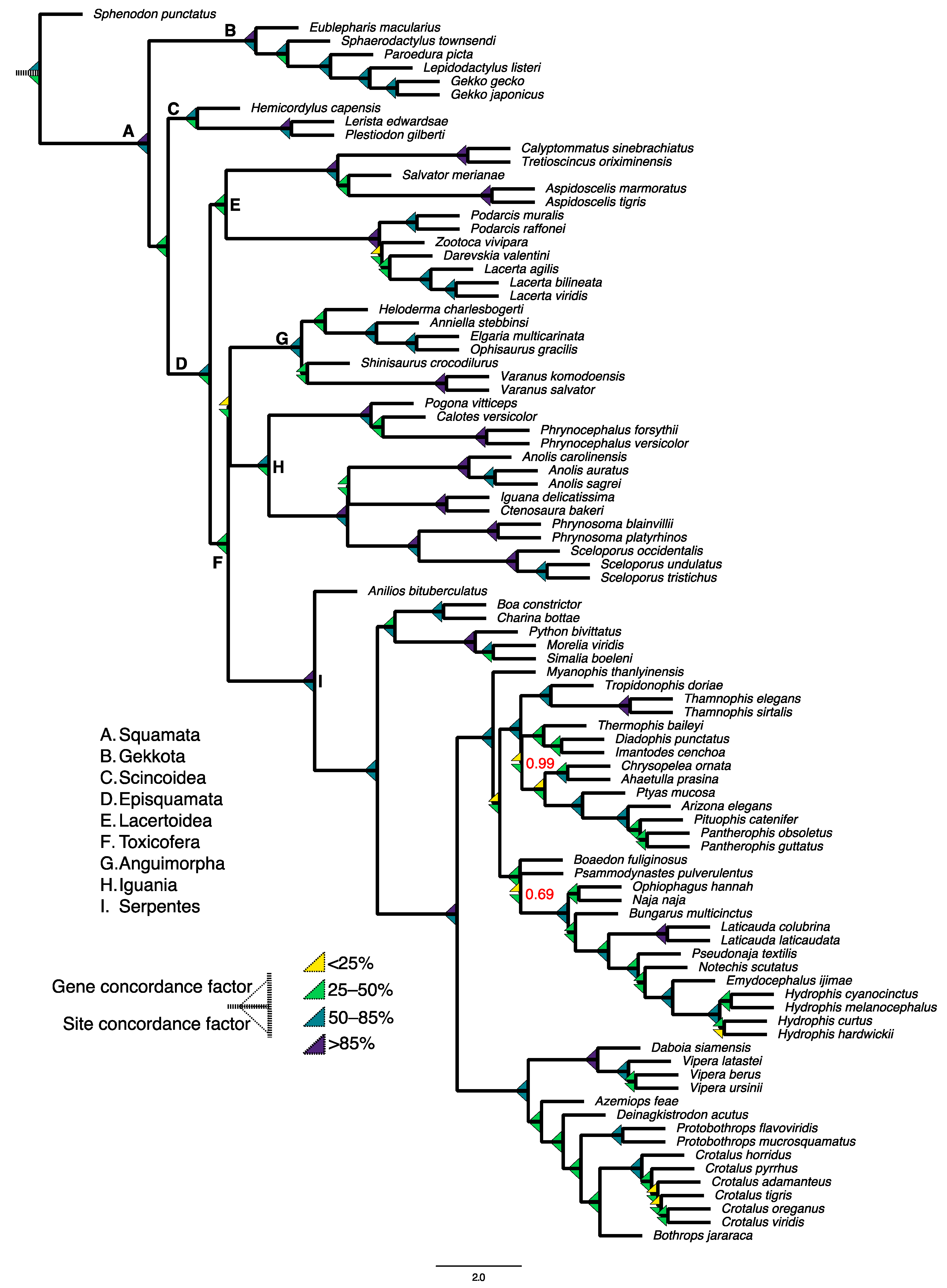
4. Putting the “Genomics” in Squamate Phylogenomics
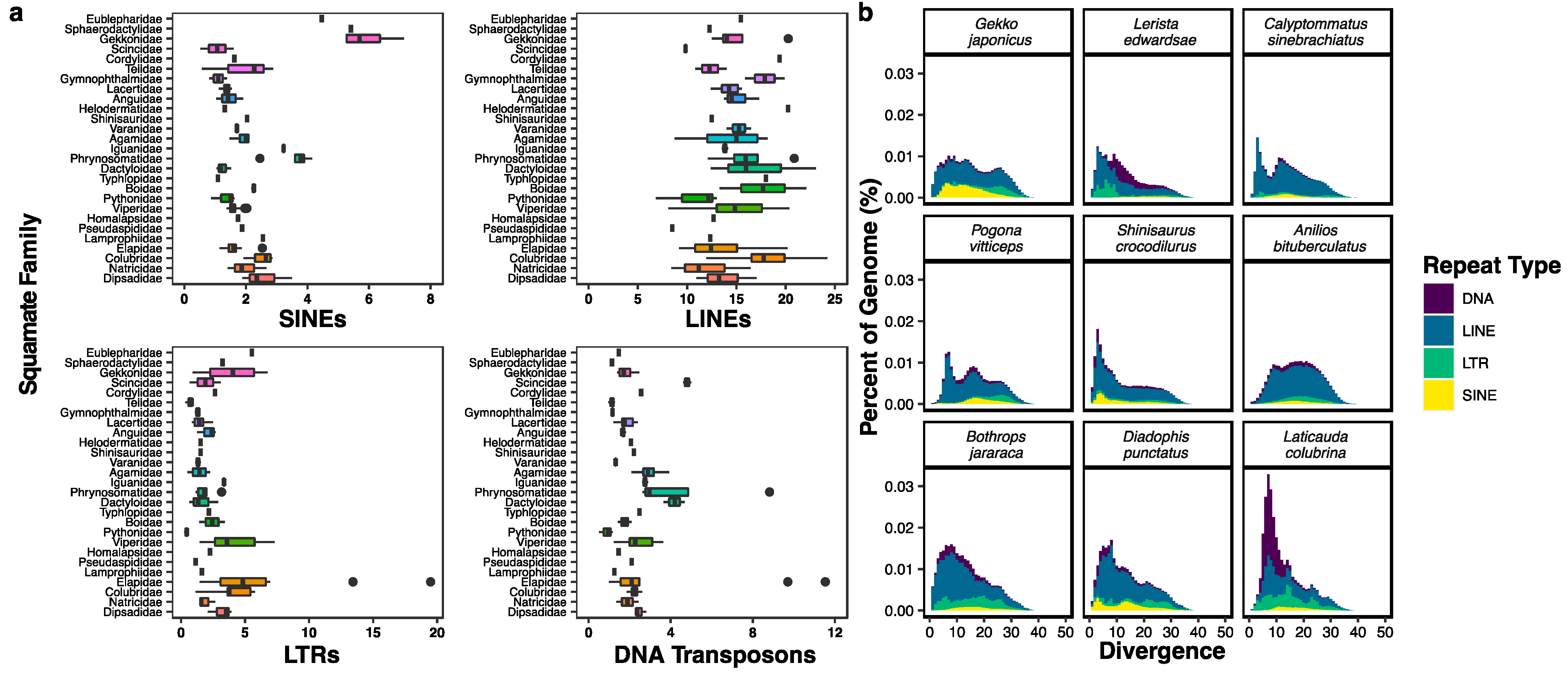
5. Transposable Elements in Squamates and Other Amniotes
Methods for Analysis of Transposable Elements
6. Genomic and Phenotypic Evolution in Squamates
7. The Contribution of Squamate Genomics to Other Diverse Fields of Research
7.1. Venomics
7.2. Sex Determination
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uetz, P.; Aguilar, R.; Brings, E.; Catenazzi, A. A Quarter Century of Reptile and Amphibian Databases. Herpetol. Rev. 2021, 52, 246–255. [Google Scholar]
- Feng, S.; Stiller, J.; Deng, Y.; Armstrong, J.; Fang, Q.; Reeve, A.H.; Xie, D.; Chen, G.; Guo, C.; Faircloth, B.C.; et al. Dense Sampling of Bird Diversity Increases Power of Comparative Genomics. Nature 2020, 587, 252–257. [Google Scholar] [CrossRef]
- Genereux, D.P.; Serres, A.; Armstrong, J.; Johnson, J.; Marinescu, V.D.; Murén, E.; Juan, D.; Bejerano, G.; Casewell, N.R.; Chemnick, L.G.; et al. A Comparative Genomics Multitool for Scientific Discovery and Conservation. Nature 2020, 587, 240–245. [Google Scholar] [CrossRef]
- Benton, M.J. Vertebrate Palaeontology, 3rd ed.; Blackwell Science: Malden, MA, USA, 2005. [Google Scholar]
- Burgin, C.J.; Colella, J.P.; Kahn, P.L.; Upham, N.S. How Many Species of Mammals Are There? J. Mammal. 2018, 99, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Modesto, S.P.; Anderson, J.S. The Phylogenetic Definition of Reptilia. Syst. Biol. 2004, 53, 815–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, N.G.; Parham, J.F.; Sellas, A.B.; Faircloth, B.C.; Glenn, T.C.; Papenfuss, T.J.; Henderson, J.B.; Hansen, M.H.; Simison, W.B. A Phylogenomic Analysis of Turtles. Mol. Phylogenet. Evol. 2015, 83, 250–257. [Google Scholar] [CrossRef]
- Gable, S.M.; Byars, M.I.; Literman, R.; Tollis, M. A Genomic Perspective on the Evolutionary Diversification of Turtles. Syst. Biol. 2022, 71, 1331–1347. [Google Scholar] [CrossRef]
- Shaffer, H.B.; Minx, P.; Warren, D.E.; Shedlock, A.M.; Thomson, R.C.; Valenzuela, N.; Abramyan, J.; Amemiya, C.T.; Badenhorst, D.; Biggar, K.K.; et al. The Western Painted Turtle Genome, a Model for the Evolution of Extreme Physiological Adaptations in a Slowly Evolving Lineage. Genome Biol. 2013, 14, R28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Pascual-Anaya, J.; Zadissa, A.; Li, W.; Niimura, Y.; Wang, J.; Huang, Z.; Li, C.; White, S.; Xiong, Z.; et al. The Draft Genomes of Soft-Shell Turtle and Green Sea Turtle Yield Insights into the Development and Evolution of the Turtle-Specific Body Plan. Nat. Genet. 2013, 45, 701–706. [Google Scholar] [CrossRef] [Green Version]
- Simões, T.R.; Caldwell, M.W.; Tałanda, M.; Bernardi, M.; Palci, A.; Vernygora, O.; Bernardini, F.; Mancini, L.; Nydam, R.L. The Origin of Squamates Revealed by a Middle Triassic Lizard from the Italian Alps. Nature 2018, 557, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.H.; Shipley, G.P. Cultural Taboos as a Factor in the Participation Rate of Native Americans in STEM. Int. J. STEM Educ. 2018, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onyishi, I.E.; Nwonyi, S.K.; Pazda, A.; Prokop, P. Attitudes and Behaviour toward Snakes on the Part of Igbo People in Southeastern Nigeria. Sci. Total Environ. 2021, 763, 143045. [Google Scholar] [CrossRef] [PubMed]
- Moazami, M. Evil Animals in the Zoroastrian Religion. Hist. Relig. 2005, 44, 300–317. [Google Scholar] [CrossRef]
- Lange, G. Cobra Deities and Divine Cobras: The Ambiguous Animality of Nāgas. Religions 2019, 10, 454. [Google Scholar] [CrossRef] [Green Version]
- Landry Yuan, F.; Ballullaya, U.P.; Roshnath, R.; Bonebrake, T.C.; Sinu, P.A. Sacred Groves and Serpent-Gods Moderate Human–Snake Relations. People Nat. 2020, 2, 111–122. [Google Scholar] [CrossRef]
- Ollonen, J.; Da Silva, F.O.; Mahlow, K.; Di-Poï, N. Skull Development, Ossification Pattern, and Adult Shape in the Emerging Lizard Model Organism Pogona Vitticeps: A Comparative Analysis with Other Squamates. Front. Physiol. 2018, 9, 278. [Google Scholar] [CrossRef] [Green Version]
- Waheed, H.; Moin, S.F.; Choudhary, M.I. Snake Venom: From Deadly Toxins to Life-Saving Therapeutics. Curr. Med. Chem. 2017, 24, 1874–1891. [Google Scholar] [CrossRef]
- Vitt, L.J.; Caldwell, J.P. Herpetology: An Introductory Biology of Amphibians and Reptiles, 4th ed.; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Mewes, H.W.; Albermann, K.; Bähr, M.; Frishman, D.; Gleissner, A.; Hani, J.; Heumann, K.; Kleine, K.; Maierl, A.; Oliver, S.G.; et al. Overview of the Yeast Genome. Nature 1997, 387 (Suppl. S6632), 7–65. [Google Scholar] [CrossRef]
- The Caenorhabditis elegans Sequencing Consortium. Genome Sequence of the Nematode C. Elegans: A Platform for Investigating Biology. Science 1998, 282, 2012–2018. [Google Scholar] [CrossRef]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The Genome Sequence of Drosophila Melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Arabidopsis Genome Initiative. Analysis of the Genome Sequence of the Flowering Plant Arabidopsis Thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The Sequence of the Human Genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily Conserved Elements in Vertebrate, Insect, Worm, and Yeast Genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, R.A.; Weinstock, G.M.; Metzker, M.L.; Muzny, D.M.; Sodergren, E.J.; Scherer, S.; Scott, G.; Steffen, D.; Worley, K.C.; Burch, P.E.; et al. Genome Sequence of the Brown Norway Rat Yields Insights into Mammalian Evolution. Nature 2004, 428, 493–521. [Google Scholar] [CrossRef] [Green Version]
- Mouse Genome Sequencing Consortium; Birney, E.; Rogers, J.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; Antonarakis, S.E.; Attwood, J.; Baertsch, R.; et al. Initial Sequencing and Comparative Analysis of the Mouse Genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, R.A.; Rogers, J.; Katze, M.G.; Bumgarner, R.; Weinstock, G.M.; Mardis, E.R.; Remington, K.A.; Strausberg, R.L.; Venter, J.C.; Wilson, R.K.; et al. Evolutionary and Biomedical Insights from the Rhesus Macaque Genome. Science 2007, 316, 222–234. [Google Scholar] [CrossRef] [Green Version]
- Waterson, R.H.; Lander, E.S.; Wilson, R.K.; The Chimpanzee Sequencing and Analysis Consortium. Initial Sequence of the Chimpanzee Genome and Comparison with the Human Genome. Nature 2005, 437, 69–87. [Google Scholar] [CrossRef] [Green Version]
- Lindblad-Toh, K.; Garber, M.; Zuk, O.; Lin, M.F.; Parker, B.J.; Washietl, S.; Kheradpour, P.; Ernst, J.; Jordan, G.; Mauceli, E.; et al. A High-Resolution Map of Human Evolutionary Constraint Using 29 Mammals. Nature 2011, 478, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Koepfli, K.-P.; Paten, B.; O’Brien, S.J. The Genome 10K Project: A Way Forward. Annu. Rev. Anim. Biosci. 2015, 3, 57–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, H.A.; Richards, S.; Lieberman Aiden, E.; Allende, M.L.; Archibald, J.M.; Bálint, M.; Barker, K.B.; Baumgartner, B.; Belov, K.; Bertorelle, G.; et al. The Earth BioGenome Project 2020: Starting the Clock. Proc. Natl. Acad. Sci. USA 2022, 119, e2115635118. [Google Scholar] [CrossRef] [PubMed]
- Christmas, M.J.; Kaplow, I.M.; Genereux, D.P.; Dong, M.X.; Hughes, G.M.; Li, X.; Sullivan, P.F.; Hindle, A.G.; Andrews, G.; Armstrong, J.C.; et al. Evolutionary Constraint and Innovation across Hundreds of Placental Mammals. Science 2023, 380, eabn3943. [Google Scholar] [CrossRef] [PubMed]
- Hillier, L.W.; Miller, W.; Birney, E.; Warren, W.; Hardison, R.C.; Ponting, C.P.; Bork, P.; Burt, D.W.; Groenen, M.A.M.; Delany, M.E.; et al. Sequence and Comparative Analysis of the Chicken Genome Provide Unique Perspectives on Vertebrate Evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef] [Green Version]
- Warren, W.C.; Clayton, D.F.; Ellegren, H.; Arnold, A.P.; Hillier, L.W.; Künstner, A.; Searle, S.; White, S.; Vilella, A.J.; Fairley, S.; et al. The Genome of a Songbird. Nature 2010, 464, 757–762. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative Genomics Reveals Insights into Avian Genome Evolution and Adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Alföldi, J.; Di Palma, F.; Grabherr, M.; Williams, C.; Kong, L.; Mauceli, E.; Russell, P.; Lowe, C.B.; Glor, R.E.; Jaffe, J.D.; et al. The Genome of the Green Anole Lizard and a Comparative Analysis with Birds and Mammals. Nature 2011, 477, 587–591. [Google Scholar] [CrossRef] [Green Version]
- Castoe, T.A.; de Koning, A.P.J.; Hall, K.T.; Card, D.C.; Schield, D.R.; Fujita, M.K.; Ruggiero, R.P.; Degner, J.F.; Daza, J.M.; Gu, W.; et al. The Burmese Python Genome Reveals the Molecular Basis for Extreme Adaptation in Snakes. Proc. Natl. Acad. Sci. USA 2013, 110, 20645–20650. [Google Scholar] [CrossRef]
- Vonk, F.J.; Casewell, N.R.; Henkel, C.V.; Heimberg, A.M.; Jansen, H.J.; McCleary, R.J.R.; Kerkkamp, H.M.E.; Vos, R.A.; Guerreiro, I.; Calvete, J.J.; et al. The King Cobra Genome Reveals Dynamic Gene Evolution and Adaptation in the Snake Venom System. Proc. Natl. Acad. Sci. USA 2013, 110, 20651–20656. [Google Scholar] [CrossRef]
- Lind, A.L.; Lai, Y.Y.Y.; Mostovoy, Y.; Holloway, A.K.; Iannucci, A.; Mak, A.C.Y.; Fondi, M.; Orlandini, V.; Eckalbar, W.L.; Milan, M.; et al. Genome of the Komodo Dragon Reveals Adaptations in the Cardiovascular and Chemosensory Systems of Monitor Lizards. Nat. Ecol. Evol. 2019, 3, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhou, Q.; Wang, Y.; Luo, L.; Yang, J.; Yang, L.; Liu, M.; Li, Y.; Qian, T.; Zheng, Y.; et al. Gekko Japonicus Genome Reveals Evolution of Adhesive Toe Pads and Tail Regeneration. Nat. Commun. 2015, 6, 10033. [Google Scholar] [CrossRef] [Green Version]
- Pinto, B.J.; Keating, S.E.; Nielsen, S.V.; Scantlebury, D.P.; Daza, J.D.; Gamble, T. Chromosome-Level Genome Assembly Reveals Dynamic Sex Chromosomes in Neotropical Leaf-Litter Geckos (Sphaerodactylidae: Sphaerodactylus). J. Hered. 2022, 113, 272–287. [Google Scholar] [CrossRef]
- Xiong, Z.; Li, F.; Li, Q.; Zhou, L.; Gamble, T.; Zheng, J.; Kui, L.; Li, C.; Li, S.; Yang, H.; et al. Draft Genome of the Leopard Gecko, Eublepharis Macularius. GigaScience 2016, 5, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roscito, J.G.; Sameith, K.; Pippel, M.; Francoijs, K.-J.; Winkler, S.; Dahl, A.; Papoutsoglou, G.; Myers, G.; Hiller, M. The Genome of the Tegu Lizard Salvator Merianae: Combining Illumina, PacBio, and Optical Mapping Data to Generate a Highly Contiguous Assembly. GigaScience 2018, 7, giy141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Feiner, N.; Pinho, C.; While, G.M.; Kaliontzopoulou, A.; Harris, D.J.; Salvi, D.; Uller, T. Extensive Introgression and Mosaic Genomes of Mediterranean Endemic Lizards. Nat. Commun. 2021, 12, 2762. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, A.A.; Recknagel, H.; Elmer, K.R. Chromosome-Level Assembly of the Common Lizard (Zootoca Vivipara) Genome. Genome Biol. Evol. 2020, 12, 1953–1960. [Google Scholar] [CrossRef]
- Fry, B.G.; Vidal, N.; Norman, J.A.; Vonk, F.J.; Scheib, H.; Ramjan, S.F.R.; Kuruppu, S.; Fung, K.; Blair Hedges, S.; Richardson, M.K.; et al. Early Evolution of the Venom System in Lizards and Snakes. Nature 2006, 439, 584–588. [Google Scholar] [CrossRef]
- Pyron, R.A.; Burbrink, F.T.; Wiens, J.J. A Phylogeny and Revised Classification of Squamata, Including 4161 Species of Lizards and Snakes. BMC Evol. Biol. 2013, 13, 93. [Google Scholar] [CrossRef] [Green Version]
- Tollis, M.; Boissinot, S. The Transposable Element Profile of the Anolis Genome. Mob. Genet. Elem. 2011, 1, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Pasquesi, G.I.M.; Adams, R.H.; Card, D.C.; Schield, D.R.; Corbin, A.B.; Perry, B.W.; Reyes-Velasco, J.; Ruggiero, R.P.; Vandewege, M.W.; Shortt, J.A.; et al. Squamate Reptiles Challenge Paradigms of Genomic Repeat Element Evolution Set by Birds and Mammals. Nat. Commun. 2018, 9, 2774. [Google Scholar] [CrossRef] [Green Version]
- Giani, A.M.; Gallo, G.R.; Gianfranceschi, L.; Formenti, G. Long Walk to Genomics: History and Current Approaches to Genome Sequencing and Assembly. Comput. Struct. Biotechnol. J. 2020, 18, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Heckenhauer, J.; Frandsen, P.B.; Sproul, J.S.; Li, Z.; Paule, J.; Larracuente, A.M.; Maughan, P.J.; Barker, M.S.; Schneider, J.V.; Stewart, R.J.; et al. Genome Size Evolution in the Diverse Insect Order Trichoptera. GigaScience 2022, 11, giac011. [Google Scholar] [CrossRef] [PubMed]
- Hotaling, S.; Sproul, J.S.; Heckenhauer, J.; Powell, A.; Larracuente, A.M.; Pauls, S.U.; Kelley, J.L.; Frandsen, P.B. Long Reads Are Revolutionizing 20 Years of Insect Genome Sequencing. Genome Biol. Evol. 2021, 13, evab138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jain, C.; Aluru, S. A Comprehensive Evaluation of Long Read Error Correction Methods. BMC Genomics 2020, 21, 889. [Google Scholar] [CrossRef] [PubMed]
- Myers, E.A.; Strickland, J.L.; Rautsaw, R.M.; Mason, A.J.; Schramer, T.D.; Nystrom, G.S.; Hogan, M.P.; Yooseph, S.; Rokyta, D.R.; Parkinson, C.L. De Novo Genome Assembly Highlights the Role of Lineage-Specific Gene Duplications in the Evolution of Venom in Fea’s Viper (Azemiops feae). Genome Biol. Evol. 2022, 14, evac082. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.-X.; Liang, X.-X.; Chen, Z.-Q.; Li, W.-M.; Mi, C.-R.; Li, M.; Wu, Z.-J.; Zhou, X.-M.; Du, W.-G. Ancient Demographics Determine the Effectiveness of Genetic Purging in Endangered Lizards. Mol. Biol. Evol. 2021, 39, msab359. [Google Scholar] [CrossRef]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W.; et al. Chromosome-Scale Shotgun Assembly Using an in Vitro Method for Long-Range Linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P.; et al. De Novo Assembly of the Aedes aegypti Genome Using Hi-C Yields Chromosome-Length Scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef] [Green Version]
- Sedlazeck, F.J.; Lee, H.; Darby, C.A.; Schatz, M.C. Piercing the Dark Matter: Bioinformatics of Long-Range Sequencing and Mapping. Nat. Rev. Genet. 2018, 19, 329–346. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Thrash, A.; Hoffmann, F.; Perkins, A. Toward a More Holistic Method of Genome Assembly Assessment. BMC Bioinform. 2020, 21, 249. [Google Scholar] [CrossRef]
- Schield, D.R.; Card, D.C.; Hales, N.R.; Perry, B.W.; Pasquesi, G.M.; Blackmon, H.; Adams, R.H.; Corbin, A.B.; Smith, C.F.; Ramesh, B.; et al. The Origins and Evolution of Chromosomes, Dosage Compensation, and Mechanisms Underlying Venom Regulation in Snakes. Genome Res. 2019, 29, 590–610. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Wang, Z.-J.; Li, Q.-Y.; Lian, J.-M.; Zhou, Y.; Lu, B.-Z.; Jin, L.-J.; Qiu, P.-X.; Zhang, P.; Zhu, W.-B.; et al. Evolutionary Trajectories of Snake Genes and Genomes Revealed by Comparative Analyses of Five-Pacer Viper. Nat. Commun. 2016, 7, 13107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blommaert, J. Genome Size Evolution: Towards New Model Systems for Old Questions. Proc. R. Soc. B Biol. Sci. 2020, 287, 20201441. [Google Scholar] [CrossRef]
- Gregory, T.R. Animal Genome Size Database. Available online: http://genomesize.com (accessed on 1 June 2023).
- Geneva, A.J.; Park, S.; Bock, D.G.; de Mello, P.L.H.; Sarigol, F.; Tollis, M.; Donihue, C.M.; Reynolds, R.G.; Feiner, N.; Rasys, A.M.; et al. Chromosome-Scale Genome Assembly of the Brown Anole (Anolis sagrei), an Emerging Model Species. Commun. Biol. 2022, 5, 1126. [Google Scholar] [CrossRef] [PubMed]
- Rasys, A.M.; Park, S.; Ball, R.E.; Alcala, A.J.; Lauderdale, J.D.; Menke, D.B. CRISPR-Cas9 Gene Editing in Lizards through Microinjection of Unfertilized Oocytes. Cell Rep. 2019, 28, 2288–2292.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grenfell, B.T.; Pybus, O.G.; Gog, J.R.; Wood, J.L.N.; Daly, J.M.; Mumford, J.A.; Holmes, E.C. Unifying the Epidemiological and Evolutionary Dynamics of Pathogens. Science 2004, 303, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Liberles, D.A.; Thoren, A.; Heijne, G.; Elofsson, A. The Use of Phylogenetic Profiles for Gene Predictions. Curr. Genom. 2002, 3, 131–137. [Google Scholar] [CrossRef]
- Freeman, S.; Herron, J.C. Evolutionary Analysis, 4th ed.; Pearson Prentice Hall: Upper Saddle River, NJ, USA, 2007. [Google Scholar]
- Tollis, M.; DeNardo, D.F.; Cornelius, J.A.; Dolby, G.A.; Edwards, T.; Henen, B.T.; Karl, A.E.; Murphy, R.W.; Kusumi, K. The Agassiz’s Desert Tortoise Genome Provides a Resource for the Conservation of a Threatened Species. PLoS ONE 2017, 12, e0177708. [Google Scholar] [CrossRef] [Green Version]
- Gemmell, N.J.; Rutherford, K.; Prost, S.; Tollis, M.; Winter, D.; Macey, J.R.; Adelson, D.L.; Suh, A.; Bertozzi, T.; Grau, J.H.; et al. The Tuatara Genome Reveals Ancient Features of Amniote Evolution. Nature 2020, 584, 403–409. [Google Scholar] [CrossRef]
- Tollis, M.; Hutchins, E.D.; Stapley, J.; Rupp, S.M.; Eckalbar, W.L.; Maayan, I.; Lasku, E.; Infante, C.R.; Dennis, S.R.; Robertson, J.A.; et al. Comparative Genomics Reveals Accelerated Evolution in Conserved Pathways during the Diversification of Anole Lizards. Genome Biol. Evol. 2018, 10, 489–506. [Google Scholar] [CrossRef] [Green Version]
- Simões, T.R.; Vernygora, O.; Caldwell, M.W.; Pierce, S.E. Megaevolutionary Dynamics and the Timing of Evolutionary Innovation in Reptiles. Nat. Commun. 2020, 11, 3322. [Google Scholar] [CrossRef]
- Gauthier, J.A.; Kearney, M.; Maisano, J.A.; Rieppel, O.; Behlke, A.D.B. Assembling the Squamate Tree of Life: Perspectives from the Phenotype and the Fossil Record. Bull. Peabody Mus. Nat. Hist. 2012, 53, 3–308. [Google Scholar] [CrossRef]
- Kumaza, Y.; Nishida, M. Variations in Mitochondrial TRNA Gene Organization of Reptiles as Phylogenetic Markers. Mol. Biol. Evol. 1995, 12, 759–772. [Google Scholar] [CrossRef] [Green Version]
- Townsend, T.M.; Larson, A.; Louis, E.; Macey, J.R. Molecular Phylogenetics of Squamata: The Position of Snakes, Amphisbaenians, and Dibamids, and the Root of the Squamate Tree. Syst. Biol. 2004, 53, 735–757. [Google Scholar] [CrossRef]
- Vidal, N.; Hedges, S.B. The Phylogeny of Squamate Reptiles (Lizards, Snakes, and Amphisbaenians) Inferred from Nine Nuclear Protein-Coding Genes. Comptes Rendus Biol. 2005, 328, 1000–1008. [Google Scholar] [CrossRef]
- Losos, J.B.; Hillis, D.M.; Greene, H.W. Who Speaks with a Forked Tongue? Science 2012, 338, 1428–1429. [Google Scholar] [CrossRef] [PubMed]
- Burbrink, F.T.; Grazziotin, F.G.; Pyron, R.A.; Cundall, D.; Donnellan, S.; Irish, F.; Keogh, J.S.; Kraus, F.; Murphy, R.W.; Noonan, B.; et al. Interrogating Genomic-Scale Data for Squamata (Lizards, Snakes, and Amphisbaenians) Shows No Support for Key Traditional Morphological Relationships. Syst. Biol. 2020, 69, 502–520. [Google Scholar] [CrossRef]
- Singhal, S.; Colston, T.J.; Grundler, M.R.; Smith, S.A.; Costa, G.C.; Colli, G.R.; Moritz, C.; Pyron, R.A.; Rabosky, D.L. Congruence and Conflict in the Higher-Level Phylogenetics of Squamate Reptiles: An Expanded Phylogenomic Perspective. Syst. Biol. 2021, 70, 542–557. [Google Scholar] [CrossRef]
- Wiens, J.J.; Hutter, C.R.; Mulcahy, D.G. Resolving the Phylogeny of Lizards and Snakes (Squamata) with Extensive Sampling of Genes and Species. Biol. Lett. 2012, 8, 1043–1046. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.V. Is a New and General Theory of Molecular Systematics Emerging? Evolution 2009, 63, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Hahn, M.W.; Lanfear, R. New Methods to Calculate Concordance Factors for Phylogenomic Datasets. Mol. Biol. Evol. 2020, 37, 2727–2733. [Google Scholar] [CrossRef]
- Kubatko, L.S.; Degnan, J.H. Inconsistency of Phylogenetic Estimates from Concatenated Data under Coalescence. Syst. Biol. 2007, 56, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Jeffroy, O.; Brinkmann, H.; Delsuc, F.; Philippe, H. Phylogenomics: The Beginning of Incongruence? Trends Genet. TIG 2006, 22, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippe, H.; Brinkmann, H.; Lavrov, D.V.; Littlewood, D.T.J.; Manuel, M.; Wörheide, G.; Baurain, D. Resolving Difficult Phylogenetic Questions: Why More Sequences Are Not Enough. PLoS Biol. 2011, 9, e1000602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. Inferring Phylogenies; Sinauer Associates: Sunderland, MA, USA, 2004. [Google Scholar]
- Linkem, C.W.; Minin, V.N.; Leaché, A.D. Detecting the Anomaly Zone in Species Trees and Evidence for a Misleading Signal in Higher-Level Skink Phylogeny (Squamata: Scincidae). Syst. Biol. 2016, 65, 465–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Edwards, S.V. Phylogenetic Analysis in the Anomaly Zone. Syst. Biol. 2009, 58, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Literman, R.; Schwartz, R. Genome-Scale Profiling Reveals Noncoding Loci Carry Higher Proportions of Concordant Data. Mol. Biol. Evol. 2021, 38, 2306–2318. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Borowiec, M.L. AMAS: A Fast Tool for Alignment Manipulation and Computing of Summary Statistics. PeerJ 2016, 4, e1660. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial Time Species Tree Reconstruction from Partially Resolved Gene Trees. BMC Bioinformatics 2018, 19 (Suppl. S6), 153. [Google Scholar] [CrossRef] [Green Version]
- Sayyari, E.; Mirarab, S. Fast Coalescent-Based Computation of Local Branch Support from Quartet Frequencies. Mol. Biol. Evol. 2016, 33, 1654–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenzweig, B.; Kern, A.; Hahn, M. Accurate Detection of Incomplete Lineage Sorting via Supervised Machine Learning. bioRxiv 2022. [Google Scholar] [CrossRef]
- Simões, T.R.; Pierce, S.E. Sustained High Rates of Morphological Evolution during the Rise of Tetrapods. Nat. Ecol. Evol. 2021, 5, 1403–1414. [Google Scholar] [CrossRef]
- Boissinot, S.; Bourgeois, Y.; Manthey, J.D.; Ruggiero, R.P. The Mobilome of Reptiles: Evolution, Structure, and Function. Cytogenet. Genome Res. 2019, 157, 21–33. [Google Scholar] [CrossRef]
- Tollis, M.; Boissinot, S. The Evolutionary Dynamics of Transposable Elements in Eukaryote Genomes. Genome Dyn. 2012, 7, 68–91. [Google Scholar] [CrossRef]
- de Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive Elements May Comprise Over Two-Thirds of the Human Genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [Green Version]
- Novick, P.A.; Basta, H.; Floumanhaft, M.; McClure, M.A.; Boissinot, S. The Evolutionary Dynamics of Autonomous Non-LTR Retrotransposons in the Lizard Anolis Carolinensis Shows More Similarity to Fish than Mammals. Mol. Biol. Evol. 2009, 26, 1811–1822. [Google Scholar] [CrossRef] [Green Version]
- Novick, P.A.; Smith, J.D.; Floumanhaft, M.; Ray, D.A.; Boissinot, S. The Evolution and Diversity of DNA Transposons in the Genome of the Lizard Anolis Carolinensis. Genome Biol. Evol. 2011, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Feschotte, C.; Pritham, E.J. DNA Transposons and the Evolution of Eukaryotic Genomes. Annu. Rev. Genet. 2007, 41, 331–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, D.D.; Korman, M.H.; Jakubczak, J.L.; Eickbush, T.H. Reverse Transcription of R2Bm RNA Is Primed by a Nick at the Chromosomal Target Site: A Mechanism for Non-LTR Retrotransposition. Cell 1993, 72, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a Database of Repetitive Elements in Eukaryotic Genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapitonov, V.V.; Jurka, J. Rolling-Circle Transposons in Eukaryotes. Proc. Natl. Acad. Sci. USA 2001, 98, 8714–8719. [Google Scholar] [CrossRef]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013. Available online: http://www.repeatmasker.org (accessed on 1 June 2023).
- Storer, J.; Hubley, R.; Rosen, J.; Wheeler, T.J.; Smit, A.F. The Dfam Community Resource of Transposable Element Families, Sequence Models, and Genome Annotations. Mob. DNA 2021, 12, 2. [Google Scholar] [CrossRef]
- Zhang, H.-H.; Peccoud, J.; Xu, M.-R.-X.; Zhang, X.-G.; Gilbert, C. Horizontal Transfer and Evolution of Transposable Elements in Vertebrates. Nat. Commun. 2020, 11, 1362. [Google Scholar] [CrossRef] [Green Version]
- Smit, A.; Hubley, R. RepeatModeler Open-1.0. 2008. Available online: http://www.repeatmasker.org (accessed on 1 June 2023).
- Bourgeois, Y.; Boissinot, S. On the Population Dynamics of Junk: A Review on the Population Genomics of Transposable Elements. Genes 2019, 10, E419. [Google Scholar] [CrossRef] [Green Version]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of Genome Size Evolution in Birds and Mammals. Proc. Natl. Acad. Sci. USA 2017, 114, E1460–E1469. [Google Scholar] [CrossRef]
- Suh, A.; Churakov, G.; Ramakodi, M.P.; Platt, R.N.; Jurka, J.; Kojima, K.K.; Caballero, J.; Smit, A.F.; Vliet, K.A.; Hoffmann, F.G.; et al. Multiple Lineages of Ancient CR1 Retroposons Shaped the Early Genome Evolution of Amniotes. Genome Biol. Evol. 2015, 7, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Manthey, J.D.; Moyle, R.G.; Boissinot, S. Multiple and Independent Phases of Transposable Element Amplification in the Genomes of Piciformes (Woodpeckers and Allies). Genome Biol. Evol. 2018, 10, 1445–1456. [Google Scholar] [CrossRef] [Green Version]
- Platt, R.N.; Vandewege, M.W.; Kern, C.; Schmidt, C.J.; Hoffmann, F.G.; Ray, D.A. Large Numbers of Novel MiRNAs Originate from DNA Transposons and Are Coincident with a Large Species Radiation in Bats. Mol. Biol. Evol. 2014, 31, 1536–1545. [Google Scholar] [CrossRef]
- Platt, R.N.; Mangum, S.F.; Ray, D.A. Pinpointing the Vesper Bat Transposon Revolution Using the Miniopterus Natalensis Genome. Mob. DNA 2016, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Bourgeois, Y.; Ruggiero, R.P.; Hariyani, I.; Boissinot, S. Disentangling the Determinants of Transposable Elements Dynamics in Vertebrate Genomes Using Empirical Evidences and Simulations. PLOS Genet. 2020, 16, e1009082. [Google Scholar] [CrossRef]
- Tollis, M.; Boissinot, S. Lizards and LINEs: Selection and Demography Affect the Fate of L1 Retrotransposons in the Genome of the Green Anole (Anolis carolinensis). Genome Biol. Evol. 2013, 5, 1754–1768. [Google Scholar] [CrossRef] [Green Version]
- Jurka, J.; Bao, W.; Kojima, K.K. Families of Transposable Elements, Population Structure and the Origin of Species. Biol. Direct 2011, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Chalopin, D.; Naville, M.; Plard, F.; Galiana, D.; Volff, J.-N. Comparative Analysis of Transposable Elements Highlights Mobilome Diversity and Evolution in Vertebrates. Genome Biol. Evol. 2015, 7, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Sotero-Caio, C.G.; Platt, R.N.; Suh, A.; Ray, D.A. Evolution and Diversity of Transposable Elements in Vertebrate Genomes. Genome Biol. Evol. 2017, 9, 161–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galbraith, J.D.; Ludington, A.J.; Sanders, K.L.; Suh, A.; Adelson, D.L. Horizontal Transfer and Subsequent Explosive Expansion of a DNA Transposon in Sea Kraits (Laticauda). Biol. Lett. 2021, 17, 20210342. [Google Scholar] [CrossRef] [PubMed]
- Nellåker, C.; Keane, T.M.; Yalcin, B.; Wong, K.; Agam, A.; Belgard, T.G.; Flint, J.; Adams, D.J.; Frankel, W.N.; Ponting, C.P. The Genomic Landscape Shaped by Selection on Transposable Elements across 18 Mouse Strains. Genome Biol. 2012, 13, R45. [Google Scholar] [CrossRef] [Green Version]
- Jangam, D.; Feschotte, C.; Betrán, E. Transposable Element Domestication As an Adaptation to Evolutionary Conflicts. Trends Genet. 2017, 33, 817–831. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory Activities of Transposable Elements: From Conflicts to Benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef] [Green Version]
- Grabundzija, I.; Messing, S.A.; Thomas, J.; Cosby, R.L.; Bilic, I.; Miskey, C.; Gogol-Döring, A.; Kapitonov, V.; Diem, T.; Dalda, A.; et al. A Helitron Transposon Reconstructed from Bats Reveals a Novel Mechanism of Genome Shuffling in Eukaryotes. Nat. Commun. 2016, 7, 10716. [Google Scholar] [CrossRef] [Green Version]
- Di-Poï, N.; Montoya-Burgos, J.I.; Miller, H.; Pourquié, O.; Milinkovitch, M.C.; Duboule, D. Changes in Hox Genes’ Structure and Function during the Evolution of the Squamate Body Plan. Nature 2010, 464, 99–103. [Google Scholar] [CrossRef]
- Feiner, N. Accumulation of Transposable Elements in Hox Gene Clusters during Adaptive Radiation of Anolis Lizards. Proc. R. Soc. B Biol. Sci. 2016, 283, 20161555. [Google Scholar] [CrossRef] [PubMed]
- Konkel, M.K.; Batzer, M.A. A Mobile Threat to Genome Stability: The Impact of Non-LTR Retrotransposons upon the Human Genome. Semin. Cancer Biol. 2010, 20, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, T.A.; Gregory, T.R. What’s in a Genome? The C-Value Enigma and the Evolution of Eukaryotic Genome Content. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140331. [Google Scholar] [CrossRef] [PubMed]
- Rech, G.E.; Radío, S.; Guirao-Rico, S.; Aguilera, L.; Horvath, V.; Green, L.; Lindstadt, H.; Jamilloux, V.; Quesneville, H.; González, J. Population-Scale Long-Read Sequencing Uncovers Transposable Elements Associated with Gene Expression Variation and Adaptive Signatures in Drosophila. Nat. Commun. 2022, 13, 1948. [Google Scholar] [CrossRef]
- Smith, S.D.; Pennell, M.W.; Dunn, C.W.; Edwards, S.V. Phylogenetics Is the New Genetics (for Most of Biodiversity). Trends Ecol. Evol. 2020, 35, 415–425. [Google Scholar] [CrossRef]
- Pyron, R.A.; Burbrink, F.T. Early Origin of Viviparity and Multiple Reversions to Oviparity in Squamate Reptiles. Ecol. Lett. 2014, 17, 13–21. [Google Scholar] [CrossRef]
- Booth, W.; Smith, C.F.; Eskridge, P.H.; Hoss, S.K.; Mendelson, J.R.; Schuett, G.W. Facultative Parthenogenesis Discovered in Wild Vertebrates. Biol. Lett. 2012, 8, 983–985. [Google Scholar] [CrossRef] [Green Version]
- Booth, W.; Schuett, G.W. The Emerging Phylogenetic Pattern of Parthenogenesis in Snakes. Biol. J. Linn. Soc. 2016, 118, 172–186. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.V. Off like a Shot: Scaling of Ballistic Tongue Projection Reveals Extremely High Performance in Small Chameleons. Sci. Rep. 2016, 6, 18625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellenreuther, M.; Hansson, B. Detecting Polygenic Evolution: Problems, Pitfalls, and Promises. Trends Genet. 2016, 32, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Margres, M.J.; Wray, K.P.; Hassinger, A.T.B.; Ward, M.J.; McGivern, J.J.; Moriarty Lemmon, E.; Lemmon, A.R.; Rokyta, D.R. Quantity, Not Quality: Rapid Adaptation in a Polygenic Trait Proceeded Exclusively through Expression Differentiation. Mol. Biol. Evol. 2017, 34, 3099–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, M.R.; Ankeny, R.A.; Crowe, N.; Green, S.; Leonelli, S. How to Choose Your Research Organism. Stud. Hist. Philos. Sci. Part C Stud. Hist. Philos. Biol. Biomed. Sci. 2020, 80, 101227. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, D.R.; Ågren, J.A.; Carbone, L.; Elde, N.C.; Hoekstra, H.E.; Kapheim, K.M.; Keller, L.; Moreau, C.S.; Toth, A.L.; Yeaman, S.; et al. Coevolution of Genome Architecture and Social Behavior. Trends Ecol. Evol. 2019, 34, 844–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, M.P.; Whittington, A.C.; Broe, M.B.; Ward, M.J.; Gibbs, H.L.; Rokyta, D.R. The Chemosensory Repertoire of the Eastern Diamondback Rattlesnake (Crotalus adamanteus) Reveals Complementary Genetics of Olfactory and Vomeronasal-Type Receptors. J. Mol. Evol. 2021, 89, 313–328. [Google Scholar] [CrossRef]
- Wagner, G.P. Homology, Genes, and Evolutionary Innovation; Princeton University Press: Princeton, NJ, USA, 2018. [Google Scholar]
- Roscito, J.G.; Sameith, K.; Kirilenko, B.M.; Hecker, N.; Winkler, S.; Dahl, A.; Rodrigues, M.T.; Hiller, M. Convergent and Lineage-Specific Genomic Differences in Limb Regulatory Elements in Limbless Reptile Lineages. Cell Rep. 2022, 38, 110280. [Google Scholar] [CrossRef]
- Marcovitz, A.; Jia, R.; Bejerano, G. “Reverse Genomics” Predicts Function of Human Conserved Noncoding Elements. Mol. Biol. Evol. 2016, 33, 1358–1369. [Google Scholar] [CrossRef] [Green Version]
- Sackton, T.B.; Grayson, P.; Cloutier, A.; Hu, Z.; Liu, J.S.; Wheeler, N.E.; Gardner, P.P.; Clarke, J.A.; Baker, A.J.; Clamp, M.; et al. Convergent Regulatory Evolution and Loss of Flight in Paleognathous Birds. Science 2019, 364, 74–78. [Google Scholar] [CrossRef] [Green Version]
- Parvez, S.; Herdman, C.; Beerens, M.; Chakraborti, K.; Harmer, Z.P.; Yeh, J.-R.J.; MacRae, C.A.; Yost, H.J.; Peterson, R.T. MIC-Drop: A Platform for Large-Scale in Vivo CRISPR Screens. Science 2021, 373, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Kaneko, M.; Kiyonari, H. A Reverse Genetic Approach in Geckos with the CRISPR/Cas9 System by Oocyte Microinjection. Dev. Biol. 2023, 497, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Koludarov, I.; Jackson, T.N.; op den Brouw, B.; Dobson, J.; Dashevsky, D.; Arbuckle, K.; Clemente, C.J.; Stockdale, E.J.; Cochran, C.; Debono, J.; et al. Enter the Dragon: The Dynamic and Multifunctional Evolution of Anguimorpha Lizard Venoms. Toxins 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, E.J.C.; Tyrrell, K.L.; Citron, D.M.; Cox, C.R.; Recchio, I.M.; Okimoto, B.; Bryja, J.; Fry, B.G. Anaerobic and Aerobic Bacteriology of the Saliva and Gingiva from 16 Captive Komodo Dragons (Varanus komodoensis): New Implications for the “Bacteria as Venom” Model. J. Zoo Wildl. Med. 2013, 44, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, A.D.; Tucker, A.S.; Mulley, J.F. A Critique of the Toxicoferan Hypothesis. In Evolution of Venomous Animals and Their Toxins: Toxinology; Gopalakrishnakone, P., Malhotra, A., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 1–15. [Google Scholar] [CrossRef]
- Calvete, J.J.; Bonilla, F.; Granados-Martínez, S.; Sanz, L.; Lomonte, B.; Sasa, M. Venomics of the Duvernoy’s Gland Secretion of the False Coral Snake Rhinobothryum bovallii (Andersson, 1916) and Assessment of Venom Lethality towards Synapsid and Diapsid Animal Models. J. Proteom. 2020, 225, 103882. [Google Scholar] [CrossRef]
- Fry, B.G.; Roelants, K.; Champagne, D.E.; Scheib, H.; Tyndall, J.D.A.; King, G.F.; Nevalainen, T.J.; Norman, J.A.; Lewis, R.J.; Norton, R.S.; et al. The Toxicogenomic Multiverse: Convergent Recruitment of Proteins into Animal Venoms. Annu. Rev. Genomics Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, S.A. Snake Venoms: A Brief Treatise on Etymology, Origins of Terminology, and Definitions. Toxicon 2015, 103, 188–195. [Google Scholar] [CrossRef]
- Kordiš, D.; Gubenšek, F. Adaptive Evolution of Animal Toxin Multigene Families. Gene 2000, 261, 43–52. [Google Scholar] [CrossRef]
- Rokyta, D.R.; Margres, M.J.; Calvin, K. Post-Transcriptional Mechanisms Contribute Little to Phenotypic Variation in Snake Venoms. G3 GenesGenomesGenetics 2015, 5, 2375–2382. [Google Scholar] [CrossRef] [Green Version]
- Harvey, A.L. Toxins and Drug Discovery. Toxicon 2014, 92, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Camargo, A.C.M.; Ianzer, D.; Guerreiro, J.R.; Serrano, S.M.T. Bradykinin-Potentiating Peptides: Beyond Captopril. Toxicon 2012, 59, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in Peptide Drug Discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Margres, M.J.; Rautsaw, R.M.; Strickland, J.L.; Mason, A.J.; Schramer, T.D.; Hofmann, E.P.; Stiers, E.; Ellsworth, S.A.; Nystrom, G.S.; Hogan, M.P.; et al. The Tiger Rattlesnake Genome Reveals a Complex Genotype Underlying a Simple Venom Phenotype. Proc. Natl. Acad. Sci. USA 2021, 118, e2014634118. [Google Scholar] [CrossRef] [PubMed]
- Dowell, N.L.; Giorgianni, M.W.; Kassner, V.A.; Selegue, J.E.; Sanchez, E.E.; Carroll, S.B. The Deep Origin and Recent Loss of Venom Toxin Genes in Rattlesnakes. Curr. Biol. 2016, 26, 2434–2445. [Google Scholar] [CrossRef] [Green Version]
- Montero, N.; dei Marcovaldi, M.A.G.; Lopez–Mendilaharsu, M.; Santos, A.S.; Santos, A.J.B.; Fuentes, M.M.P.B. Warmer and Wetter Conditions Will Reduce Offspring Production of Hawksbill Turtles in Brazil under Climate Change. PLoS ONE 2018, 13, e0204188. [Google Scholar] [CrossRef] [PubMed]
- Vicoso, B.; Emerson, J.J.; Zektser, Y.; Mahajan, S.; Bachtrog, D. Comparative Sex Chromosome Genomics in Snakes: Differentiation, Evolutionary Strata, and Lack of Global Dosage Compensation. PLoS Biol. 2013, 11, e1001643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schield, D.R.; Perry, B.W.; Card, D.C.; Pasquesi, G.I.M.; Westfall, A.K.; Mackessy, S.P.; Castoe, T.A. The Rattlesnake W Chromosome: A GC-Rich Retroelement Refugium with Retained Gene Function Across Ancient Evolutionary Strata. Genome Biol. Evol. 2022, 14, evac116. [Google Scholar] [CrossRef]
- Gamble, T.; Geneva, A.J.; Glor, R.E.; Zarkower, D. Anolis Sex Chromosomes Are Derived from a Single Ancestral Pair. Evol. Int. J. Org. Evol. 2014, 68, 1027–1041. [Google Scholar] [CrossRef] [Green Version]
- Steele, A.L.; Warner, D.A. Sex-Specific Effects of Developmental Temperature on Morphology, Growth and Survival of Offspring in a Lizard with Temperature-Dependent Sex Determination. Biol. J. Linn. Soc. 2020, 130, 320–335. [Google Scholar] [CrossRef]
- Pen, I.; Uller, T.; Feldmeyer, B.; Harts, A.; While, G.M.; Wapstra, E. Climate-Driven Population Divergence in Sex-Determining Systems. Nature 2010, 468, 436–438. [Google Scholar] [CrossRef]
- Gamble, T.; Coryell, J.; Ezaz, T.; Lynch, J.; Scantlebury, D.P.; Zarkower, D. Restriction Site-Associated DNA Sequencing (RAD-Seq) Reveals an Extraordinary Number of Transitions among Gecko Sex-Determining Systems. Mol. Biol. Evol. 2015, 32, 1296–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IUCN. The IUCN Red List of Threatened Species. IUCN Red List of Threatened Species. Available online: https://www.iucnredlist.org/en (accessed on 23 March 2021).





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gable, S.M.; Mendez, J.M.; Bushroe, N.A.; Wilson, A.; Byars, M.I.; Tollis, M. The State of Squamate Genomics: Past, Present, and Future of Genome Research in the Most Speciose Terrestrial Vertebrate Order. Genes 2023, 14, 1387. https://doi.org/10.3390/genes14071387
Gable SM, Mendez JM, Bushroe NA, Wilson A, Byars MI, Tollis M. The State of Squamate Genomics: Past, Present, and Future of Genome Research in the Most Speciose Terrestrial Vertebrate Order. Genes. 2023; 14(7):1387. https://doi.org/10.3390/genes14071387
Chicago/Turabian StyleGable, Simone M., Jasmine M. Mendez, Nicholas A. Bushroe, Adam Wilson, Michael I. Byars, and Marc Tollis. 2023. "The State of Squamate Genomics: Past, Present, and Future of Genome Research in the Most Speciose Terrestrial Vertebrate Order" Genes 14, no. 7: 1387. https://doi.org/10.3390/genes14071387
APA StyleGable, S. M., Mendez, J. M., Bushroe, N. A., Wilson, A., Byars, M. I., & Tollis, M. (2023). The State of Squamate Genomics: Past, Present, and Future of Genome Research in the Most Speciose Terrestrial Vertebrate Order. Genes, 14(7), 1387. https://doi.org/10.3390/genes14071387