
Cardiac and Nephrological Complications Related to the Use of Antiangiogenic and Anti-Programmed Cell Death Protein 1 Receptor/Programmed Cell Death Protein 1 Ligand Therapy

Abstract
:1. Introduction
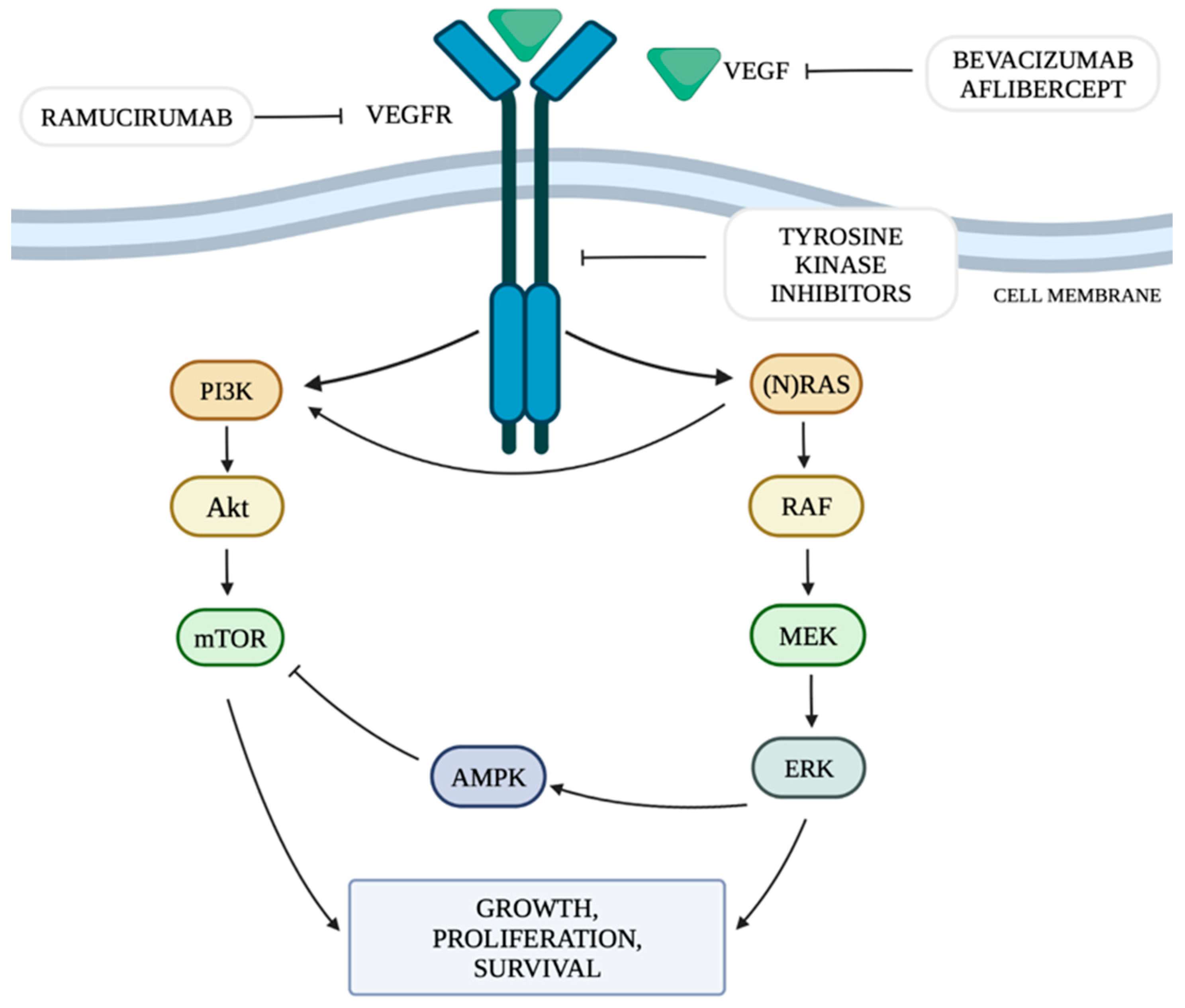
2. Mechanism of Angiogenesis in Tumors and Immune Checkpoints
3. Cardiotoxicity Associated with the Use of Anti-VEGF Therapy
4. Mechanism of Cardiotoxicity Associated with the Use of Anti-VEGF Therapy
5. Cardiotoxicity Associated with the Use of Anti-PD-1/PD-L1 Therapy
6. Mechanism of Cardiotoxicity Associated with the Use of Anti-PD-1/PD-L1 Therapy
7. Nephrotoxicity Associated with the Use of Anti-VEGF Therapy
8. Mechanism of Nephrotoxicity Associated with the Use of Anti-VEGF Therapy
9. Nephrotoxicity Associated with the Use of Anti-PD-1/PD-L1 Therapy
10. Mechanism of Nephrotoxicity Associated with the Use of Anti-PD-1/PD-L1 Therapy
11. Combination Treatment with Anti-PD-1/PD-L1 and Anti-VEGF Drugs
12. Single Nucleotide Polymorphisms and Adverse Events of Targeted Therapies
13. Conclusions and Future Direction
Author Contributions
Funding
Conflicts of Interest
References
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Morikawa, S.; Haskell, A.; Mancuso, M.; McDonald, D.M. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2003, 163, 1801–1815. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Hicklin, D.J.; Ellis, L.M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Geindreau, M.; Ghiringhelli, F.; Bruchard, M. Vascular Endothelial Growth Factor, a Key Modulator of the Anti-Tumor Immune Response. Int. J. Mol. Sci. 2021, 22, 4871. [Google Scholar] [CrossRef]
- Verheul, H.M.W.; Pinedo, H.M. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat. Rev. Cancer 2007, 7, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Roodhart, J.; Langenberg, M.; Witteveen, E.; Voest, E. The Molecular Basis of Class Side Effects Due to Treatment with Inhibitors of the VEGF/VEGFR Pathway. Curr. Clin. Pharmacol. 2008, 3, 132–143. [Google Scholar] [CrossRef]
- Ghosh, C.; Luong, G.; Sun, Y. A snapshot of the PD-1/PD-L1 pathway. J. Cancer 2021, 12, 2735–2746. [Google Scholar] [CrossRef]
- Touyz, R.M.; Herrmann, J. Cardiotoxicity with vascular endothelial growth factor inhibitor therapy. NPJ Precis Oncol. 2018, 2, 13. [Google Scholar] [CrossRef]
- Chebotareva, N.; Grechukhina, K.; Mcdonnell, V.; Zhukova, L.; Krasnova, T. Early biomarkers of nephrotoxicity associated with the use of anti-VEGF drugs. Biomed. Rep. 2022, 16, 46. [Google Scholar] [CrossRef]
- Stein-Merlob, A.F.; Rothberg, M.V.; Holman, P.; Yang, E.H. Immunotherapy-Associated Cardiotoxicity of Immune Checkpoint Inhibitors and Chimeric Antigen Receptor T Cell Therapy: Diagnostic and Management Challenges and Strategies. Curr. Cardiol. Rep. 2021, 23, 11. [Google Scholar] [CrossRef] [PubMed]
- Sise, M.E.; Seethapathy, H.; Reynolds, K.L. Diagnosis and Management of Immune Checkpoint Inhibitor-Associated Renal Toxicity: Illustrative Case and Review. Oncologist 2019, 24, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Turner, S.; Peters, K.B.; Desjardins, A.; Gururangan, S.; Sampson, J.H.; McLendon, R.E.; Herndon, J.E., 2nd; Jones, L.W.; Kirkpatrick, J.P.; et al. A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J. Natl. Compr. Canc. Netw. 2011, 9, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Bolcaen, J.; Nair, S.; Driver, C.H.S.; Boshomane, T.M.G.; Ebenhan, T.; Vandevoorde, C. Novel Receptor Tyrosine Kinase Pathway Inhibitors for Targeted Radionuclide Therapy of Glioblastoma. Pharmaceuticals 2021, 14, 626. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct. Funct. 2001, 26, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Stachyra-Strawa, P.; Ciesielka, M.; Janiszewski, M.; Grzybowska-Szatkowska, L. The role of immunotherapy and molecular-targeted therapy in the treatment of melanoma (Review). Oncol. Rep. 2021, 46, 158. [Google Scholar] [CrossRef]
- Regad, T. Targeting RTK signaling pathways in cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef]
- Figueras, A.; Arbos, M.A.; Quiles, M.T.; Viñals, F.; Germà, J.R.; Capellà, G. The impact of KRAS mutations on VEGF-A production and tumour vascular network. BMC Cancer 2013, 13, 125. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.E. Structural basis for regulation of phosphoinositide kinases and their involvement in human disease. Mol. Cell. 2018, 71, 653–673. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends. Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Akiba, H.; Iwai, H.; Matsuda, H.; Aoki, M.; Tanno, Y.; Shin, T.; Tsuchiya, H.; Pardoll, D.M.; Okumura, K. Expression of program-med death 1 ligands by murine T cells and APC. J. Immunol. 2002, 169, 5538–5545. [Google Scholar] [CrossRef]
- Dobbin, S.J.H.; Petrie, M.C.; Myles, R.C.; Touyz, R.M.; Lang, N.N. Cardiotoxic effects of angiogenesis inhibitors. Clin. Sci. 2021, 135, 71–100. [Google Scholar] [CrossRef]
- Small, H.Y.; Montezano, A.C.; Rios, F.J.; Savoia, C.; Touyz, R.M. Hypertension due to antiangiogenic cancer therapy with vascular endothelial growth factor inhibitors: Understanding and managing a new syndrome. Can. J. Cardiol. 2014, 30, 534–543. [Google Scholar] [CrossRef]
- Abdel-Qadir, H.; Ethier, J.L.; Lee, D.S.; Thavendiranathan, P.; Amir, E. Cardio- vascular toxicity of angiogenesis inhibitors in treatment of malignancy: A systematic review and meta-analysis. Cancer Treat. Rev. 2017, 53, 120–127. [Google Scholar] [CrossRef]
- Bernard, G. Mayo Clinic Heart Book; William Morrow & Co.: New York, NY, USA, 2000; pp. 120–125. [Google Scholar]
- Yamaguchi, H.; Ishinura, T.; Nishiyam; Zakaria, N.; Guerard, N.; Emanuelli, A.; Duge, P.; Watts, J.; Liew, M.; Gekkieva, M.; et al. Evaluation of cardiac parameters and other safety outcomes of brolucizumab treatment in patients with neovascular age-related macular degeneration. Pharmacol. Res. Perspect. 2022, 10, e00897. [Google Scholar]
- Wilk, M.; Szmit, S. Cardiovascular complications of antiangiogenic therapy in ovarian cancer patients. Oncol. Clin. Pract. 2017, 13, 49–56. [Google Scholar]
- Pewsner, D.; Jüni, P.; Egger, M.; Battaglia, M.; Sundström, J.; Bachmann, L.M. Accuracy of electrocardiography in diagnosis of left ventricular hypertrophy in arterial hypertension: Systematic review. BMJ 2007, 6, 711. [Google Scholar] [CrossRef]
- Vallerio, P.; Orenti, A.; Tosi, F.; Maistrello, M.; Palazzini, M.; Cingarlini, S.; Colombo, P.; Bertuzzi, M.; Spina, F.; Amatu, A.; et al. Major adverse cardiovascular events associated with VEGF-targeted anticancer tyrosine kinase inhibitors: A real-life study and proposed algorithm for proactive management. ESMO Open 2022, 7, 100338. [Google Scholar] [CrossRef]
- Billemont, B.; Medioni, J.; Taillade, L.; Helley, D.; Meric, J.B.; Rixe, O.; Oudard, S. Blood glucose levels in patients with metastatic renal cell carcinoma treated with sunitinib. Br. J. Cancer 2008, 99, 1380–1382. [Google Scholar] [CrossRef]
- National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. Available online: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcae_v5_quick_reference_8.5x11.pdf (accessed on 9 July 2023).
- Choueiri, T.K.; Mayer, E.L.; Je, Y.; Rosenberg, J.E.; Nguyen, P.L.; Azzi, G.R.; Bellmunt, J.; Burstein, H.J.; Schutz, F.A. Congestive heart failure risk in patients with breast cancer treated with bevacizumab. J. Clin. Oncol. 2011, 29, 632–638. [Google Scholar] [CrossRef]
- Ghatalia, P.; Morgan, C.J.; Je, Y.; Nguyen, P.L.; Trinh, Q.D.; Choueiri, T.K.; Sonpavde, G. Congestive heart failure with vascular endothelial growth factor receptor tyrosine kinase inhibitors. Crit. Rev. Oncol. Hematol. 2015, 94, 228–237. [Google Scholar] [CrossRef]
- Santoni, M.; Guerra, F.; Conti, A.; Lucarelli, A.; Rinaldi, S.; Belvederesi, L.; Capucci, A.; Berardi, R. Incidence and risk of cardiotoxicity in cancer patients treated with targeted therapies. Cancer Treat. Rev. 2017, 59, 123–131. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; de Souza, P.; Merchan, J.R. Pazopanib versus Sunitinib in metastatic renal-cell carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef]
- Chen, X.L.; Lei, Y.H.; Liu, C.F.; Yang, Q.F.; Zuo, P.Y.; Liu, C.Y.; Chen, C.Z.; Liu, Y.W. Angiogenesis inhibitor bevacizumab increases the risk of ischemic heart disease associated with chemotherapy: A meta-analysis. PLoS ONE 2013, 8, e66721. [Google Scholar] [CrossRef] [PubMed]
- Faruque, L.I.; Lin, M.; Battistella, M.; Wiebe, N.; Reiman, T.; Hemmelgarn, B.; Thomas, C.; Tonelli, M. Systematic review of the risk of adverse outcomes associated with vascular endothelial growth factor inhibitors for the treatment of cancer. PLoS ONE 2014, 9, e101145. [Google Scholar] [CrossRef] [PubMed]
- Maison-Blanche, P.; Vermorken, J.B.; Goksel, T.; Machiels, J.P.; Agarwala, S.; Rottey, S.; Daugaard, G.; Volovat, C.; Scheulen, M.; Sengeløv, L. A randomized, double-blind, placebo-controlled study to assess QTc interval prolongation of standard dose aflibercept in cancer patients treated with docetaxel. J. Cardiovasc. Pharmacol. 2013, 61, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Zang, J.; Wu, S.; Tang, L.; Xu, X.; Bai, J.; Ding, C.; Chang, Y.; Yue, L.; Kang, E.; He, J. Incidence and risk of QTc interval prolongation among cancer patients treated with vandetanib: A systematic review and meta-analysis. PLoS ONE 2012, 7, e30353. [Google Scholar] [CrossRef]
- Lobenwein, D.; Kocher, F.; Dobner, S.; Gollmann-Tepeköylü, C.; Holfeld, J. Cardiotoxic mechanisms of cancer immunotherapy—A systematic review. Int. J. Cardiol. 2021, 323, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Oren, O.; Yang, E.H.; Molina, J.R.; Bailey, K.R.; Blumenthal, R.S.; Kopecky, S.L. Cardiovascular health and outcomes in cancer patients receiving immune checkpoint inhibitors. Am. J. Cardiol. 2020, 125, 1920–1926. [Google Scholar] [CrossRef]
- Van Wynsberghe, M.; Flejeo, J.; Sakhi, H.; Ollero, M.; Sahali, D.; Izzedine, H.; Henique, C. Nephrotoxicity of Anti-Angiogenic Therapies. Diagnostics 2021, 11, 640. [Google Scholar] [CrossRef]
- Hayman, S.R.; Leung, N.; Grande, J.P.; Garovic, V.D. VEGF Inhibition, Hypertension, and Renal Toxicity. Curr. Oncol. Rep. 2012, 14, 285–294. [Google Scholar] [CrossRef]
- Yang, J.C.; Haworth, L.; Sherry, R.M.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Steinberg, S.M.; Chen, H.X.; Rosenberg, S.A. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N. Engl. J. Med. 2003, 349, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wu, S.; Dahut, W.L.; Parikh, C.R. Risks of proteinuria and hypertension with bevacizumab, an antibody against vascular endothelial growth factor: Systematic review and meta-analysis. Am. J. Kidney Dis. 2007, 49, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.V.; Morgan, J.A.; Demetri, G.D.; George, S.; Maki, R.G.; Quigley, M.; Humphreys, B.D. A preeclampsia-like syndrome characterized by reversible hypertension and proteinuria induced by the multitargeted kinase inhibitors sunitinib and sorafenib. J. Natl. Cancer Inst. 2008, 100, 282–284. [Google Scholar] [CrossRef]
- Garovic, V.D.; Wagner, S.J.; Petrovic, L.M.; Gray, C.E.; Hall, P.; Sugimoto, H.; Kalluri, R.; Grande, J.P. Glomerular expression of nephrin and synaptopodin, but not podocin, is decreased in kidney sections from women with preeclampsia. Nephrol. Dial. Transplant. 2007, 22, 1136–1143. [Google Scholar] [CrossRef]
- Borówka, M.; Łącki-Zynzeling, S.; Nicze, M.; Kozak, S.; Chudek, J. Adverse Renal Effects of Anticancer Immunotherapy: A Review. Cancers 2022, 14, 4086. [Google Scholar] [CrossRef]
- Cortazar, F.B.; Marrone, K.A.; Troxell, M.L.; Ralto, K.M.; Hoenig, M.P.; Brahmer, J.R.; Le, D.T.; Lipson, E.J.; Glezerman, I.G.; Wolchok, J.; et al. Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int. 2016, 90, 638–647. [Google Scholar] [CrossRef]
- Gupta, S.; Cortazar, F.B.; Riella, L.V.; Leaf, D.E. Immune Checkpoint Inhibitor Nephrotoxicity: Update 2020. Kidney360 2020, 1, 130–140. [Google Scholar] [CrossRef]
- Li, Y.-L.; Zhao, H.; Ren, X.-B. Relationship of vegf/vegfr with immune and cancer cells: Staggering or forward? Cancer Biol. Med. 2016, 13, 206–214. [Google Scholar] [CrossRef]
- Ziogas, A.C.; Gavalas, N.G.; Tsiatas, M.; Tsitsilonis, O.; Politi, E.; Terpos, E.; Rodolakis, A.; Vlahos, G.; Thomakos, N.; Haidopoulos, D.; et al. Vegf directly suppresses activation of t cells from ovarian cancer patients and healthy individuals via vegf receptor type 2. Int. J. Cancer 2012, 130, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Cabozantinib plus Nivolumab: A Review in Advanced Renal Cell Carcinoma. Target Oncol. 2022, 17, 193–201. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Zhou, X.; Yao, Z.; Bai, H.; Duan, J.; Wang, Z.; Wang, X.; He, J. Treatment-related adverse events of PD-1 and PD-L1 inhibitor-based combination therapies in clinical trials: A systematic review and meta-analysis. Lancet Oncol. 2021, 22, 1265–1274. [Google Scholar] [CrossRef]
- Tzuri, N.; Yegodayev, K.M.; Novoplansky, O. Developing a dual VEGF/PDL1 inhibitor based on high-affinity scFv heterodimers as an anti-cancer therapeutic strategy. Sci. Rep. 2023, 13, 11923. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Mao, Y.; Wu, T.; Kang, N.; Zhao, M.; Di, R.; Li, X.; Ji, X.; Liu, Y. Optimized expression and characterization of a novel fully human bispecific single-chain diabody targeting vascular endothelial growth factor165 and programmed death-1 in Pichia pastoris and evaluation of antitumor activity in vivo. Int. J. Mol. Sci. 2018, 19, 2900. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.K.; Dao, F.; Olvera, N.; Konner, J.A.; Dickler, M.; Levine, D.A. Genetic predisposition to bevacizumab-induced hypertension. Gynecol. Oncol. 2017, 147, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Quintanilha, J.C.F.; Wang, J.; Sibley, A.B.; Jiang, C.; Etheridge, A.S.; Shen, F.; Jiang, G.; Mulkey, F.; Patel, J.N.; Hertz, D.L.; et al. Bevacizumab-induced hypertension and proteinuria: A genome-wide study of more than 1000 patients. Br. J. Cancer 2022, 126, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Moisse, M.; Delmar, P.; Miles, D.W.; Leighl, N.; Escudier, B.; Van Cutsem, E.; Bansal, A.T.; Carmeliet, P.; Scherer, S.J.; et al. Genetic markers of bevacizumab-induced hypertension. Angiogenesis 2014, 17, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Eechoute, K.; van der Veldt, A.A.; Oosting, S.; Kappers, M.H.; Wessels, J.A.; Gelderblom, H.; Guchelaar, H.J.; Reyners, A.K.; van Herpen, C.M.; Haanen, J.B.; et al. Polymorphisms in Endothelial Nitric Oxide Synthase (eNOS) and Vascular Endothelial Growth Factor (VEGF) predict sunitinib-induced hypertension. Clin. Pharmacol. Ther. 2012, 92, 503–510. [Google Scholar] [CrossRef]
- Diekstra, M.H.; Belaustegui, A.; Swen, J.J.; Boven, E.; Castellano, D.; Gelderblom, H.; Mathijssen, R.H.; García-Donas, J.; Rodríguez-Antona, C.; Rini, B.I. Sunitinib-induced hypertension in CYP3A4 rs4646437 A-allele carriers with metastatic renal cell carcinoma. Pharmacogenom. J. 2017, 17, 42–46. [Google Scholar] [CrossRef]
- Qin, C.; Cao, Q.; Li, P.; Wang, S.; Wang, J.; Wang, M.; Chu, H.; Zhou, L.; Li, X.; Ye, D.; et al. The influence of genetic variants of sorafenib on clinical outcomes and toxic effects in patients with advanced renal cell carcinoma. Sci. Rep. 2016, 6, 20089–20103. [Google Scholar] [CrossRef]
- What Are Genome-Wide Association Studies (GWAS)? EMBL-EBI Train Online. 2020. Available online: https://www.ebi.ac.uk/training-β/online/courses/gwas-catalogue-exploring-snp-trait-associations/what-is-gwas-catalog/what-are-genome-wide-association-studies-gwas/ (accessed on 20 August 2023).
- Groha, S.; Alaiwi, S.A.; Xu, W.; Naranbhai, V.; Nassar, A.H.; Bakouny, Z.; El Zarif, T.; Saliby, R.M.; Wan, G.; Rajeh, A.; et al. Germline variants associated with toxicity to immune checkpoint blockade. Nat. Med. 2022, 28, 2584–2591. [Google Scholar] [CrossRef]
- Udagawa, C.; Nakano, M.H.; Yoshida, T.; Ohe, Y.; Kato, K.; Mushiroda, T.; Zembutsu, H. Association between genetic variants and the risk of nivolumab-induced immune-related adverse events. Pharmacogenomics 2022, 23, 887–901. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Angiogenesis Inhibitors | |||||
|---|---|---|---|---|---|
| Drug | Type of Agent | Target | Approval by FDA in Cancer Treatment | Main Adverse Effects | References |
| Bevacizumab | Humanized monoclonal antibody | VEGF-A | Colorectal cancer, glioblastoma, cervical cancer, NSCLC, ovarian cancer, RCC | hypertension, bleeding, thrombo-embolism, gastro-intestinal perforations, proteinuria | [6,7,8] |
| Aflibercept | Soluble decoy receptor | VEGF-A, VEGF-B, PIGF | Colorectal cancer | Bleeding, perforation of the digestive tract, hypertension, thrombo-embolism formation of a fistula, heart failure, decreased ejection fraction | [6,7] |
| Ramucirumab | Human monoclonal antibody | VEGFR-2 | Gastric cancer, NSCLC, colorectal cancer | hypertension, diarrhea, headache, hyponatremia, anaemia, intestinal obstruction | [6] |
| Sunitinib | Small-molecule TKI | VEGFR-1, VEGFR-2, PDGFR, KIT, FLT3 | Gastrointestinal stromal tumor, pancreatic neuroendocrine tumor, RCC | hypertension, nausea, vomiting, diarrhea, fatigue, lymphopenia, neutropenia | [6,7,8] |
| Sorafenib | Small-molecule TKI | VEGFR-1, VEGFR-2, Raf, PDGFR, KIT, RET | Hepatocellular carcinoma, RCC, differentiated thyroid cancer | hypertension, nausea, vomiting, diarrhea, fatigue, anorexia, hand-food syndrome and rash | [6,7,8] |
| Pazopanib | Small-molecule TKI | VEGFR-1, VEGFR-2, VEGFR-3 | RCC, soft tissue sarcoma | hypertension, nausea, vomiting, diarrhea | [6,7] |
| Axitinib | Small-molecule TKI | VEGFR-1, VEGFR-2, VEGFR-3 | RCC | hypertension, nausea, vomiting, diarrhea, fatigue, stomatitis | [7,8] |
| Vantedanib | Small-molecule TKI | VEGFR-2, EGFR, FGFR1, RET | Medullary thyroid cancer | hypertension, nausea, vomiting, diarrhea, fatigue, weight loss | [7,8] |
| Immune checkpoint inhibitors | |||||
| Nivolumab | Human monoclonal antibody | PD-1 | melanoma, NSCLC, pleural mesothelioma, renal cell carcinoma, HNSCC, urothelial carcinoma, colorectal cancer, HCC, esophageal cancer, gastroesophageal junction cancer, gastric cancer | pruritus, fatigue, loss of appetite, immune related-adverse events (irAE)—dermatitis, hypophysitis, colitis and hepatitis | [9] |
| Pembrolizumab | Humanized monoclonal antibody | PD-1 | melanoma, NSCLC, HNSCC, urothelial carcinoma, gastric cancer, colorectal cancer, esophageal cancer, cervical cancer, HCC, RCC, endometrial carcinoma, triple-negative breast cancer, cutaneous squamous cell carcinoma, Merkel cell carcinoma | [9] | |
| Cemiplimab | Human monoclonal antibody | PD-1 | NSCLC, squamous cell skin cancer | [9] | |
| Atezolizumab | Humanized monoclonal antibody | PD-L1 | NSCLC, SCLC, HCC, melanoma, alveolar soft part sarcoma | [9] | |
| Avelumab | Humanized monoclonal antibody | PD-L1 | Merkel Cell carcinoma, urothelial carcinoma, RCC | [9] | |
| Durvalumab | Humanized monoclonal antibody | PD-L1 | NSCLC, SCLC, biliary tract cancer, HCC | [9] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stachyra-Strawa, P.; Szatkowska-Sieczek, L.; Cisek, P.; Gołębiowski, P.; Grzybowska-Szatkowska, L. Cardiac and Nephrological Complications Related to the Use of Antiangiogenic and Anti-Programmed Cell Death Protein 1 Receptor/Programmed Cell Death Protein 1 Ligand Therapy. Genes 2024, 15, 177. https://doi.org/10.3390/genes15020177
Stachyra-Strawa P, Szatkowska-Sieczek L, Cisek P, Gołębiowski P, Grzybowska-Szatkowska L. Cardiac and Nephrological Complications Related to the Use of Antiangiogenic and Anti-Programmed Cell Death Protein 1 Receptor/Programmed Cell Death Protein 1 Ligand Therapy. Genes. 2024; 15(2):177. https://doi.org/10.3390/genes15020177
Chicago/Turabian StyleStachyra-Strawa, Paulina, Lidia Szatkowska-Sieczek, Paweł Cisek, Paweł Gołębiowski, and Ludmiła Grzybowska-Szatkowska. 2024. "Cardiac and Nephrological Complications Related to the Use of Antiangiogenic and Anti-Programmed Cell Death Protein 1 Receptor/Programmed Cell Death Protein 1 Ligand Therapy" Genes 15, no. 2: 177. https://doi.org/10.3390/genes15020177
APA StyleStachyra-Strawa, P., Szatkowska-Sieczek, L., Cisek, P., Gołębiowski, P., & Grzybowska-Szatkowska, L. (2024). Cardiac and Nephrological Complications Related to the Use of Antiangiogenic and Anti-Programmed Cell Death Protein 1 Receptor/Programmed Cell Death Protein 1 Ligand Therapy. Genes, 15(2), 177. https://doi.org/10.3390/genes15020177