
DNA Damage, Genome Stability, and Adaptation: A Question of Chance or Necessity?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
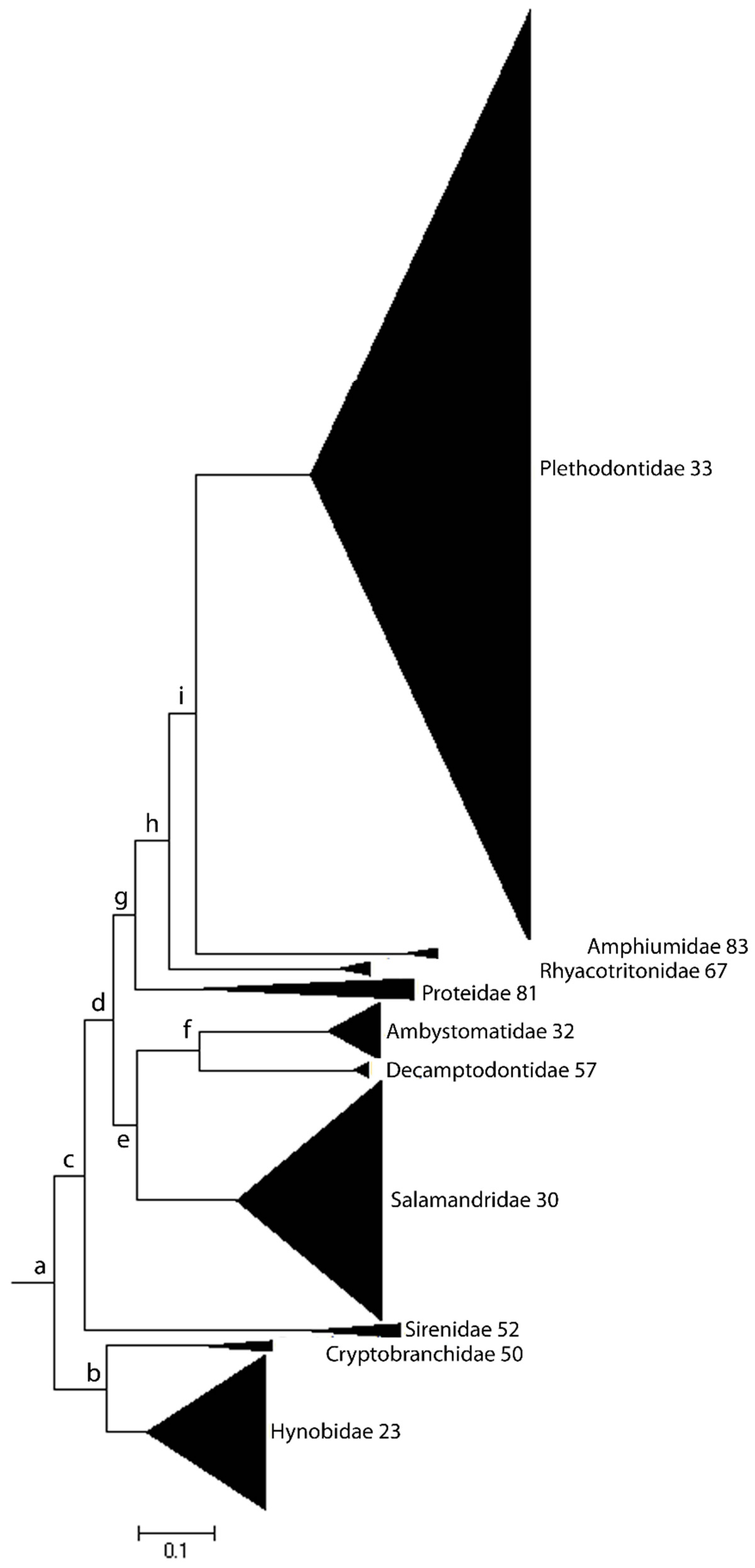
1.1. Karyotype Diversity and Species Richness
1.2. Framing the Question: What Is the Role of Genome Stability in Karyotype Evolution and Species Diversity?
2. Non-Adaptive Radiation: Ecological Selection vs. Genetic Drift
- (1)
- Relaxation of a selective constraint (a weakened negative, or purifying, selection) resulting in a burst in the number of new gene and genotype variants;
- (2)
- Differential fixation of variants in a population, or subpopulations, under the force of genetic drift;
- (3)
- Rapid habitat-driven diversification into new niches and environments (ecological selection);
- (4)
- Competitive exclusion between related groups leading to extensive adaptive evolution and radically different taxa following successful adaptation to new ecological niches.
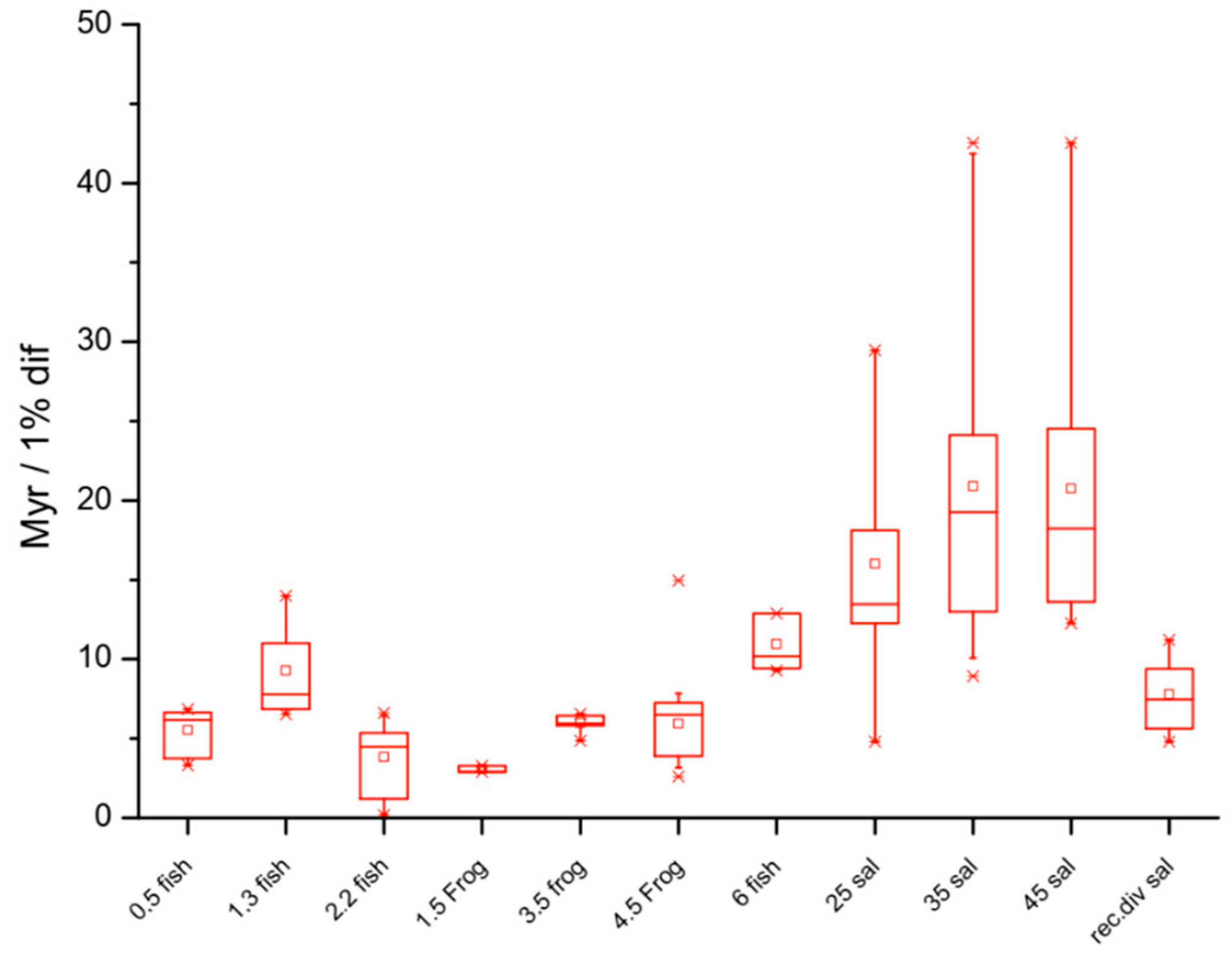
3. Genome Stability and Rates of Speciation: Karyotype Diversity versus Gene Diversity in Determining Species Richness
4. DNA Damage Detection and Repair Systems (DDR) and Chromatin Structure
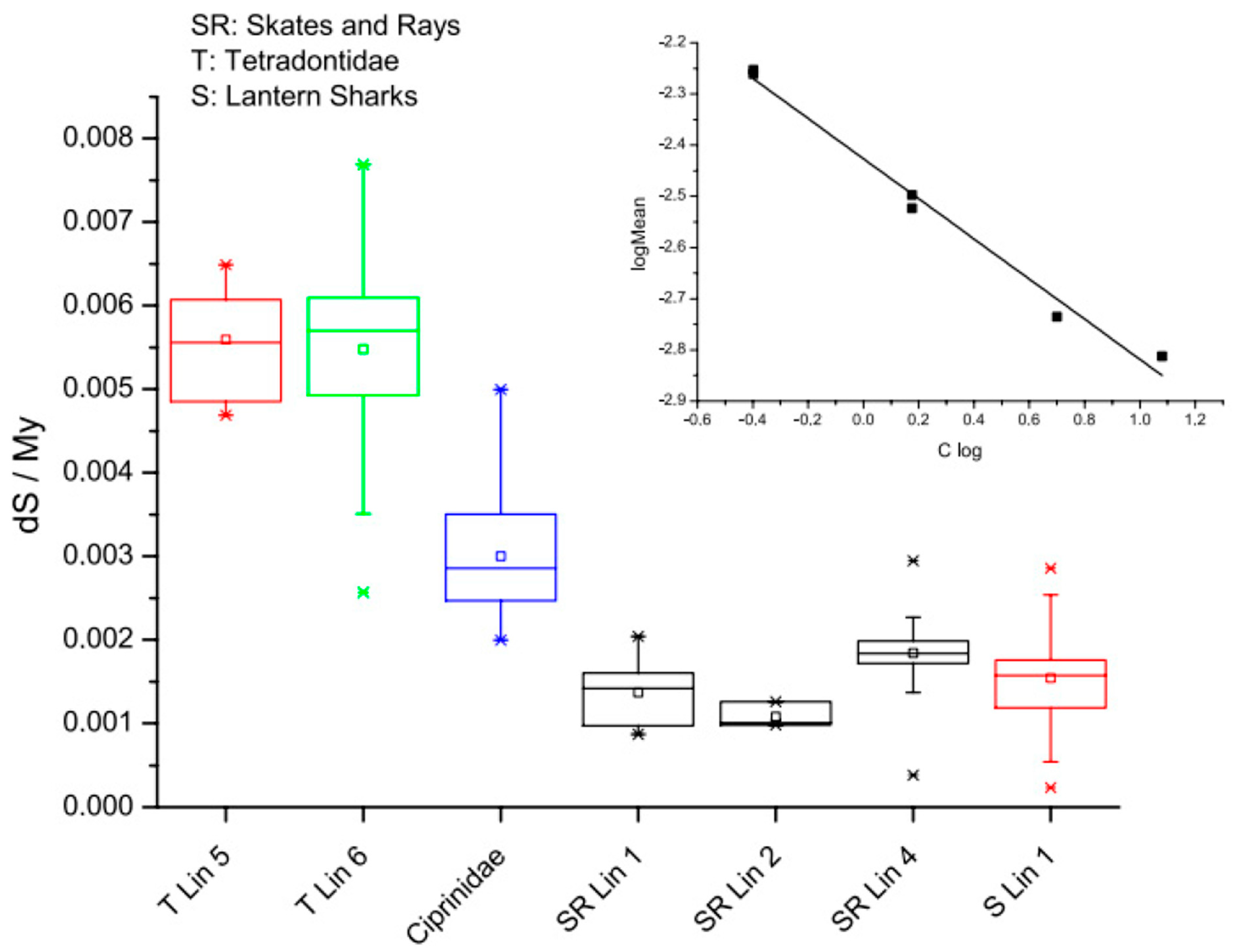
5. Mutation–Substitution Balance and Replication Timing
6. Life History Traits and the DDR System: Limb Regeneration and Maximum Lifespan
7. Life History Traits and the DDR System: Cellular Differentiation and Development
8. Life History Traits and the DDR system: Sharks, Salamanders, and Resistance to Genetic Diseases
9. Discussion
10. Does Evolution Proceed by Repeated Cycles of Genetic Drift and Ecological Speciation?
11. Conclusions
Funding
Conflicts of Interest
References
- Soltis, P.S.; Folk, R.A.; Soltis, D.E. Darwin review: Angiosperm phylogeny and evolutionary radiations. Proc. R. Soc. B Biol. Sci. 2019, 286, 20190099. [Google Scholar] [CrossRef]
- Lynch, M. Streamlining and Simplification of Microbial Genome Architecture. Annu. Rev. Microbiol. 2006, 60, 327–349. [Google Scholar] [CrossRef]
- Pellicer, J.; Hidalgo, O.; Dodsworth, S.; Leitch, I.J. Genome Size Diversity and Its Impact on the Evolution of Land Plants. Genes 2018, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Pellicer, J.; Fay, M.F.; Leitch, I.J. The largest eukaryotic genome of them all? Bot. J. Linn. Soc. 2010, 164, 10–15. [Google Scholar] [CrossRef]
- Rodríguez-Gijón, A.; Buck, M.; Andersson, A.F.; Izabel-Shen, D.; A Nascimento, F.J.; Garcia, S.L. Linking prokaryotic genome size variation to metabolic potential and environment. ISME Commun. 2023, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, O.; Pellicer, J.; Christenhusz, M.; Schneider, A.; RLeitch, A.R.; Leitch, I.J. Is There an Upper Limit to Genome Size? Trends Plant Sci. 2017, 22, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Prachumwat, A.; Li, W.-H. Gene number expansion and contraction in vertebrate genomes with respect to invertebrate genomes. Genome Res. 2008, 18, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Demuth, J.P.; De Bie, T.; Stajich, J.E.; Cristianini, N.; Hahn, M.W. The Evolution of Mammalian Gene Families. PLoS ONE 2006, 1, e85. [Google Scholar] [CrossRef] [PubMed]
- Damas, J.; Corbo, M.; Kim, J.; Turner-Maier, J.; Farré, M.; Larkin, D.M.; Ryder, O.A.; Steiner, C.; Houck, M.L.; Hall, S.; et al. Evolution of the ancestral mammalian karyotype and syntenic regions. Proc. Natl. Acad. Sci. USA 2022, 119, e2209139119. [Google Scholar] [CrossRef]
- Graphodatsky, A.S.; A Trifonov, V.; Stanyon, R. The genome diversity and karyotype evolution of mammals. Mol. Cytogenet. 2011, 4, 22. [Google Scholar] [CrossRef]
- Gregory, T.R. The C-value Enigma in Plants and Animals: A Review of Parallels and an Appeal for Partnership. Ann. Bot. 2005, 95, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Puttick, M.N.; Clark, J.; Donoghue, P.C. Size is not everything: Rates of genome size evolution, not C-value, correlate with speciation in angiosperms. Proc. R. Soc. B Biol. Sci. 2015, 282, 20152289. [Google Scholar] [CrossRef] [PubMed]
- Voss, S.R.; Kump, D.K.; Putta, S.; Pauly, N.; Reynolds, A.; Henry, R.J.; Basa, S.; Walker, J.A.; Smith, J.J. Origin of amphibian and avian chromosomes by fission, fusion, and retention of ancestral chromosomes. Genome Res. 2011, 21, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ali, M.; Zhou, Q. Establishment and evolution of heterochromatin. Ann. N. Y. Acad. Sci. 2020, 1476, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. The neutral theory of molecular evolution: A review of recent evidence. Jpn. J. Genet. 1991, 66, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Schluter, D.; Conte, G.L. Genetics and ecological speciation. Proc. Natl. Acad. Sci. USA 2009, 106 (Suppl. S1), 9955–9962. [Google Scholar] [CrossRef] [PubMed]
- Ravinet, M.; Faria, R.; Butlin, R.K.; Galindo, J.; Bierne, N.; Rafajlović, M.; Noor, M.A.F.; Mehlig, B.; Westram, A.M. Interpreting the genomic landscape of speciation: A road map for finding barriers to gene flow. J. Evol. Biol. 2017, 30, 1450–1477. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.H.; Richards, E.J. The Paradox Behind the Pattern of Rapid Adaptive Radiation: How Can the Speciation Process Sustain Itself Through an Early Burst? Annu. Rev. Ecol. Evol. Syst. 2019, 50, 569–593. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, R.G.; Bennett, G.M.; De Meester, L.; Feder, J.L.; Fleischer, R.C.; Harmon, L.J.; Hendry, A.P.; Knope, M.L.; Mallet, J.; Martin, C.; et al. Comparing Adaptive Radiations Across Space, Time, and Taxa. J. Hered. 2020, 111, 1–20. [Google Scholar] [CrossRef]
- Czekanski-Moir, J.E.; Rundell, R.J. The Ecology of Nonecological Speciation and Nonadaptive Radiations. Trends Ecol. Evol. 2019, 34, 400–415. [Google Scholar] [CrossRef]
- Schenk, J.J. The Next Generation of Adaptive Radiation Studies in Plants. Int. J. Plant Sci. 2021, 182, 245–262. [Google Scholar] [CrossRef]
- Kozak, K.H.; Weisrock, D.W.; Larson, A. Rapid lineage accumulation in a non-adaptive radiation: Phylogenetic analysis of diversification rates in eastern North American woodland salamanders (Plethodontidae: Plethodon). Proc. R. Soc. B Biol. Sci. 2006, 273, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Murrell, B.; Smith, M.D.; Pond, S.L.K.; Scheffler, K. RELAX: Detecting Relaxed Selection in a Phylogenetic Framework. Mol. Biol. Evol. 2015, 32, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Hunt, B.G.; Ometto, L.; Wurm, Y.; Shoemaker, D.; Yi, S.V.; Keller, L.; Goodisman, M.A.D. Relaxed selection is a precursor to the evolution of phenotypic plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 15936–15941. [Google Scholar] [CrossRef] [PubMed]
- Persi, E.; Wolf, Y.I.; Koonin, E.V. Positive and strongly relaxed purifying selection drive the evolution of repeats in proteins. Nat. Commun. 2016, 7, 13570. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Conery, J.S. The Origins of Genome Complexity. Science 2003, 302, 1401–1404. [Google Scholar] [CrossRef] [PubMed]
- Fuselli, S.; Greco, S.; Biello, R.; Palmitessa, S.; Lago, M.; Meneghetti, C.; McDougall, C.; Trucchi, E.; Stabelli, O.R.; Biscotti, A.M.; et al. Relaxation of Natural Selection in the Evolution of the Giant Lungfish Genomes. Mol. Biol. Evol. 2023, 40, msad193. [Google Scholar] [CrossRef] [PubMed]
- Mohlhenrich, E.R.; Mueller, R.L. Genetic drift and mutational hazard in the evolution of salamander genomic gigantism. Evolution 2016, 70, 2865–2878. [Google Scholar] [CrossRef] [PubMed]
- Ai, B.; Wang, Z.-S.; Ge, S. Genome size is not correlated with effective population size in the oryzas pecies. Evolution 2012, 66, 3302–3310. [Google Scholar] [CrossRef]
- Whitney, K.D.; Baack, E.J.; Hamrick, J.L.; Godt, M.J.W.; Barringer, B.C.; Bennett, M.D.; Eckert, C.G.; Goodwillie, C.; Kalisz, S.; Leitch, I.J.; et al. A role for nonadaptive processes in plant genome size evolution? Evolution 2010, 64, 2097–2109. [Google Scholar] [CrossRef]
- Blommaert, J. Genome size evolution: Towards new model systems for old questions. Proc. R. Soc. B Biol. Sci. 2020, 287, 20201441. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, Y.; Boissinot, S. On the Population Dynamics of Junk: A Review on the Population Genomics of Transposable Elements. Genes 2019, 10, 419. [Google Scholar] [CrossRef]
- Llaurens, V.; Whibley, A.; Joron, M. Genetic architecture and balancing selection: The life and death of differentiated variants. Mol. Ecol. 2017, 26, 2430–2448. [Google Scholar] [CrossRef]
- Ding, G.; Hasselmann, M.; Huang, J.; Roberts, J.; Oldroyd, B.P.; Gloag, R. Global allele polymorphism indicates a high rate of allele genesis at a locus under balancing selection. Heredity 2021, 126, 163–177. [Google Scholar] [CrossRef]
- Avise, J.C. Genic heterozygosity and rate of speciation. Paleobiology 1977, 3, 422–432. [Google Scholar] [CrossRef]
- Charlesworth, D. Balancing Selection and Its Effects on Sequences in Nearby Genome Regions. PLoS Genet. 2006, 2, e64. [Google Scholar] [CrossRef] [PubMed]
- Bitarello, B.D.; Brandt, D.Y.C.; Meyer, D.; Andrés, A.M. Inferring Balancing Selection From Genome-Scale Data. Genome Biol. Evol. 2023, 15, evad032. [Google Scholar] [CrossRef]
- Harvey, M.G.; Seeholzer, G.F.; Smith, B.T.; Rabosky, D.L.; Cuervo, A.M.; Brumfield, R.T. Positive association between population genetic differentiation and speciation rates in New World birds. Proc. Natl. Acad. Sci. USA 2017, 114, 6328–6333. [Google Scholar] [CrossRef]
- Mani, G.S.; Clarke, B.C. Mutational order: A major stochastic process in evolution. Proc. R. Soc. London. Ser. B. Biol. Sci. 1990, 240, 29–37. [Google Scholar] [CrossRef]
- Uyeda, J.C.; Hansen, T.F.; Arnold, S.J.; Pienaar, J. The million-year wait for macroevolutionary bursts. Proc. Natl. Acad. Sci. USA 2011, 108, 15908–15913. [Google Scholar] [CrossRef]
- Folk, R.A.; Stubbs, R.L.; Mort, M.E.; Cellinese, N.; Allen, J.M.; Soltis, P.S.; Soltis, D.E.; Guralnick, R.P. Rates of niche and phenotype evolution lag behind diversification in a temperate radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 10874–10882. [Google Scholar] [CrossRef] [PubMed]
- Aguilée, R.; Gascuel, F.; Lambert, A.; Ferriere, R. Clade diversification dynamics and the biotic and abiotic controls of speciation and extinction rates. Nat. Commun. 2018, 9, 3013. [Google Scholar] [CrossRef] [PubMed]
- Rezazadegan, R.; Reidys, C. Degeneracy and genetic assimilation in RNA evolution. BMC Bioinform. 2018, 19, 543. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, V.; Greenbury, S.F.; Sarkany, T.; Narayanan, S.; Dingle, K.; Ahnert, S.E.; Louis, A.A. Maximum mutational robustness in genotype—Phenotype maps follows a self-similar blancmange-like curve. J. R. Soc. Interface 2023, 20, 20230169. [Google Scholar] [CrossRef] [PubMed]
- Manrubia, S.; Cuesta, J.A.; Aguirre, J.; Ahnert, S.E.; Altenberg, L.; Cano, A.V.; Catalán, P.; Diaz-Uriarte, R.; Elena, S.F.; García-Martín, J.A.; et al. From genotypes to organisms: State-of-the-art and perspectives of a cornerstone in evolutionary dynamics. Phys. Life Rev. 2021, 38, 55–106. [Google Scholar] [CrossRef] [PubMed]
- Whitacre, J.M.; Atamas, S.P. Degeneracy allows for both apparent homogeneity and diversification in populations. Biosystems 2012, 110, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Paaby, A.B.; Rockman, M.V. Cryptic genetic variation: Evolution’s hidden substrate. Nat. Rev. Genet. 2014, 15, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Wideman, J.G.; Novick, A.; A Muñoz-Gómez, S.; Doolittle, W.F. Neutral evolution of cellular phenotypes. Curr. Opin. Genet. Dev. 2019, 58–59, 87–94. [Google Scholar] [CrossRef]
- Maleszka, R.; Mason, P.H.; Barron, A.B. Epigenomics and the concept of degeneracy in biological systems. Briefings Funct. Genom. 2014, 13, 191–202. [Google Scholar] [CrossRef]
- Milosavljevic, A. Emerging patterns of epigenomic variation. Trends Genet. 2011, 27, 242–250. [Google Scholar] [CrossRef]
- Bertucci-Richter, E.M.; Parrott, B.B. The rate of epigenetic drift scales with maximum lifespan across mammals. Nat. Commun. 2023, 14, 7731. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, D.; Barton, N.H.; Charlesworth, B. The sources of adaptive variation. Proc. R. Soc. B Biol. Sci. 2017, 284, 20162864. [Google Scholar] [CrossRef] [PubMed]
- Afanasyeva, A.; Bockwoldt, M.; Cooney, C.R.; Heiland, I.; Gossmann, T.I. Human long intrinsically disordered protein regions are frequent targets of positive selection. Genome Res. 2018, 28, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.C.; Sarich, V.M.; Maxson, L.R. The Importance of Gene Rearrangement in Evolution: Evidence from Studies on Rates of Chromosomal, Protein, and Anatomical Evolution. Proc. Natl. Acad. Sci. USA 1974, 71, 3028–3030. [Google Scholar] [CrossRef] [PubMed]
- Bush, G.L.; Case, S.M.; Wilson, A.C.; Patton, J.L. Rapid speciation and chromosomal evolution in mammals. Proc. Natl. Acad. Sci. USA 1977, 74, 3942–3946. [Google Scholar] [CrossRef] [PubMed]
- Maxson, L.E.R.; Wilson, A.C. Rates of molecular and chromosomal evolution in salamanders. Evolution 1979, 33, 734–740. [Google Scholar] [CrossRef]
- Coyne, J.A. Correlation between Heterozygosity and Rate of Chromosome Evolution in Animals. Am. Nat. 1984, 123, 725–729. [Google Scholar] [CrossRef]
- Lynch, M.; Ali, F.; Lin, T.; Wang, Y.; Ni, J.; Long, H. The divergence of mutation rates and spectra across the Tree of Life. Embo Rep. 2023, 24, e57561. [Google Scholar] [CrossRef]
- Roddy, A.B.; Alvarez-Ponce, D.; Roy, S.W. Mammals with Small Populations Do Not Exhibit Larger Genomes. Mol. Biol. Evol. 2021, 38, 3737–3741. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Li, Z.; Van de Peer, Y.; Ingvarsson, P.K. Contrasting Rates of Molecular Evolution and Patterns of Selection among Gymnosperms and Flowering Plants. Mol. Biol. Evol. 2017, 34, 1363–1377. [Google Scholar] [CrossRef]
- Bengtsson, B.O. Rates of karyotype evolution in placental mammals. Hereditas 1980, 92, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Schoch, R.R.; Werneburg, R.; Voigt, S. A Triassic stem-salamander from Kyrgyzstan and the origin of salamanders. Proc. Natl. Acad. Sci. USA 2020, 117, 11584–11588. [Google Scholar] [CrossRef] [PubMed]
- Portik, D.M.; Streicher, J.W.; Wiens, J.J. Frog phylogeny: A time-calibrated, species-level tree based on hundreds of loci and 5242 species. Mol. Phylogenet. Evol. 2023, 188, 107907. [Google Scholar] [CrossRef] [PubMed]
- Hunter, P. The rise of the mammals: Fossil discoveries combined with dating advances give insight into the great mammal expansion. EMBO Rep. 2020, 21, e51617. [Google Scholar] [CrossRef] [PubMed]
- Bredeson, J.V.; Mudd, A.B.; Medina-Ruiz, S.; Mitros, T.; Smith, O.K.; Miller, K.E.; Lyons, J.B.; Batra, S.S.; Park, J.; Berkoff, K.C.; et al. Conserved chromatin and repetitive patterns reveal slow genome evolution in frogs. Nat. Commun. 2024, 15, 579. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, H.C.; Gower, D.J.; Wilkinson, M.; Gomez-Mestre, I. Macroevolutionary shift in the size of amphibian genomes and the role of life history and climate. Nat. Ecol. Evol. 2018, 2, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Rundell, R.J.; Price, T.D. Adaptive radiation, nonadaptive radiation, ecological speciation and nonecological speciation. Trends Ecol. Evol. 2009, 24, 394–399. [Google Scholar] [CrossRef]
- Janssen, A.; Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Biol. 2018, 34, 265–288. [Google Scholar] [CrossRef]
- Zylicz, J.J.; Heard, E. Molecular Mechanisms of Facultative Heterochromatin Formation: An X-Chromosome Perspective. Annu. Rev. Biochem. 2020, 89, 255–282. [Google Scholar] [CrossRef]
- Nakatani, T.; Schauer, T.; Altamirano-Pacheco, L.; Klein, K.N.; Ettinger, A.; Pal, M.; Gilbert, D.M.; Torres-Padilla, M.-E. Emergence of replication timing during early mammalian development. Nature 2024, 625, 401–409. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tyler, J.K. The Chromatin Landscape Channels DNA Double-Strand Breaks to Distinct Repair Pathways. Front. Cell Dev. Biol. 2022, 10, 909696. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, E.; Hochegger, H.; Saberi, A.; Taniguchi, Y.; Takeda, S. Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair 2006, 5, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E. Intron—Genome Size Relationship on a Large Evolutionary Scale. J. Mol. Evol. 1999, 49, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Farlow, A.; Meduri, E.; Schlötterer, C. DNA double-strand break repair and the evolution of intron density. Trends Genet. 2011, 27, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kin, C.; Dmitry, A. Gordenin Clusters of Multiple Mutations: Incidence and Molecular Mechanisms. Annu. Rev. Genet. 2015, 49, 243–267. [Google Scholar] [CrossRef]
- Stamatoyannopoulos, J.A.; Adzhubei, I.; Thurman, R.E.; Kryukov, G.V.; Mirkin, S.M.; Sunyaev, S.R. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009, 41, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.I.; Murray, A.W. Mutation Rates across Budding Yeast Chromosome VI Are Correlated with Replication Timing. Genome Biol. Evol. 2011, 3, 799–811. [Google Scholar] [CrossRef]
- Pink, C.J.; Hurst, L.D. Timing of Replication Is a Determinant of Neutral Substitution Rates but Does Not Explain Slow Y Chromosome Evolution in Rodents. Mol. Biol. Evol. 2009, 27, 1077–1086. [Google Scholar] [CrossRef]
- Weber, C.C.; Pink, C.J.; Hurst, L.D. Late-Replicating Domains Have Higher Divergence and Diversity in Drosophila melanogaster. Mol. Biol. Evol. 2012, 29, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Agier, N.; Fischer, G. The Mutational Profile of the Yeast Genome Is Shaped by Replication. Mol. Biol. Evol. 2012, 29, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Rappailles, A.; Duquenne, L.; Huvet, M.; Guilbaud, G.; Farinelli, L.; Audit, B.; D’Aubenton-Carafa, Y.; Arneodo, A.; Hyrien, O.; et al. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 2010, 20, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Murat, P.; Perez, C.; Crisp, A.; van Eijk, P.; Reed, S.H.; Guilbaud, G.; Sale, J.E. DNA replication initiation shapes the mutational landscape and expression of the human genome. Sci. Adv. 2022, 8, eadd3686. [Google Scholar] [CrossRef] [PubMed]
- Bomblies, K.; Peichel, C.L. Genetics of adaptation. Proc. Natl. Acad. Sci. USA 2022, 119, e2122152119. [Google Scholar] [CrossRef] [PubMed]
- Bracci, A.N.; Dallmann, A.; Ding, Q.; Hubisz, M.J.; Caballero, M.; Koren, A. The evolution of the human DNA replication timing program. Proc. Natl. Acad. Sci. USA 2023, 120, e2213896120. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-I.; Ting, C.-T. Genes and speciation. Nat. Rev. Genet. 2004, 5, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.H.; Li, H. Functional Bias and Spatial Organization of Genes in Mutational Hot and Cold Regions in the Human Genome. PLoS Biol. 2004, 2, e29. [Google Scholar] [CrossRef] [PubMed]
- Monroe, J.G.; Srikant, T.; Carbonell-Bejerano, P.; Becker, C.; Lensink, M.; Exposito-Alonso, M.; Klein, M.; Hildebrandt, J.; Neumann, M.; Kliebenstein, D.; et al. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 2022, 602, 101–105. [Google Scholar] [CrossRef]
- Cohn, M.; Mitchison, N.A.; Paul, W.E.; Silverstein, A.M.; Talmage, D.W.; Weigert, M. Reflections on the clonal-selection theory. Nat. Rev. Immunol. 2007, 7, 823–830. [Google Scholar] [CrossRef]
- Heyn, P.; Kalinka, A.T.; Tomancak, P.; Neugebauer, K.M. Introns and gene expression: Cellular constraints, transcriptional regulation, and evolutionary consequences. BioEssays 2015, 37, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Chakra, M.A.; Isserlin, R.; Tran, T.N.; Bader, G.D. Control of tissue development and cell diversity by cell cycle-dependent transcriptional filtering. eLife 2021, 10, e64951. [Google Scholar] [CrossRef] [PubMed]
- Sessions, S.K.; Wake, D.B. Forever young: Linking regeneration and genome size in salamanders. Dev. Dyn. 2021, 250, 768–778. [Google Scholar] [CrossRef]
- Joven, A.; Elewa, A.; Simon, A. Model systems for regeneration: Salamanders. Development 2019, 146, dev167700. [Google Scholar] [CrossRef]
- Sousounis, K.; Bryant, D.M.; Fernandez, J.M.; Eddy, S.S.; Tsai, S.L.; Gundberg, G.C.; Han, J.; Courtemanche, K.; Levin, M.; Whited, J.L. Eya2 promotes cell cycle progression by regulating DNA damage response during vertebrate limb regeneration. eLife 2020, 9, e51217. [Google Scholar] [CrossRef]
- Lemaitre, J.-M.; Danis, E.; Pasero, P.; Vassetzky, Y.; Méchali, M. Mitotic Remodeling of the Replicon and Chromosome Structure. Cell 2005, 123, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Gregory, T.R. Genome size and developmental complexity. Genetica 2002, 115, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Mueller, R.L.; E Cressler, C.; Schwartz, R.S.; A Chong, R.; A Butler, M. Metamorphosis Imposes Variable Constraints on Genome Expansion through Effects on Development. Integr. Org. Biol. 2023, 5, obad015. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat. Res. Mol. Mech. Mutagen. 2019, 780, 37–47. [Google Scholar] [CrossRef]
- Tian, X.; Firsanov, D.; Zhang, Z.; Cheng, Y.; Luo, L.; Tombline, G.; Tan, R.; Simon, M.; Henderson, S.; Steffan, J.; et al. SIRT6 Is Responsible for More Efficient DNA Double-Strand Break Repair in Long-Lived Species. Cell 2019, 177, 622–638.e22. [Google Scholar] [CrossRef]
- Crofts, S.J.C.; Latorre-Crespo, E.; Chandra, T. DNA methylation rates scale with maximum lifespan across mammals. Nat. Aging 2024, 4, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Pértille, F.; Da Silva, V.H.; Johansson, A.M.; Lindström, T.; Wright, D.; Coutinho, L.L.; Jensen, P.; Guerrero-Bosagna, C. Mutation dynamics of CpG dinucleotides during a recent event of vertebrate diversification. Epigenetics 2019, 14, 685–707. [Google Scholar] [CrossRef] [PubMed]
- Venney, C.J.; Anastasiadi, D.; Wellenreuther, M.; Bernatchez, L. The Evolutionary Complexities of DNA Methylation in Animals: From Plasticity to Genetic Evolution. Genome Biol. Evol. 2023, 15, evad216. [Google Scholar] [CrossRef] [PubMed]
- Sjakste, N.; Riekstiņa, U. DNA damage and repair in differentiation of stem cells and cells of connective cell lineages: A trigger or a complication? Eur. J. Histochem. 2021, 65, 3236. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Bassing, C.H.; Teitell, M.A. Regulation of cell differentiation by the DNA damage response. Trends Cell Biol. 2011, 21, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Meier, P.; Finch, A.; Evan, G. Apoptosis in development. Nature 2000, 407, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kothari, P.; Mullins, M.; Lampson, M.A. Regulation of zygotic genome activation and DNA damage checkpoint acquisition at the mid-blastula transition. Cell Cycle 2014, 13, 3828–3838. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.A.; Shermoen, A.W.; Yuan, K.; O’Farrell, P.H. Embryonic onset of late replication requires Cdc25 down-regulation. Genes Dev. 2012, 26, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Jockusch, E.L.; Kruuk, L.E.B.; Gilchrist, J.S. An evolutionary correlate of genome size change in plethodontid salamanders. Proc. R. Soc. B Biol. Sci. 1997, 264, 597–604. [Google Scholar] [CrossRef]
- Jablonski, D. Developmental bias, macroevolution, and the fossil record. Evol. Dev. 2020, 22, 103–125. [Google Scholar] [CrossRef]
- Arthur, W. The emerging conceptual framework of evolutionary developmental biology. Nature 2002, 415, 757–764. [Google Scholar] [CrossRef]
- Uesaka, M.; Kuratani, S.; Irie, N. The developmental hourglass model and recapitulation: An attempt to integrate the two models. J. Exp. Zool. Part B Mol. Dev. Evol. 2022, 338, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, C.D.; MacGuigan, D.J.; Kim, D.; Orr, O.; Yang, L.; David, S.R.; Kreiser, B.; Near, T.J. The genomic signatures of evolutionary stasis. Evolution 2024, qpae028. [Google Scholar] [CrossRef] [PubMed]
- Herrick, J. Genetic variation and dna replication timing, or why is there late replicating dna? Evolution 2011, 65, 3031–3047. [Google Scholar] [CrossRef] [PubMed]
- Sclavi, B.; Herrick, J. Slow Evolution of rag1 and pomc Genes in Vertebrates with Large Genomes. Submitted on 9 February 2013. Available online: https://arxiv.org/ftp/arxiv/papers/1302/1302.2182.pdf (accessed on 17 March 2024).
- Sendell-Price, A.T.; Tulenko, F.J.; Pettersson, M.; Kang, D.; Montandon, M.; Winkler, S.; Kulb, K.; Naylor, G.P.; Phillippy, A.; Fedrigo, O.; et al. Low mutation rate in epaulette sharks is consistent with a slow rate of evolution in sharks. Nat. Commun. 2023, 14, 6628. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J. From DNA damage to mutations: All roads lead to aging. Ageing Res. Rev. 2021, 68, 101316. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, M.; Yamashita, Y.M. Defective Satellite DNA Clustering into Chromocenters Underlies Hybrid Incompatibility in Drosophila. Mol. Biol. Evol. 2021, 38, 4977–4986. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.; Peona, V.; Guichard, E.; Taccioli, C.; Boattini, A. Transposable Elements Activity is Positively Related to Rate of Speciation in Mammals. J. Mol. Evol. 2018, 86, 303–310. [Google Scholar] [CrossRef]
- Quadrana, L.; Etcheverry, M.; Gilly, A.; Caillieux, E.; Madoui, M.-A.; Guy, J.; Silveira, A.B.; Engelen, S.; Baillet, V.; Wincker, P.; et al. Transposition favors the generation of large effect mutations that may facilitate rapid adaption. Nat. Commun. 2019, 10, 3421. [Google Scholar] [CrossRef]
- Baduel, P.; Quadrana, L.; Hunter, B.; Bomblies, K.; Colot, V. Relaxed purifying selection in autopolyploids drives transposable element over-accumulation which provides variants for local adaptation. Nat. Commun. 2019, 10, 5818. [Google Scholar] [CrossRef]
- Zeh, D.W.; Zeh, J.A.; Ishida, Y. Transposable elements and an epigenetic basis for punctuated equilibria. BioEssays 2009, 31, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.E.; Hawley, R.S. Heterochromatin: A Rapidly Evolving Species Barrier. PLOS Biol. 2009, 7, e1000233. [Google Scholar] [CrossRef] [PubMed]
- Clouaire, T.; Legube, G. DNA double strand break repair pathway choice: A chromatin based decision? Nucleus 2015, 6, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Krenning, L.; Berg, J.v.D.; Medema, R.H. Life or Death after a Break: What Determines the Choice? Mol. Cell 2019, 76, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Halliday, T.J.D.; dos Reis, M.; Tamuri, A.U.; Ferguson-Gow, H.; Yang, Z.; Goswami, A. Rapid morphological evolution in placental mammals post-dates the origin of the crown group. Proc. R. Soc. B Biol. Sci. 2019, 286, 20182418. [Google Scholar] [CrossRef] [PubMed]
- Budd, G.E.; Mann, R.P. The dynamics of stem and crown groups. Sci. Adv. 2020, 6, eaaz1626. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Molecular clocks: Four decades of evolution. Nat. Rev. Genet. 2005, 6, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.C.; Gordon, R. Differentiation trees, a junk DNA molecular clock, and the evolution of neoteny in salamanders. J. Evol. Biol. 1995, 8, 339–354. [Google Scholar] [CrossRef]
- Bock, D.G.; Cai, Z.; Elphinstone, C.; González-Segovia, E.; Hirabayashi, K.; Huang, K.; Keais, G.L.; Kim, A.; Owens, G.L.; Rieseberg, L.H. Genomics of plant speciation. Plant Commun. 2023, 4, 100599. [Google Scholar] [CrossRef]
- Wolfsberger, W.W.; Battistuzzi, F.U.; Oleksyk, T.K. Genomics of Adaptation and Speciation. Genes 2022, 13, 1187. [Google Scholar] [CrossRef]
- Campbell, C.R.; Poelstra, J.W.; Yoder, A.D. What is Speciation Genomics? The roles of ecology, gene flow, and genomic architecture in the formation of species. Biol. J. Linn. Soc. 2018, 124, 561–583. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrick, J. DNA Damage, Genome Stability, and Adaptation: A Question of Chance or Necessity? Genes 2024, 15, 520. https://doi.org/10.3390/genes15040520
Herrick J. DNA Damage, Genome Stability, and Adaptation: A Question of Chance or Necessity? Genes. 2024; 15(4):520. https://doi.org/10.3390/genes15040520
Chicago/Turabian StyleHerrick, John. 2024. "DNA Damage, Genome Stability, and Adaptation: A Question of Chance or Necessity?" Genes 15, no. 4: 520. https://doi.org/10.3390/genes15040520
APA StyleHerrick, J. (2024). DNA Damage, Genome Stability, and Adaptation: A Question of Chance or Necessity? Genes, 15(4), 520. https://doi.org/10.3390/genes15040520