
When Livestock Genomes Meet Third-Generation Sequencing Technology: From Opportunities to Applications

Abstract
:1. Introduction
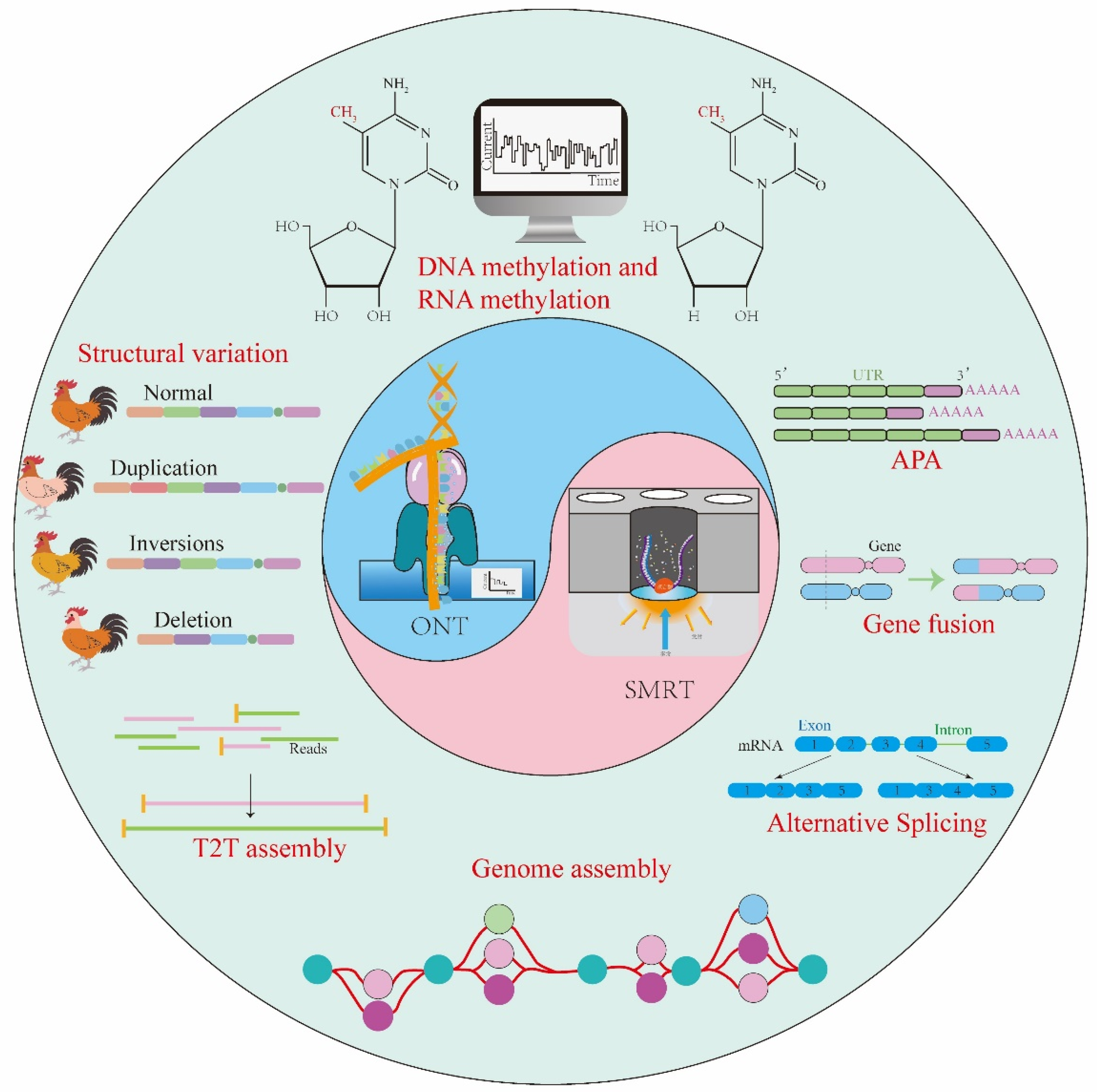
2. Advances of TGS in the Genomic Research of Livestock
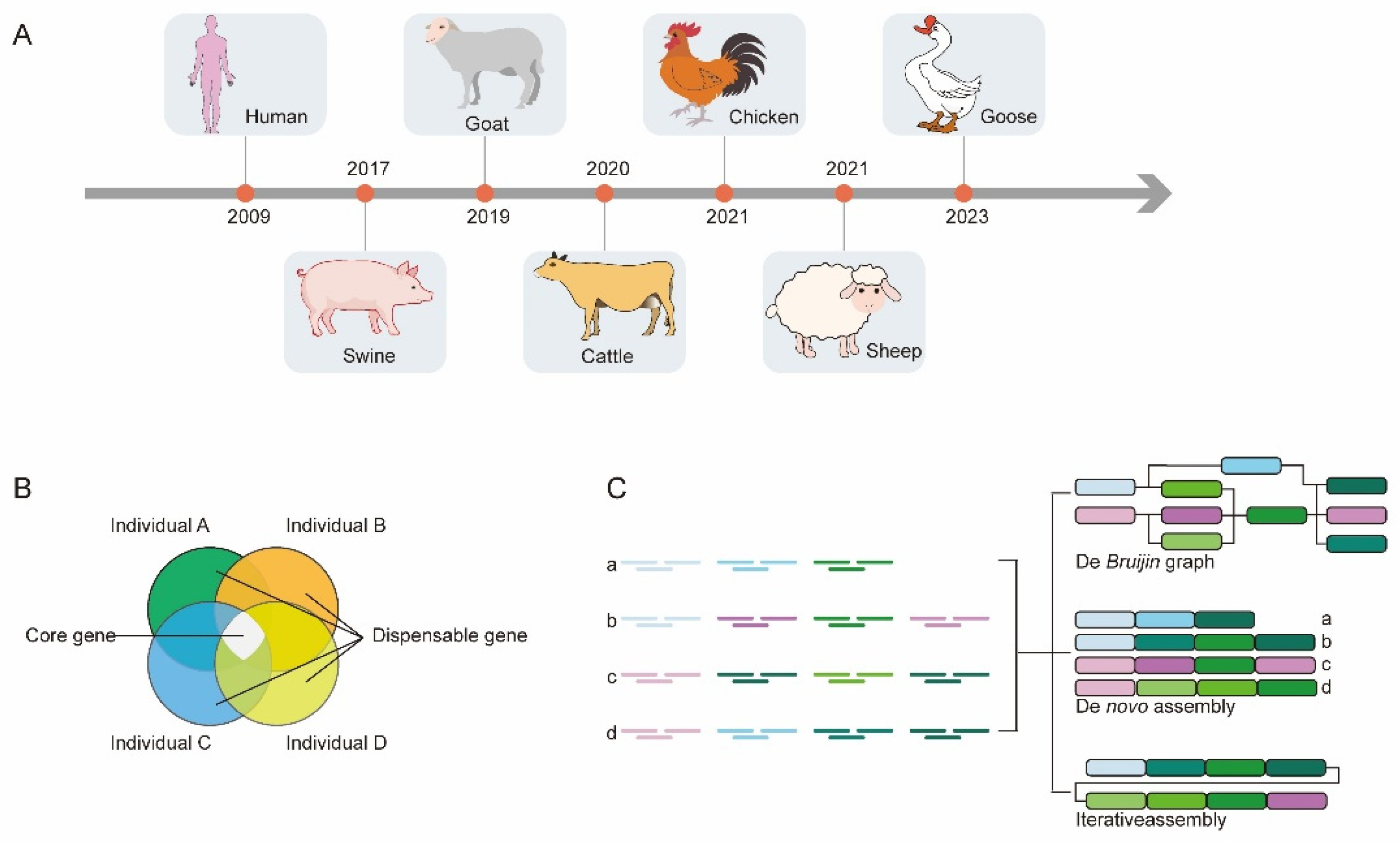
2.1. Progress in Genome Assembly Using TGS in Livestock
2.2. Progress in Pan-Genome Using TGS in Livestock
2.3. Progress in Telomere-to-Telomere Assembly Using TGS

{kind=link}
{kind=link}
| Species | Feature | Breed | Sequencing Platform | Key Findings | Publication Year | Reference |
|---|---|---|---|---|---|---|
| Bos taurus | Cattle | 1.OⅹO F1 2.NⅹB F1 3.GⅹP F | ONT | Constructed haplotype-resolved genomes for cattle and related species, established a pan-genome for cattle, and quantified structural diversity | 2022 | [14] |
| B. taurus | Cattle | Southern Yellow Cattle | PacBio-SMRT | Confirmed genetic diversity in the southern yellow cattle population in China, identified gene introgression events from five different wild cattle species | 2023 | [15] |
| B. taurus | Cattle | Hainan Cattle, Mongolian Cattle | ONT | Discovered significant structural variations influencing environmental adaptability in Chinese yellow cattle | 2023 | [49] |
| Bos frontalis | Cattle | Gayal | PacBio-SMRT | Conducted chromosome-level genome assembly for Dulong cattle | 2023 | [16] |
| Bos grunniens | Yak | Yak | ONT | Obtained high-quality chromosome-level genomes for wild and domestic yaks, a structural variation catalog for yaks, and a single-cell transcriptome atlas of lung tissues | 2022 | [17] |
| B. grunniens | Yak | White Yak | ONT | Revealed genetic introgression of unique structural variations in the color-sided yellow cattle, resulting in the creation of the color-sided Yak. Subsequent genetic variations gave rise to the white Yak | 2023 | [18] |
| Bubalus bubalis | Buffalo | Water Buffalo | PacBio-SMRT | Generated a detailed genomic map for water buffalo (2n = 50) and performed chromosome-level genome assembly | 2019 | [19] |
| B. bubalis | Buffalo | Swamp-type Water Buffalo, River-type Water Buffalo | PacBio-SMRT | Attained high-quality chromosome-level reference genomes for swamp-type water buffalo (2n = 48) and river-type water buffalo (2n = 50) | 2020 | [20] |
| Ovis aries | Sheep | Dorper Sheep | ONT | Revealed the genetic basis of allele-specific expression (ASE) genes and specific phenotypic traits in Dorper sheep | 2022 | [21] |
| O. aries | Sheep | 15different breeds of sheep | PacBio-SMRT | Constructed high-quality pan-genome maps for different sheep breeds | 2023 | [22] |
| Capra hircus | Goat | Saanen Dairy Goat | PacBio-SMRT | Assembled the reference genome Saanen_v1 for Saanen dairy goats | 2021 | [23] |
| C. hircus | Goat | Tibetan Goat | PacBio-SMRT | Unveiled PAPSS2 as a key gene not only for high-altitude adaptation in goats but also a significant gene in genetic introgression analysis | 2022 | [24] |
| Sus scrofa | Pig | Tibetan Pig, Jinhua Pig, and 8 other breeds | ONT | Completed pan-genome maps for Anqing Liubai Pig, Laiwu Pig, Meishan Pig, Min Pig, Rongchang Pig, Wuzhishan Pig, Yorkshire Pig, European Wild Boar, etc. | 2023 | [25] |
| S. scrofa | Pig | Duroc | PacBio-SMRT | Assembled the reference genome Sscrofa11.1 for pigs from scratch | 2020 | [26] |
| Gallus gallus | Chicken | Huxu Chicken | ONT | First published complete genome atlas (T2T) for vertebrates; characterized the epigenetics of the W chromosome; elucidated the origin, sequence structure, and diversity of chicken centromeres | 2023 | [28] |
| G. gallus | Chicken | Chickens from Four Continents | PacBio-SMRT | Established the pan-genome of chickens, identified new coding genes, long non-coding RNAs, and new gene families; identified new gene clusters for studying collinearity | 2022 | [50] |
| G. gallus | Chicken | Wenshang Lu Hua Chicken | PacBio-SMRT | Obtained a high-quality chromosome-level reference genome for the Wenshang Lu Hua chicken | 2023 | [51] |
| Anas platyrhynchos | Duck | Peking Duck, Shaoxing Duck, and Mallard | PacBio-SMRT | Assembled chromosome-level high-quality genomes for Peking Duck, Shaoxing Duck, and mallard, refuting the “missing gene hypothesis” in birds | 2021 | [29] |
2.4. Understanding the Genetic Mechanisms of Livestock Traits Using TGS
2.4.1. Understanding the Genetic Mechanisms of Traits in Ruminants
2.4.2. Understanding the Genetic Mechanisms of Traits in Monogastric Animals
3. Application of TGS in the Transcriptome of Livestock
3.1. Application of TGS in the Transcriptome of Ruminant Animals
3.2. Application of TGS in the Transcriptome of Monogastric Animals
| Species | Feature | Breed | Sequencing Platform | Key Findings | Publication Year | References |
|---|---|---|---|---|---|---|
| B. taurus | Cattle | Hereford Cattle | ONT | Discovered tissue-specific transcripts in cattle, with the testes exhibiting the most complex transcriptome | 2021 | [69] |
| B. taurus | Cattle | Simmental Cattle | PacBio-SMRT | Analyzed the full-length transcriptome of Simmental cattle, providing a foundation for refining the cattle draft genome annotation, optimizing genome structure, and comprehensively characterizing the cattle transcriptome | 2021 | [76] |
| O. aries | Sheep | (Dorper × Hu) × Hu sheep; Dorper × (Dorper × Hu sheep) | PacBio-SMRT | Revealed the transcriptome complexity and identified many candidate transcripts in tail fat, which could enhance the understanding of molecular mechanisms behind tail fat deposition | 2021 | [82] |
| C. hircus | Goat | Chinese Cashmere Goat | PacBio-SMRT | Showed the superiority of full-length transcriptome data in gene annotation; more such data are required to improve the gene annotation for goat genome and that of other species | 2023 | [83] |
| S. scrofa | Pig | Large White Pig × Min Pig F2 Generation | ONT | Discovered differentially expressed mRNA isoforms involved in skeletal muscle development and fatty acid metabolism | 2022 | [71] |
| G. gallus | Chicken | White Leghorn Chicken | ONT | Identified the most tissue-specific transcripts in reproductive tissues (testes and ovaries) of chickens | 2022 | [78] |
| G. gallus | Chicken | Hy-Line Brown Chicken | ONT | Revealed mRNA and lncRNA expression differences between pre-GCs and post-GCs during chicken follicle selection; discovered significant estrogen-induced expression of three DHCR7 isoforms | 2023 | [72,79] |
| Cairina moschata | Duck | Muscovy Duck | ONT | Obtained the full-length transcriptome of Muscovy duck follicles, providing structural and functional annotations for new transcripts | 2021 2022 | [80,81] |
4. Advances of TGS in Epigenetic Studies of Livestock
4.1. Application of TGS in DNA Methylation Modification
4.2. Application of TGS in RNA Epigenetic Modifications
5. Prospects
Author Contributions
Funding
Conflicts of Interest
References
- van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, J.; Tan, L.; Lu, C.; Fu, G.; Fu, L.; Zhang, X.J.; Wang, Q.; Ma, C.L.; Cong, B.; et al. Highly accurate mtGenome haplotypes from long-read SMRT sequencing can distinguish between monozygotic twins. Forensic Sci. Int. Genet. 2020, 47, 102306. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2019, 374, 20190097. [Google Scholar] [CrossRef] [PubMed]
- Delahaye, C.; Nicolas, J. Sequencing DNA with nanopores: Troubles and biases. PLoS ONE 2021, 16, e0257521. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, E.L.; Naquin, D.; Gorrichon, K.; Jaszczyszyn, Y.; Ouazahrou, R.; Thermes, C.; Hernandez, C. Genomics in the long-read sequencing era. Trends Genet. 2023, 39, 649–671. [Google Scholar] [CrossRef]
- Hu, T.; Li, J.; Long, M.; Wu, J.; Zhang, Z.; Xie, F.; Zhao, J.; Yang, H.P.; Song, Q.Q.; Lian, S.; et al. Detection of Structural Variations and Fusion Genes in Breast Cancer Samples Using Third-Generation Sequencing. Front. Cell Dev. Biol. 2022, 10, 854640. [Google Scholar] [CrossRef]
- Nakano, K.; Shiroma, A.; Shimoji, M.; Tamotsu, H.; Ashimine, N.; Ohki, S.; Shinzato, M.; Minami, M.; Nakanishi, T.; Teruya, K.; et al. Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum. Cell 2017, 30, 149–161. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Y.; Jiang, D.; Du, S.; Deng, Z.; Wang, L.; Chen, S. Recent Advances in the Genomic Profiling of Bacterial Epigenetic Modifications. Biotechnol. J. 2019, 14, e1800001. [Google Scholar] [CrossRef]
- Gurgul, A.; Jasielczuk, I.; Szmatoła, T.; Sawicki, S.; Semik-Gurgul, E.; Długosz, B.; Bugno-Poniewierska, M. Application of Nanopore Sequencing for High Throughput Genotyping in Horses. Animals 2023, 13, 2227. [Google Scholar] [CrossRef]
- Yang, C.; Chowdhury, D.; Zhang, Z.; Cheung, W.K.; Lu, A.; Bian, Z.; Zhang, L. A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. Comput. Struct. Biotechnol. J. 2021, 19, 6301–6314. [Google Scholar] [CrossRef]
- Sohn, J.-I.; Nam, J.-W. The present and future ofde novowhole-genome assembly. Brief. Bioinform. 2016, 19, 23–40. [Google Scholar] [CrossRef]
- Leonard, A.S.; Crysnanto, D.; Fang, Z.H.; Heaton, M.P.; Vander Ley, B.L.; Herrera, C.; Bollwein, H.; Bickhart, D.M.; Kuhn, K.L.; Smith, T.P.L.; et al. Structural variant-based pangenome construction has low sensitivity to variability of haplotype-resolved bovine assemblies. Nat. Commun. 2022, 13, 3012. [Google Scholar] [CrossRef]
- Dai, X.; Bian, P.; Hu, D.; Luo, F.; Huang, Y.; Jiao, S.; Wang, X.H.; Gong, M.; Li, R.; Cai, Y.D.; et al. A Chinese indicine pangenome reveals a wealth of novel structural variants introgressed from other Bos species. Genome Res. 2023, 33, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, S.; Zhang, Z.; Luo, J.; Lin, G.L.; Deng, W.D.; Guo, Z.F.; Han, F.M.; Wang, L.L.; Li, J.; et al. Large-Scale Chromosomal Changes Lead to Genome-Level Expression Alterations, Environmental Adaptation, and Speciation in the Gayal (Bos frontalis). Mol. Biol. Evol. 2023, 40, msad006. [Google Scholar] [CrossRef]
- Gao, X.; Wang, S.; Wang, Y.F.; Li, S.; Wu, S.X.; Yan, R.G.; Zhang, Y.W.; Wan, R.D.; He, Z.; Song, R.D.; et al. Long read genome assemblies complemented by single cell RNA-sequencing reveal genetic and cellular mechanisms underlying the adaptive evolution of yak. Nat. Commun. 2022, 13, 4887. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, W.; Lenstra, J.A.; Zheng, Z.; Wu, X.; Yang, J.; Li, B.W.; Yang, Y.Z.; Qiu, Q.; Liu, H.Y.; et al. Evolutionary origin of genomic structural variations in domestic yaks. Nat. Commun. 2023, 14, 5617. [Google Scholar] [CrossRef]
- Low, W.Y.; Tearle, R.; Bickhart, D.M.; Rosen, B.D.; Kingan, S.B.; Swale, T.; Thibaud-Nissen, F.; Murphy, T.D.; Young, R.; Lefevre, L.; et al. Chromosome-level assembly of the water buffalo genome surpasses human and goat genomes in sequence contiguity. Nat. Commun. 2019, 10, 260. [Google Scholar] [CrossRef]
- Luo, X.; Zhou, Y.; Zhang, B.; Zhang, Y.; Wang, X.; Feng, T.; Li, Z.P.; Cui, K.Q.; Wang, Z.Q.; Luo, C.; et al. Understanding divergent domestication traits from the whole-genome sequencing of swamp- and river-buffalo populations. Natl. Sci. Rev. 2020, 7, 686–701. [Google Scholar] [CrossRef] [PubMed]
- Qiao, G.; Xu, P.; Guo, T.; Wu, Y.; Lu, X.; Zhang, Q.; He, X.; Zhu, S.H.; Zhao, H.C.; Lei, Z.H.; et al. Genetic Basis of Dorper Sheep (Ovis aries) Revealed by Long-Read De Novo Genome Assembly. Front. Genet. 2022, 13, 846449. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Gong, M.; Zhang, X.; Wang, F.; Liu, Z.; Zhang, L.; Yang, Q.M.; Xu, Y.; Xu, M.S.; Zhang, H.H.; et al. A sheep pangenome reveals the spectrum of structural variations and their effects on tail phenotypes. Genome Res. 2023, 33, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yang, P.; Dai, X.; Asadollahpour Nanaei, H.; Fang, W.; Yang, Z.; Cai, Y.D.; Zheng, Z.Q.; Wang, X.H.; Jiang, Y. A near complete genome for goat genetic and genomic research. Genet. Sel. Evol. 2021, 53, 74. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, Y.; Chen, B.; Cai, Y.; Guo, J.; Leonard, A.S.; Kalds, P.; Zhou, S.W.; Zhang, J.C.; Ping, Z.; et al. Markhor-derived Introgression of a Genomic Region Encompassing PAPSS2 Confers High-altitude Adaptability in Tibetan Goats. Mol. Biol. Evol. 2022, 39, msac253. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.F.; Wang, S.; Wang, C.L.; Xu, R.H.; Wang, W.W.; Jiang, Y.; Wang, M.S.; Jiang, L.; Dai, L.H.; Wang, J.R.; et al. Pangenome obtained by long-read sequencing of 11 genomes reveal hidden functional structural variants in pigs. iScience 2023, 26, 106119. [Google Scholar] [CrossRef] [PubMed]
- Warr, A.; Affara, N.; Aken, B.; Beiki, H.; Bickhart, D.M.; Billis, K.; Chow, W.; Eory, L.; Finlayson, H.A.; Flicek, P.; et al. An improved pig reference genome sequence to enable pig genetics and genomics research. Gigascience 2020, 9, giaa051. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Yin, Z.-T.; Zhao, Q.-S.; Sun, Y.-X.; Jie, Y.-C.; Smith, J.; Yang, Y.Z.; Burt, D.W.; Hincke, M.; Zhang, Z.D.; et al. A chromosome-level genome assembly for the Silkie chicken resolves complete sequences for key chicken metabolic, reproductive, and immunity genes. Commun. Biol. 2023, 6, 1233. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xu, Z.; Bai, H.; Huang, Y.; Kang, N.; Ding, X.; Liu, J.; Luo, H.R.; Yang, C.T.; Chen, W.J.; et al. Evolutionary analysis of a complete chicken genome. Proc. Natl. Acad. Sci. USA 2023, 120, e2216641120. [Google Scholar] [CrossRef]
- Zhu, F.; Yin, Z.T.; Wang, Z.; Smith, J.; Zhang, F.; Martin, F.; Ogeh, D.; Hincke, M.; Lin, F.B.; Burt, D.W.; et al. Three chromosome-level duck genome assemblies provide insights into genomic variation during domestication. Nat. Commun. 2021, 12, 5932. [Google Scholar] [CrossRef]
- Gao, G.; Zhang, H.; Ni, J.; Zhao, X.; Zhang, K.; Wang, J.; Kong, X.D.; Wang, Q.G. Insights into genetic diversity and phenotypic variations in domestic geese through comprehensive population and pan-genome analysis. J. Anim. Sci. Biotechnol. 2023, 14, 150. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef]
- Smith, T.P.L.; Bickhart, D.M.; Boichard, D.; Chamberlain, A.J.; Djikeng, A.; Jiang, Y.; Low, W.Y.; Pausch, H.; Demyda-Peyras, S.; Prendergast, J.; et al. The Bovine Pangenome Consortium: Democratizing production and accessibility of genome assemblies for global cattle breeds and other bovine species. Genome Biol. 2023, 24, 139. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, Y.; Zheng, H.; Luo, R.; Zhu, H.; Li, Q.; Qian, W.B.; Ren, Y.Y.; Tian, G.; Li, J.X.; et al. Building the sequence map of the human pan-genome. Nat. Biotechnol. 2010, 28, 57–63. [Google Scholar] [CrossRef]
- Li, M.; Chen, L.; Tian, S.; Lin, Y.; Tang, Q.; Zhou, X.; Li, D.Y.; Yeung, C.K.L.; Che, T.D.; Jin, L.; et al. Comprehensive variation discovery and recovery of missing sequence in the pig genome using multiple de novo assemblies. Genome Res. 2017, 27, 865–874. [Google Scholar] [CrossRef]
- Li, Z.; Liu, X.; Wang, C.; Li, Z.; Jiang, B.; Zhang, R.; Tong, L.; Qu, Y.; He, S.; Chen, H.; et al. The pig pangenome provides insights into the roles of coding structural variations in genetic diversity and adaptation. Genome Res. 2023, 33, 1833–1847. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, R.; Fu, W.; Li, Y.; Wang, X.; Li, M.; Du, D.; Tang, Q.Z.; Cai, Y.D.; Long, Y.M.; et al. Building a sequence map of the pig pan-genome from multiple de novo assemblies and Hi-C data. Sci. China Life Sci. 2019, 63, 750–763. [Google Scholar] [CrossRef]
- Li, R.; Fu, W.; Su, R.; Tian, X.; Du, D.; Zhao, Y.; Zheng, Z.Q.; Chen, Q.M.; Gao, S.; Cai, Y.D.; et al. Towards the Complete Goat Pan-Genome by Recovering Missing Genomic Segments from the Reference Genome. Front. Genet. 2019, 10, 1169. [Google Scholar] [CrossRef]
- Crysnanto, D.; Pausch, H. Bovine breed-specific augmented reference graphs facilitate accurate sequence read mapping and unbiased variant discovery. Genome Biol. 2020, 21, 184. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, L.; Han, X.; Han, J.; Hu, Y.; Li, F.; Xia, H.; Peng, L.W.; Boschiero, C.; Rosen, B.D.; et al. Assembly of a pangenome for global cattle reveals missing sequences and novel structural variations, providing new insights into their diversity and evolutionary history. Genome Res. 2022, 32, 1585–1601. [Google Scholar] [CrossRef]
- Wang, K.; Hu, H.; Tian, Y.; Li, J.; Scheben, A.; Zhang, C.; Li, Y.Y.; Wu, J.F.; Yang, L.; Fan, X.W.; et al. The Chicken Pan-Genome Reveals Gene Content Variation and a Promoter Region Deletion in IGF2BP1 Affecting Body Size. Mol. Biol. Evol. 2021, 38, 5066–5081. [Google Scholar] [CrossRef]
- Li, R.; Yang, P.; Li, M.; Fang, W.; Yue, X.; Nanaei, H.A.; Gan, S.Q.; Du, D.; Cai, Y.D.; Dai, X.L.; et al. A Hu sheep genome with the first ovine Y chromosome reveal introgression history after sheep domestication. Sci. China Life Sci. 2021, 64, 1116–1130. [Google Scholar] [CrossRef]
- Golicz, A.A.; Bayer, P.E.; Barker, G.C.; Edger, P.P.; Kim, H.; Martinez, P.A.; Chan, C.K.K.; Severn-Ellis, A.; McCombie, W.R.; Parkin, I.A.P.; et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 2016, 7, 13390. [Google Scholar] [CrossRef]
- Rosconi, F.; Rudmann, E.; Li, J.; Surujon, D.; Anthony, J.; Frank, M.; Jones, D.S.; Rock, C.; Rosch, J.W.; Johnston, C.D.; et al. A bacterial pan-genome makes gene essentiality strain-dependent and evolvable. Nat. Microbiol. 2022, 7, 1580–1592. [Google Scholar] [CrossRef]
- Golicz, A.A.; Batley, J.; Edwards, D. Towards plant pangenomics. Plant Biotechnol. J. 2016, 14, 1099–1105. [Google Scholar] [CrossRef]
- Kille, B.; Balaji, A.; Sedlazeck, F.J.; Nute, M.; Treangen, T.J. Multiple genome alignment in the telomere-to-telomere assembly era. Genome Biol. 2022, 23, 182. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Z.; Tan, K.; Huang, W.; Shi, J.; Li, T.; Hu, J.; Wang, K.; Wang, C.; Xin, B.B.; et al. A complete telomere-to-telomere assembly of the maize genome. Nat. Genet. 2023, 55, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G.A.; Vollger, M.R.; Hsieh, P.; Mao, Y.; Liskovykh, M.A.; Koren, S.; Nurk, S.; Mercuri, L.; Dishuck, P.C.; Rhie, A.; et al. The structure, function and evolution of a complete human chromosome 8. Nature 2021, 593, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A.; et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zhang, F.; Li, S.; Luo, X.; Peng, L.; Dong, Z.; Pausch, H.; Leonard, A.S.; Crysnanto, D.; Wang, S.K.; et al. Structural variation and introgression from wild populations in East Asian cattle genomes confer adaptation to local environment. Genome Biol. 2023, 24, 211. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, C.; Xu, N.; Bian, P.; Tian, X.; Wang, X.; Wang, Y.Z.; Jia, X.Z.; Heller, R.; Wang, M.S.; et al. De Novo Assembly of 20 Chicken Genomes Reveals the Undetectable Phenomenon for Thousands of Core Genes on Microchromosomes and Subtelomeric Regions. Mol. Biol. Evol. 2022, 39, msac066. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Xing, W.J.; Li, G.H.; Zhou, C.H.; Zhang, H.Y.; Yin, J.Z.; Jiang, Y.X.; Zhu, Y.F.; Han, W. Assembly of high-quality Wenshang barred chickens genome based on PacBio third-generation sequencing. China Anim. Husb. Vet. Med. 2023, 50, 3869–3881. [Google Scholar]
- Bickhart, D.M.; Liu, G.E. The challenges and importance of structural variation detection in livestock. Front. Genet. 2014, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Deng, E.; Wang, J.; Zhou, W.; Ao, J.; Liu, R.; Su, D.; Fan, X.Y. Single-cell third-generation sequencing-based multi-omics uncovers gene expression changes governed by ecDNA and structural variants in cancer cells. Clin. Transl. Med. 2023, 13, e1351. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ma, L.; Liu, G.E. Initial Analysis of Structural Variation Detections in Cattle Using Long-Read Sequencing Methods. Genes 2022, 13, 828. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Liu, Y.Z.; Jiang, Y.; Li, J.Y.; Gao, Y.; Cui, Z.; Liu, Y.D.; Liu, B.; Wang, Y.D. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. 2020, 21, 189. [Google Scholar] [CrossRef] [PubMed]
- Stancu, M.C.; van Roosmalen, M.J.; Renkens, I.; Nieboer, M.M.; Middelkamp, S.; de Ligt, J.; Pregno, G.; Giachino, D.; Mandrile, G.; Valle-lnclan, J.E.; et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 2017, 8, 1326. [Google Scholar] [CrossRef] [PubMed]
- Tham, C.Y.; Tirado-Magallanes, R.; Goh, Y.; Fullwood, M.J.; Koh, B.T.H.; Wang, W.; Ng, C.H.; Chng, W.J.; Thiery, A.; Tenen, D.G.; et al. NanoVar: Accurate characterization of patients’ genomic structural variants using low-depth nanopore sequencing. Genome Biol. 2020, 21, 56. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Heller, D.; Vingron, M. SVIM: Structural variant identification using mapped long reads. Bioinformatics 2019, 35, 2907–2915. [Google Scholar] [CrossRef]
- Pös, O.; Radvanszky, J.; Buglyó, G.; Pös, Z.; Rusnakova, D.; Nagy, B.; Szemes, T. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef]
- Magini, P.; Mingrino, A.; Gega, B.; Mattei, G.; Semeraro, R.; Bolognini, D.; Mongelli, P.; Desiderio, L.; Pittalis, M.C.; Pippucci, T.; et al. Third-Generation Cytogenetic Analysis: Diagnostic Application of Long-Read Sequencing. J. Mol. Diagn. 2022, 24, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Lamb, H.J.; Ross, E.M.; Nguyen, L.T.; Lyons, R.E.; Moore, S.S.; Hayes, B.J. Characterization of the poll allele in Brahman cattle using long-read Oxford Nanopore sequencing. J. Anim. Sci. 2020, 98, skaa127. [Google Scholar] [CrossRef]
- Viluma, A.; Mikko, S.; Hahn, D.; Skow, L.; Andersson, G.; Bergstrom, T.F. Genomic structure of the horse major histocompatibility complex class II region resolved using PacBio long-read sequencing technology. Sci. Rep. 2017, 7, 45518. [Google Scholar] [CrossRef]
- Rice, E.S.; Alberdi, A.; Alfieri, J.; Athrey, G.; Balacco, J.R.; Bardou, P.; Blackmon, H.; Charles, M.; Cheng, H.H.; Fedrigo, O.; et al. A pangenome graph reference of 30 chicken genomes allows genotyping of large and complex structural variants. BMC Biol. 2023, 21, 267. [Google Scholar] [CrossRef]
- Sun, Y.H.; Cui, H.; Song, C.; Shen, J.T.; Zhuo, X.; Wang, R.H.; Yu, X.H.; Ndamba, R.; Mu, Q.; Gu, H.W.; et al. Amniotes co-opt intrinsic genetic instability to protect germ-line genome integrity. Nat. Commun. 2023, 14, 812. [Google Scholar] [CrossRef] [PubMed]
- Tilgner, H.; Grubert, F.; Sharon, D.; Snyder, M.P. Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. USA 2014, 111, 9869–9874. [Google Scholar] [CrossRef] [PubMed]
- Glinos, D.A.; Garborcauskas, G.; Hoffman, P.; Ehsan, N.; Jiang, L.H.; Gokden, A.; Dai, X.G.; Aguet, F.; Brown, K.L.; Garimella, K.; et al. Transcriptome variation in human tissues revealed by long-read sequencing. Nature 2022, 608, 353–359. [Google Scholar] [CrossRef]
- Ren, Y.; Tseng, E.; Smith, T.P.L.; Hiendleder, S.; Williams, J.L.; Low, W.Y. Long read isoform sequencing reveals hidden transcriptional complexity between cattle subspecies. BMC Genom. 2023, 24, 108. [Google Scholar] [CrossRef]
- Halstead, M.M.; Islas-Trejo, A.; Goszczynski, D.E.; Medrano, J.F.; Zhou, H.; Ross, P.J. Large-Scale Multiplexing Permits Full-Length Transcriptome Annotation of 32 Bovine Tissues From a Single Nanopore Flow Cell. Front. Genet. 2021, 12, 664260. [Google Scholar] [CrossRef]
- Liu, J.; Lang, K.; Tan, S.; Jie, W.; Zhu, Y.; Huang, S.; Huang, W. A web-based database server using 43,710 public RNA-seq samples for the analysis of gene expression and alternative splicing in livestock animals. BMC Genom. 2022, 23, 706. [Google Scholar] [CrossRef]
- Shu, Z.; Wang, L.; Wang, J.; Zhang, L.; Hou, X.; Yan, H.; Wang, L. Integrative Analysis of Nanopore and Illumina Sequencing Reveals Alternative Splicing Complexity in Pig Longissimus Dorsi Muscle. Front. Genet. 2022, 13, 877646. [Google Scholar] [CrossRef]
- Li, D.; Zhong, C.; Sun, Y.; Kang, L.; Jiang, Y. Identification of genes involved in chicken follicle selection by ONT sequencing on granulosa cells. Front. Genet. 2022, 13, 1090603. [Google Scholar] [CrossRef]
- Jin, Y.; Dong, H.; Shi, Y.; Bian, L. Mutually exclusive alternative splicing of pre-mRNAs. Wiley Interdiscip. Rev. RNA 2018, 9, e1468. [Google Scholar] [CrossRef]
- Lopez, A.J. Alternative splicing of pre-mRNA: Developmental consequences and mechanisms of regulation. Annu. Rev. Genet. 1998, 32, 279–305. [Google Scholar] [CrossRef]
- Peng, L.; Zhang, X.; Du, Y.; Li, F.; Han, J.; Liu, O.; Dai, S.L.; Zhang, X.; Liu, G.E.; Yang, L.G.; et al. New insights into transcriptome variation during cattle adipocyte adipogenesis by direct RNA sequencing. iScience 2023, 26, 107753. [Google Scholar] [CrossRef]
- Chang, T.; An, B.; Liang, M.; Duan, X.; Du, L.; Cai, W.; Zhu, B.; Gao, X.; Chen, Y.; Xu, L.Y.; et al. PacBio Single-Molecule Long-Read Sequencing Provides New Light on the Complexity of Full-Length Transcripts in Cattle. Front. Genet. 2021, 12, 664974. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Ge, L.; Zhang, W.; Lv, X.; Wang, S.; Cao, X.; Sun, W. Preliminary Results about Lamb Meat Tenderness Based on the Study of Novel Isoforms and Alternative Splicing Regulation Pathways Using Iso-seq, RNA-seq and CTCF ChIP-seq Data. Foods 2022, 11, 1068. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Halstead, M.M.; Islas-Trejo, A.D.; Goszczynski, D.E.; Cheng, H.H.; Ross, P.J.; Zhou, H. Prediction of transcript isoforms in 19 chicken tissues by Oxford Nanopore long-read sequencing. Front. Genet. 2022, 13, 997460. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Liu, Z.; Li, D.; Kang, L.; Jiang, Y. Long-read sequencing reveals the effect of follicle-stimulating hormone on the mRNA profile of chicken granulosa cells from prehierarchical follicles. Poult. Sci. 2023, 102, 102600. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Guan, L.; Ge, L.; Liu, G.; Bai, Y.; Liu, X. Nanopore-based full-length transcriptome sequencing of Muscovy duck (Cairina moschata) ovary. Poult. Sci. 2021, 100, 101246. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Ge, L.; Mei, X.; Niu, Y.; Chen, C.; Hou, S.; Liu, X. Integrated ONT Full-Length Transcriptome and Metabolism Reveal the Mechanism Affecting Ovulation in Muscovy Duck (Cairina moschata). Front. Vet. Sci. 2022, 9, 890979. [Google Scholar] [CrossRef]
- Yuan, Z.; Ge, L.; Sun, J.; Zhang, W.; Wang, S.; Cao, X.; Sun, W. Integrative analysis of Iso-Seq and RNA-seq data reveals transcriptome complexity and differentially expressed transcripts in sheep tail fat. PeerJ 2021, 9, e12454. [Google Scholar] [CrossRef]
- Zhang, H.; Liang, Y.; Chen, S.; Xuan, Z.; Jiang, Y.; Li, R.; Cao, Y. Full-length transcriptome sequencing reveals extreme incomplete annotation of the goat genome. Anim. Genet. 2023, 54, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Hajkova, P.; Ecker, J.R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef]
- Rand, A.C.; Jain, M.; Eizenga, J.M.; Musselman-Brown, A.; Olsen, H.E.; Akeson, M.; Paten, B. Mapping DNA methylation with high-throughput nanopore sequencing. Nat. Methods 2017, 14, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, R.; Xu, M.; Wang, X.; Zhou, B.; Wu, X.; Leone, V.; Chang, E.B.; Zhong, X. The regulatory role of N(6) -methyladenosine modification in the interaction between host and microbes. Wiley Interdiscip. Rev. RNA 2022, 13, e1725. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Munera, D.; Friedman, D.I.; Mandlik, A.; Chao, M.C.; Banerjee, O.; Feng, Z.X.; Losic, B.; Mahajan, M.C.; Jabado, O.J.; et al. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 2012, 30, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Mangiavacchi, P.M.; Caldas-Bussiere, M.C.; Mendonca, M.D.S.; Rumpf, R.; Lemos Junior, P.E.S.; Alves, C.S.; Carneiro, W.S.; Dias, A.J.B.; Riso, A.F.L. Multi-locus DNA methylation analysis of imprinted genes in cattle from somatic cell nuclear transfer. Theriogenology 2022, 186, 95–107. [Google Scholar] [CrossRef]
- Lee, I.; Razaghi, R.; Gilpatrick, T.; Molnar, M.; Gershman, A.; Sadowski, N.; Sedlazeck, F.J.; Hansen, K.D.; Simpson, J.T. Timp, W. Simultaneous profiling of chromatin accessibility and methylation on human cell lines with nanopore sequencing. Nat. Methods 2020, 17, 1191–1199. [Google Scholar] [CrossRef]
- Katsman, E.; Orlanski, S.; Martignano, F.; Fox-Fisher, I.; Shemer, R.; Dor, Y.; Zick, A.; Eden, A.; Petrini, L.; Conticello, S.G.; et al. Detecting cell-of-origin and cancer-specific methylation features of cell-free DNA from Nanopore sequencing. Genome Biol. 2022, 23, 158. [Google Scholar] [CrossRef]
- Dunin-Horkawicz, S.; Czerwoniec, A.; Gajda, M.J.; Feder, M.; Grosjean, H.; Bujnicki, J.M. MODOMICS: A database of RNA modification pathways. Nucleic Acids Res. 2006, 34, D145–D149. [Google Scholar] [CrossRef]
- Hengwei, Y.; Raza, S.H.A.; Wenzhen, Z.; Xinran, Y.; Almohaimeed, H.M.; Alshanwani, A.R.; Assiri, R.; Aggad, W.S.; Zan, L.S. Research progress of m(6)A regulation during animal growth and development. Mol. Cell. Probes 2022, 65, 101851. [Google Scholar] [CrossRef]
- Jenjaroenpun, P.; Wongsurawat, T.; Wadley, T.D.; Wassenaar, T.M.; Liu, J.; Dai, Q.; Wanchai, V.; Akel, N.S.; Jamshidi-Parsian, A.; Franco, A.T.; et al. Decoding the epitranscriptional landscape from native RNA sequences. Nucleic Acids Res. 2021, 49, e7. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Sun, B.; Liu, Q.; Cai, M.; Wu, R.; Wang, F.; Yao, Y.X.; Wang, Y.Z.; Wang, X.X. MTCH2 promotes adipogenesis in intramuscular preadipocytes via an m(6)A-YTHDF1-dependent mechanism. FASEB J. 2019, 33, 2971–2981. [Google Scholar] [CrossRef] [PubMed]
- Ibeagha-Awemu, E.M.; Kiefer, H.; McKay, S.; Liu, G.E. Editorial: Epigenetic Variation Influences on Livestock Production and Disease Traits. Front. Genet. 2022, 13, 942747. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Cowley, M.; Wetterstrand, K.; Watson, J.; Crick, F.; Sanger, F.; Nicklen, S.; Coulson, A.; Metzker, M.; Schadt, E.; et al. Next-Generation Sequencing and Emerging Technologies. Semin. Thromb. Hemost. 2019, 45, 661–673. [Google Scholar] [PubMed]
- Low, W.Y.; Tearle, R.; Liu, R.; Koren, S.; Rhie, A.; Bickhart, D.M.; Rosen, B.D.; Kronenberg, Z.N.; Kingan, S.B.; Tseng, E.; et al. Haplotype-resolved genomes provide insights into structural variation and gene content in Angus and Brahman cattle. Nat. Commun. 2020, 11, 2071. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admasu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Zheng, J.; Ding, J.; Wu, J.; Zuo, F.; Zhang, G. When Livestock Genomes Meet Third-Generation Sequencing Technology: From Opportunities to Applications. Genes 2024, 15, 245. https://doi.org/10.3390/genes15020245
Liu X, Zheng J, Ding J, Wu J, Zuo F, Zhang G. When Livestock Genomes Meet Third-Generation Sequencing Technology: From Opportunities to Applications. Genes. 2024; 15(2):245. https://doi.org/10.3390/genes15020245
Chicago/Turabian StyleLiu, Xinyue, Junyuan Zheng, Jialan Ding, Jiaxin Wu, Fuyuan Zuo, and Gongwei Zhang. 2024. "When Livestock Genomes Meet Third-Generation Sequencing Technology: From Opportunities to Applications" Genes 15, no. 2: 245. https://doi.org/10.3390/genes15020245
APA StyleLiu, X., Zheng, J., Ding, J., Wu, J., Zuo, F., & Zhang, G. (2024). When Livestock Genomes Meet Third-Generation Sequencing Technology: From Opportunities to Applications. Genes, 15(2), 245. https://doi.org/10.3390/genes15020245