Playing Hide-and-Seek in Beta-Globin Genes: Gene Conversion Transferring a Beneficial Mutation between Differentially Expressed Gene Duplicates





Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Genotyping
2.3. Testing for Genotype-Environment Correlation
2.4. Gene Sequencing
2.5. Sequence Analysis
3. Results
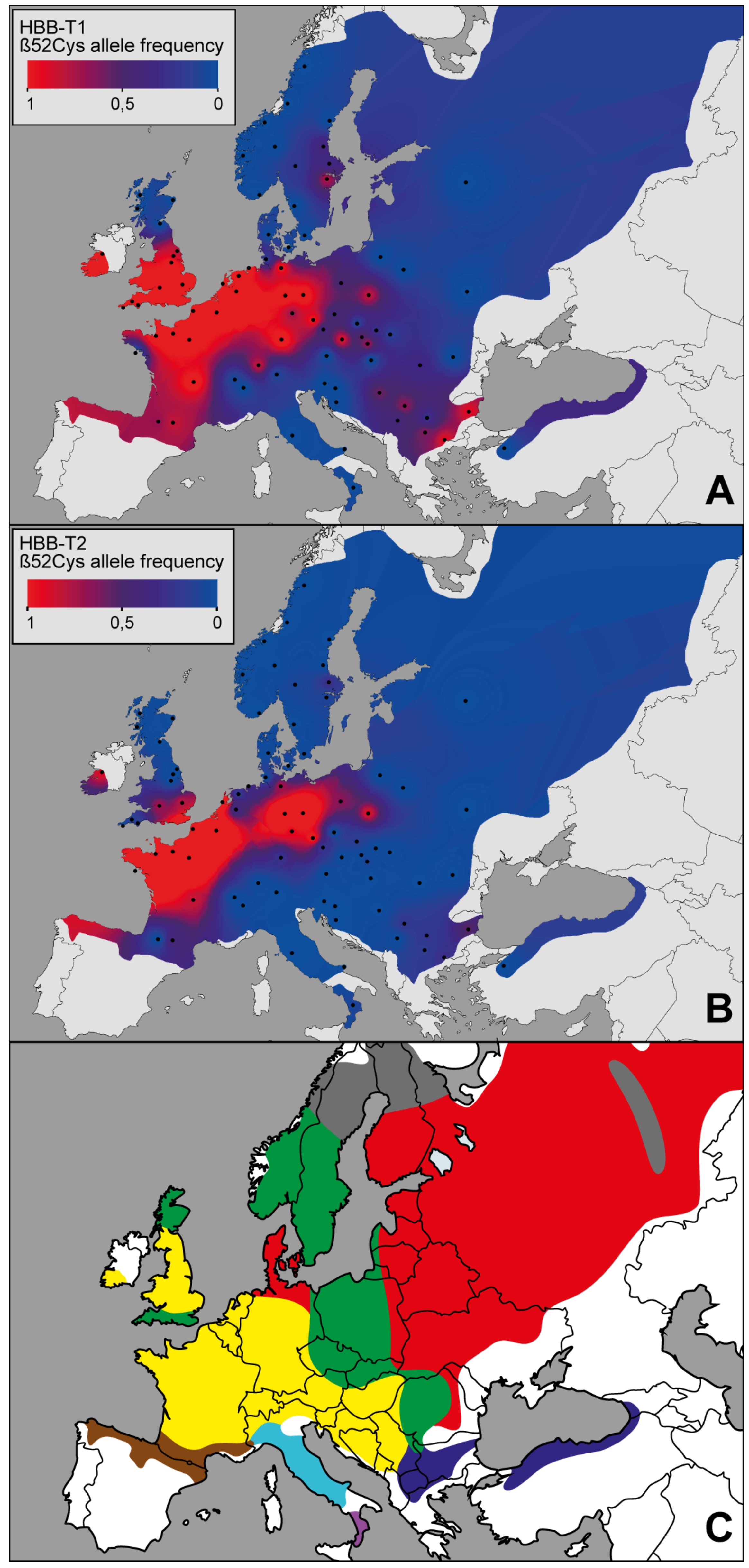
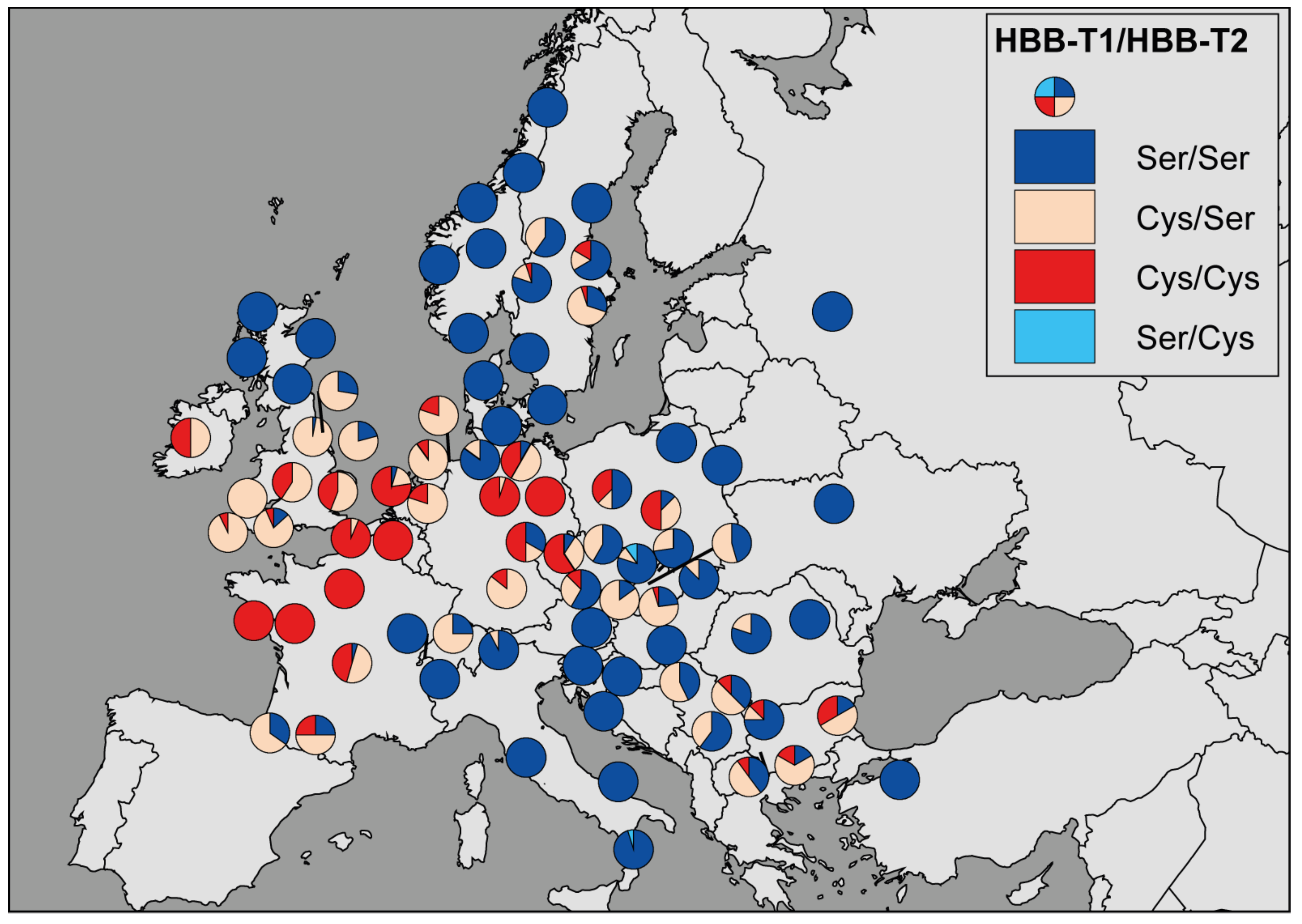
3.1. The Geographic Pattern
3.2. The Genotype-Environment Correlation
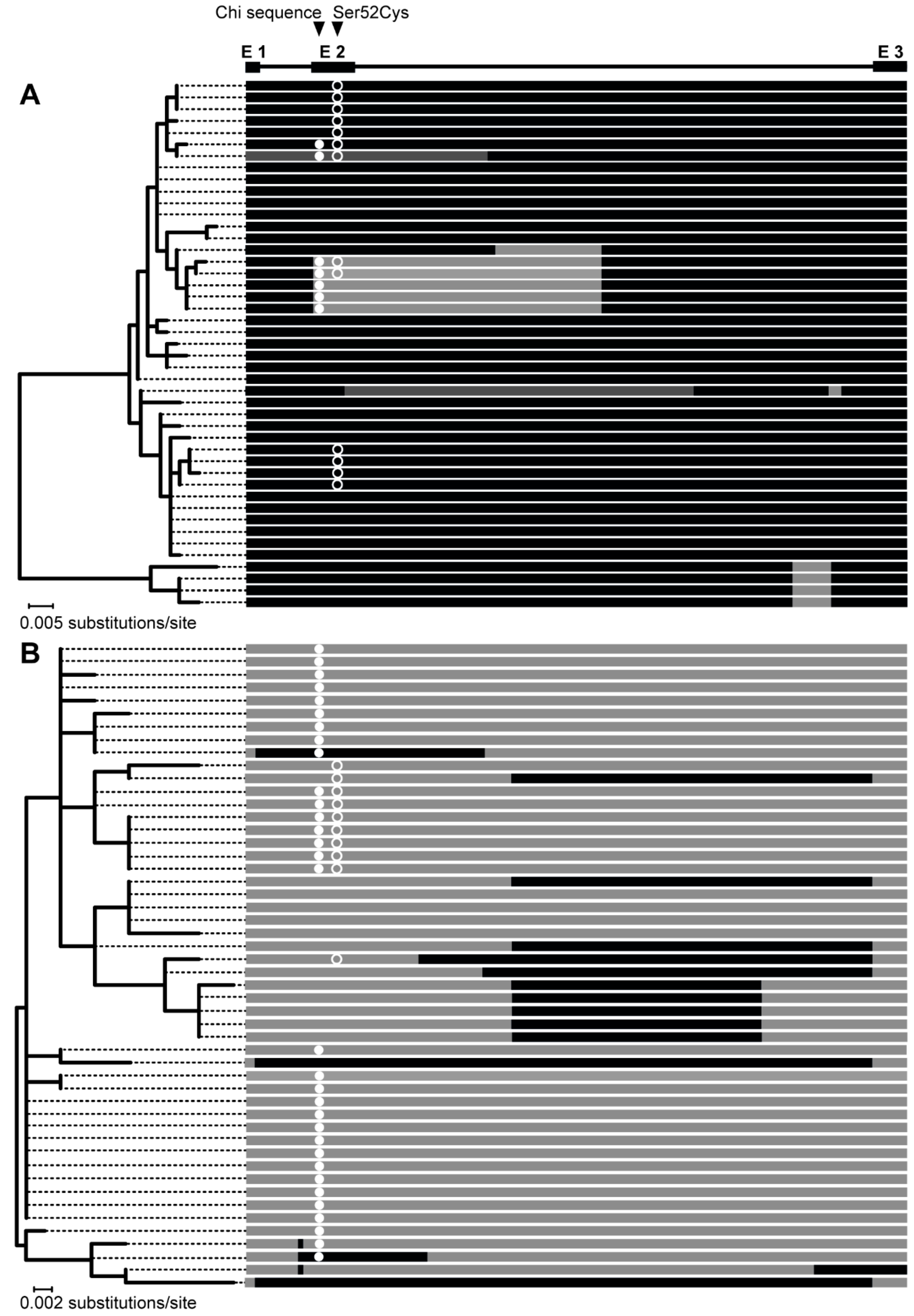
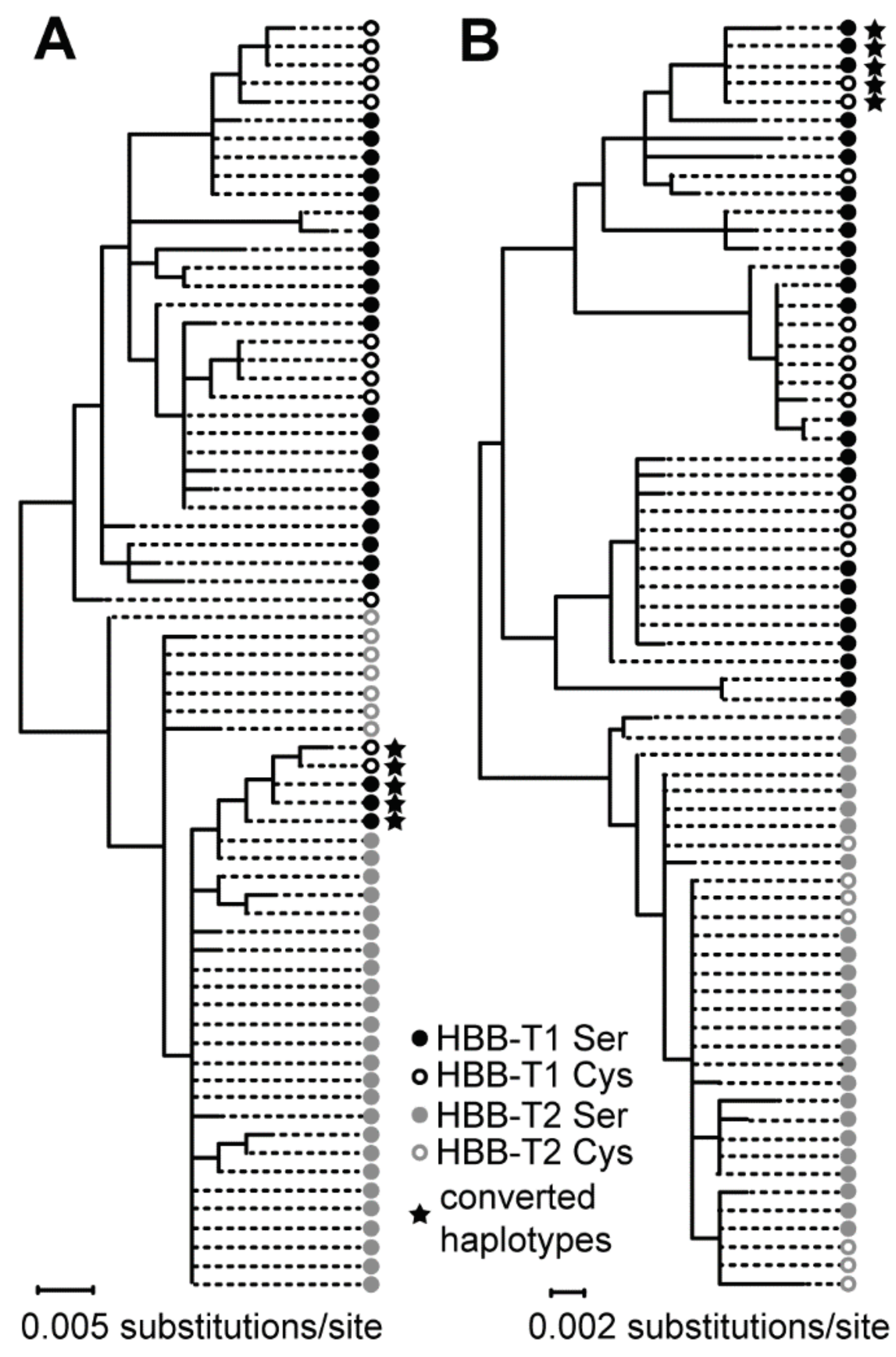
3.3. The Bidirectional Gene Conversion
4. Discussion
4.1. Is the Polymorphism under Environmental Selection?
4.2. Gene Conversion as a Possible Function-Altering Mechanism
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bergland, A.O.; Behrman, E.L.; O’Brien, K.R.; Schmidt, P.S.; Petrov, D.A. Genomic evidence of rapid and stable adaptive oscillations over seasonal time scales in Drosophila. PLoS Genet. 2014, 10, e1004775. [Google Scholar] [CrossRef] [PubMed]
- de Filippo, C.; Key, F.M.; Ghirotto, S.; Benazzo, A.; Meneu, J.R.; Weihmann, A.; NISC Comparative Sequence Program; Parra, G.; Green, E.D.; Andrés, A.M. Recent selection changes in human genes under long-term balancing selection. Mol. Biol. Evol. 2016, 33, 1435–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermisson, J.; Pennings, P.S. Soft sweeps and beyond: Understanding the patterns and probabilities of selection footprints under rapid adaptation. Methods Ecol. Evol. 2017, 8, 700–716. [Google Scholar] [CrossRef]
- Llaurens, V.; Whibley, A.; Joron, M. Genetic architecture and balancing selection: The life and death of differentiated variants. Mol. Ecol. 2017, 26, 2430–2448. [Google Scholar] [CrossRef] [PubMed]
- Mackinnon, M.J.; Ndila, C.; Uyoga, S.; Macharia, A.; Snow, R.W.; Band, G.; Rautanen, A.; Rockett, K.A.; Kwiatkowski, D.P.; Williams, T.N. Environmental correlation analysis for genes associated with protection against malaria. Mol. Biol. Evol. 2016, 33, 1188–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnaire, P.-A.; Normandeau, E.; Côté, C.; Hansen, M.M.; Bernatchez, L. The genetic consequences of spatially varying selection in the panmictic American eel (Anguilla rostrata). Genetics 2012, 190, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Hermisson, J.; Pennings, P.S. Soft sweeps: Molecular population genetics of adaptation from standing genetic variation. Genetics 2005, 169, 2335–2352. [Google Scholar] [CrossRef] [PubMed]
- Colosimo, P.F.; Hosemann, K.E.; Balabhadra, S.; Villarreal, G.; Dickson, M.; Grimwood, J.; Schmutz, J.; Myers, R.M.; Schluter, D.; Kingsley, D.M. Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science 2005, 307, 1928–1933. [Google Scholar] [CrossRef] [PubMed]
- Pelz, H.-J.; Rost, S.; Hünerberg, M.; Fregin, A.; Heiberg, A.-C.; Baert, K.; MacNicoll, A.D.; Prescott, C.V.; Walker, A.-S.; Oldenburg, J.; et al. The genetic basis of resistance to anticoagulants in rodents. Genetics 2005, 170, 1839–1847. [Google Scholar] [CrossRef] [PubMed]
- Steiner, C.C.; Weber, J.N.; Hoekstra, H.E. Adaptive variation in beach mice produced by two interacting pigmentation genes. PLoS Biol. 2007, 5, e219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Lorenzen, E.D.; Fumagalli, M.; Li, B.; Harris, K.; Xiong, Z.; Zhou, L.; Korneliussen, T.S.; Somel, M.; Babbitt, C.; et al. Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 2014, 157, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Bataillon, T.; Galtier, N.; Bernard, A.; Cryer, N.; Faivre, N.; Santoni, S.; Severac, D.; Mikkelsen, T.N.; Larsen, K.S.; Beier, C.; et al. A replicated climate change field experiment reveals rapid evolutionary response in an ecologically important soil invertebrate. Glob. Chang. Biol. 2016, 22, 2370–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, R.E.; Ostojic, H.; Fago, A.; Dewilde, S.; Van Hauwaert, M.-L.; Moens, L.; Monge, C. Novel mechanism for high-altitude adaptation in hemoglobin of the Andean frog Telmatobius peruvianus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R1052–R1060. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F.; Sabatino, S.J.; Hoffmann, F.G.; Gering, E.J.; Moriyama, H.; Ferrand, N.; Monteiro, B.; Nachman, M.W. The molecular basis of high-altitude adaptation in deer mice. PLoS Genet. 2007, 3, e45. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F.; Runck, A.M.; Sabatino, S.J.; Kelly, J.K.; Ferrand, N.; Moriyama, H.; Weber, R.E.; Fago, A. Evolutionary and functional insights into the mechanism underlying high-altitude adaptation of deer mouse hemoglobin. Proc. Natl. Acad. Sci. USA 2009, 106, 14450–14455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCracken, K.G.; Barger, C.P.; Bulgarella, M.; Johnson, K.P.; Sonsthagen, S.A.; Trucco, J.; Valqui, T.H.; Wilson, R.E.; Winker, K.; Sorenson, M.D. Parallel evolution in the major haemoglobin genes of eight species of Andean waterfowl. Mol. Ecol. 2009, 18, 3992–4005. [Google Scholar] [CrossRef] [PubMed]
- McCracken, K.G.; Barger, C.P.; Bulgarella, M.; Johnson, K.P.; Kuhner, M.K.; Moore, A.V.; Peters, J.L.; Trucco, J.; Valqui, T.H.; Winker, K.; et al. Signatures of high-altitude adaptation in the major hemoglobin of five species of Andean dabbling ducks. Am. Nat. 2009, 174, 631–650. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.L.; Storz, J.F.; Signore, A.V.; Moriyama, H.; Catania, K.C.; Payson, A.P.; Bonaventura, J.; Stetefeld, J.; Weber, R.E. Molecular basis of a novel adaptation to hypoxic-hypercapnia in a strictly fossorial mole. BMC Evol. Biol. 2010, 10, 214. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.L.; Roberts, J.E.E.; Watson, L.N.; Stetefeld, J.; Sloan, A.M.; Signore, A.V.; Howatt, J.W.; Tame, J.R.H.; Rohland, N.; Shen, T.-J.; et al. Substitutions in woolly mammoth hemoglobin confer biochemical properties adaptive for cold tolerance. Nat. Genet. 2010, 42, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, C.; Hoffmann, F.G.; Lanier, H.C.; Wolf, C.J.; Cheviron, Z.A.; Spangler, M.L.; Weber, R.E.; Fago, A.; Storz, J.F. Intraspecific polymorphism, interspecific divergence, and the origins of function-altering mutations in deer mouse hemoglobin. Mol. Biol. Evol. 2015, 32, 978–997. [Google Scholar] [CrossRef] [PubMed]
- Di Simplicio, P.; Cacace, M.G.; Lusini, L.; Giannerini, F.; Giustarini, D.; Rossi, R. Role of protein -SH groups in redox homeostasis—The erythrocyte as a model system. Arch. Biochem. Biophys. 1998, 355, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Dalle-Donne, I.; Cavarra, E.; Fineschi, S.; Lungarella, G.; Milzani, A.; Rossi, R. Metabolism of oxidants by blood from different mouse strains. Biochem. Pharmacol. 2006, 71, 1753–1764. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F.; Weber, R.E.; Fago, A. Oxygenation properties and oxidation rates of mouse hemoglobins that differ in reactive cysteine content. Comp. Biochem. Physiol. A. Mol. Integr. Physiol. 2012, 161, 265–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitturi, D.A.; Sun, C.-W.; Harper, V.M.; Thrash-Williams, B.; Cantu-Medellin, N.; Chacko, B.K.; Peng, N.; Dai, Y.; Wyss, J.M.; Townes, T.; et al. Antioxidant functions for the hemoglobin β93 cysteine residue in erythrocytes and in the vascular compartment in vivo. Free Radic. Biol. Med. 2013, 55, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotlík, P.; Marková, S.; Vojtek, L.; Stratil, A.; Šlechta, V.; Hyršl, P.; Searle, J.B. Adaptive phylogeography: Functional divergence between haemoglobins derived from different glacial refugia in the bank vole. Proc. R. Soc. B 2014, 281, 20140021. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.J.G. Haemoglobin polymorphism in the bank vole, Clethrionomys glareolus, in Britain. J. Zool. 1979, 187, 153–160. [Google Scholar] [CrossRef]
- Rossi, R.; Barra, D.; Bellelli, A.; Boumis, G.; Canofeni, S.; Di Simplicio, P.; Lusini, L.; Pascarella, S.; Amiconi, G. Fast-reacting thiols in rat hemoglobins can intercept damaging species in erythrocytes more efficiently than glutathione. J. Biol. Chem. 1998, 273, 19198–19206. [Google Scholar] [CrossRef] [PubMed]
- Miranda, J.J. Highly reactive cysteine residues in rodent hemoglobins. Biochem. Biophys. Res. Commun. 2000, 275, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Pörtner, H.O. Physiological basis of temperature-dependent biogeography: Trade-offs in muscle design and performance in polar ectotherms. J. Exp. Biol. 2002, 205, 2217–2230. [Google Scholar] [PubMed]
- Losdat, S.; Helfenstein, F.; Blount, J.D.; Richner, H. Resistance to oxidative stress shows low heritability and high common environmental variance in a wild bird. J. Evol. Biol. 2014, 27, 1990–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novembre, J.; Di Rienzo, A. Spatial patterns of variation due to natural selection in humans. Nat. Rev. Genet. 2009, 10, 745–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manel, S.; Conord, C.; Després, L. Genome scan to assess the respective role of host-plant and environmental constraints on the adaptation of a widespread insect. BMC Evol. Biol. 2009, 9, 288. [Google Scholar] [CrossRef] [PubMed]
- Stucki, S.; Orozco-terWengel, P.; Forester, B.R.; Duruz, S.; Colli, L.; Masembe, C.; Negrini, R.; Landguth, E.; Jones, M.R.; The NEXTGEN Consortium; et al. High performance computation of landscape genomic models including local indicators of spatial association. Mol. Ecol. Resour. 2016, 17, 1072–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deffontaine, V.; Libois, R.; Kotlík, P.; Sommer, R.; Nieberding, C.; Paradis, E.; Searle, J.B.; Michaux, J.R. Beyond the Mediterranean peninsulas: Evidence of central European glacial refugia for a temperate forest mammal species, the bank vole (Clethrionomys glareolus). Mol. Ecol. 2005, 14, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Kotlík, P.; Deffontaine, V.; Mascheretti, S.; Zima, J.; Michaux, J.R.; Searle, J.B. A northern glacial refugium for bank voles (Clethrionomys glareolus). Proc. Natl. Acad. Sci. USA 2006, 103, 14860–14864. [Google Scholar] [CrossRef] [PubMed]
- Filipi, K.; Marková, S.; Searle, J.B.; Kotlík, P. Mitogenomic phylogenetics of the bank vole (Clethrionomys glareolus), a model system for studying end-glacial colonization of Europe. Mol. Phylogenet. Evol. 2015, 82, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, J.M.; Kawałko, A.; Marková, S.; Searle, J.B.; Kotlík, P. Phylogeographic signatures of northward post-glacial colonization from high-latitude refugia: A case study of bank voles using museum specimens. J. Zool. 2010, 281, 249–262. [Google Scholar] [CrossRef]
- Runck, A.M.; Weber, R.E.; Fago, A.; Storz, J.F. Evolutionary and functional properties of a two-locus β-globin polymorphism in Indian house mice. Genetics 2010, 184, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F.; Natarajan, C.; Cheviron, Z.A.; Hoffmann, F.G.; Kelly, J.K. Altitudinal variation at duplicated β-globin genes in deer mice: Effects of selection, recombination, and gene conversion. Genetics 2012, 190, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. Genepop’007: A complete re-implementation of the Genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; Smith, N.J.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; Donnelly, P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2003, 73, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Haldane, J.B.S. An exact test for randomness of mating. J. Genet. 1954, 52, 631–635. [Google Scholar] [CrossRef]
- Rousset, F.; Raymond, M. Testing heterozygote excess and deficiency. Genetics 1995, 140, 1413–1419. [Google Scholar] [PubMed]
- Asmussen, M.A.; Basten, C.J. Constraints and normalized measures for cytonuclear disequilibria. Heredity 1996, 76, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basten, C.J.; Asmussen, M.A. The exact test for cytonuclear disequilibria. Genetics 1997, 146, 1165–1171. [Google Scholar] [PubMed]
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 2005, 25, 1965–1978. [Google Scholar] [CrossRef] [Green Version]
- Stuart, P.; Mirimin, L.; Cross, T.F.; Sleeman, D.P.; Buckley, N.J.; Telfer, S.; Birtles, R.J.; Kotlík, P.; Searle, J.B. The origin of Irish bank voles (Clethrionomys glareolus) assessed by mitochondrial DNA analysis. Ir. Nat. J. 2007, 28, 440–446. [Google Scholar]
- Joost, S.; Bonin, A.; Bruford, M.W.; Després, L.; Conord, C.; Erhardt, G.; Taberlet, P. A spatial analysis method (SAM) to detect candidate loci for selection: Towards a landscape genomics approach to adaptation. Mol. Ecol. 2007, 16, 3955–3969. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Frost, S.D. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Muse, S.V. HyPhy: Hypothesis Testing Using Phylogenies. In Statistical Methods in Molecular Evolution; Nielsen, R., Ed.; Springer: New York, NY, USA, 2005; pp. 125–181. ISBN 978-0-387-27733-2. [Google Scholar]
- Pond, S.L.K.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Betrán, E.; Rozas, J.; Navarro, A.; Barbadilla, A. The estimation of the number and the length distribution of gene conversion tracts from population DNA sequence data. Genetics 1997, 146, 89–99. [Google Scholar] [PubMed]
- Sawyer, S. Statistical tests for detecting gene conversion. Mol. Biol. Evol. 1989, 6, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S. GENECONV: A Computer Package for The Statistical Detection of Gene Conversion; Department of Mathematics, Washington University in St. Louis: St. Louis, MO, USA, 1999. [Google Scholar]
- Storz, J.F.; Baze, M.; Waite, J.L.; Hoffmann, F.G.; Opazo, J.C.; Hayes, J.P. Complex signatures of selection and gene conversion in the duplicated globin genes of house mice. Genetics 2007, 177, 481–500. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Prevodnik, A.; Gardestrom, J.; Lilja, K.; Elfwing, T.; McDonagh, B.; Petrovic, N.; Tedengren, M.; Sheehan, D.; Bollner, T. Oxidative stress in response to xenobiotics in the blue mussel Mytilus edulis L.: Evidence for variation along a natural salinity gradient of the Baltic Sea. Aquat. Toxicol. 2007, 82, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Costantini, D.; Dell’Omo, G.; De Filippis, S.P.; Marquez, C.; Snell, H.L.; Snell, H.M.; Tapia, W.; Brambilla, G.; Gentile, G. Temporal and spatial covariation of gender and oxidative stress in the Galápagos land iguana Conolophus subcristatus. Physiol. Biochem. Zool. 2009, 82, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Searle, J.B.; Kotlík, P.; Rambau, R.V.; Marková, S.; Herman, J.S.; McDevitt, A.D. The Celtic fringe of Britain: Insights from small mammal phylogeography. Proc. R. Soc. Lond. B 2009, 276, 4287–4294. [Google Scholar] [CrossRef] [PubMed]
- Frichot, E.; Schoville, S.D.; de Villemereuil, P.; Gaggiotti, O.E.; François, O. Detecting adaptive evolution based on association with ecological gradients: Orientation matters! Heredity 2015, 115, 22. [Google Scholar] [CrossRef] [PubMed]
- Stier, A.; Dupoué, A.; Picard, D.; Angelier, F.; Brischoux, F.; Lourdais, O. Oxidative stress in a capital breeder (Vipera aspis) facing pregnancy and water constraints. J. Exp. Biol. 2017, 220, 1792–1796. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Mitchell-Olds, T. Environmental adaptation contributes to gene polymorphism across the Arabidopsis thaliana genome. Mol. Biol. Evol. 2012, 29, 3721–3728. [Google Scholar] [CrossRef] [PubMed]
- Tiffin, P.; Ross-Ibarra, J. Advances and limits of using population genetics to understand local adaptation. Trends Ecol. Evol. 2014, 29, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Outridge, P.M.; Hutchinson, T.C. Induction of cadmium tolerance by acclimation transferred between ramets of the clonal fern Salvinia minima Baker. New Phytol. 1991, 117, 597–605. [Google Scholar] [CrossRef]
- Marino, S.M.; Gladyshev, V.N. Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J. Mol. Biol. 2010, 404, 902–916. [Google Scholar] [CrossRef] [PubMed]
- Mano, S.; Innan, H. The evolutionary rate of duplicated genes under concerted evolution. Genetics 2008, 180, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Hallast, P.; Nagirnaja, L.; Margus, T.; Laan, M. Segmental duplications and gene conversion: Human luteinizing hormone/chorionic gonadotropin β gene cluster. Genome Res. 2005, 15, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- von Salomé, J.; Kukkonen, J.P. Sequence features of HLA-DRB1 locus define putative basis for gene conversion and point mutations. BMC Genom. 2008, 9, 228. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.T.; Stahl, M.M.; McMilin, K.D.; Stahl, F.W. Rec-mediated recombinational hot spot activity in bacteriophage lambda. II. A mutation which causes hot spot activity. Genetics 1974, 77, 425–433. [Google Scholar] [PubMed]
- Henderson, D.; Weil, J. Recombination-deficient deletions in bacteriophage lambda and their interaction with chi mutations. Genetics 1975, 79, 143–174. [Google Scholar] [PubMed]
- Smith, G.R. How RecBCD enzyme and Chi promote DNA break repair and recombination: A molecular biologist’s view. Microbiol. Mol. Biol. Rev. 2012, 76, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Kenter, A.L.; Birshtein, B.K. Chi, a promoter of generalized recombination in λ phage, is present in immunoglobulin genes. Nature 1981, 293, 402–404. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, Y.; Yamashiro, Y.; Yamamoto, K.; Hattori, Y.; Yamamoto, K.; Ohba, Y.; Miyaji, T. A possible example of gene conversion with a common β-thalassemia mutation and Chi sequence present in the β-globin gene. Hum. Genet. 1992, 88, 357–358. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-M.; Ferec, C. Gene conversion-like missense mutations in the human cationic trypsinogen gene and insights into the molecular evolution of the human trypsinogen family. Mol. Genet. Metab. 2000, 71, 463–469. [Google Scholar] [CrossRef] [PubMed]
- López-Correa, C.; Dorschner, M.; Brems, H.; Lázaro, C.; Clementi, M.; Upadhyaya, M.; Dooijes, D.; Moog, U.; Kehrer-Sawatzki, H.; Rutkowski, J.L.; et al. Recombination hotspot in NF1 microdeletion patients. Hum. Mol. Genet. 2001, 10, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cai, W.-W.; Zhou, W.-P.; Li, H.-P.; Li, L.; Yan, W.; Deng, Q.-K.; Zhang, Y.-P.; Fu, Y.-X.; Xu, X.-M. Evidence of gene conversion in the evolutionary process of the codon 41/42 (-CTTT) mutation causing β-thalassemia in southern China. J. Mol. Evol. 2008, 66, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Innan, H. A two-locus gene conversion model with selection and its application to the human RHCE and RHD genes. Proc. Natl. Acad. Sci. USA 2003, 100, 8793–8798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafontaine, G.; Napier, J.D.; Petit, R.J.; Hu, F.S. Invoking adaptation to decipher the genetic legacy of past climate change. Ecology 2018, 99, 1530–1546. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Variable | Variable 2 | Variable 3 | Log Likelihood | G Score | Wald Score |
|---|---|---|---|---|---|---|
| Univariate | Isotherm 1 | −307.98 | 93.63 | 67.76 | ||
| PwetQ 2 | −304.82 | 99.94 | 58.08 | |||
| Pwet 3 | −308.24 | 93.11 | 55.01 | |||
| AP 4 | −318.05 | 73.49 | 47.24 | |||
| MeanTcoldQ 5 | −330.26 | 49.07 | 42.95 | |||
| AMT 6 | −330.70 | 48.19 | 41.25 | |||
| MinTcold 7 | −332.83 | 43.93 | 39.27 | |||
| LONG 8 | −334.14 | 41.31 | 37.01 | |||
| Pseason 9 | −334.78 | 40.04 | 36.33 | |||
| PopStr 10 | −255.33 | 198.93 | 34.57 | |||
| Bivariate | PopStr | AP | −221.04 | 68.57 | 39.40 | |
| PopStr | PcoldQ 11 | −230.18 | 50.29 | 30.66 | ||
| PopStr | PwetQ | −222.44 | 65.78 | 29.65 | ||
| PopStr | Pwet | −225.44 | 59.78 | 29.37 | ||
| PopStr | Pdry 12 | −235.88 | 38.89 | 27.09 | ||
| PopStr | PdryQ 13 | −235.97 | 38.72 | 26.82 | ||
| PopStr | PwarmQ 14 | −239.11 | 32.44 | 25.23 | ||
| PopStr | MeanTwetQ 15 | −243.04 | 24.58 | 21.76 | ||
| Trivariate | PopStr | Pseason | PdryQ | −223.97 | 24.00 | 22.21 |
| PopStr | Pseason | PcoldQ | −217.72 | 24.93 | 22.06 | |
| PopStr | Tseason 16 | AP | −207.47 | 27.15 | 21.64 | |
| PopStr | Tseason | PcoldQ | −210.55 | 39.28 | 21.16 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strážnická, M.; Marková, S.; Searle, J.B.; Kotlík, P. Playing Hide-and-Seek in Beta-Globin Genes: Gene Conversion Transferring a Beneficial Mutation between Differentially Expressed Gene Duplicates. Genes 2018, 9, 492. https://doi.org/10.3390/genes9100492
Strážnická M, Marková S, Searle JB, Kotlík P. Playing Hide-and-Seek in Beta-Globin Genes: Gene Conversion Transferring a Beneficial Mutation between Differentially Expressed Gene Duplicates. Genes. 2018; 9(10):492. https://doi.org/10.3390/genes9100492
Chicago/Turabian StyleStrážnická, Michaela, Silvia Marková, Jeremy B. Searle, and Petr Kotlík. 2018. "Playing Hide-and-Seek in Beta-Globin Genes: Gene Conversion Transferring a Beneficial Mutation between Differentially Expressed Gene Duplicates" Genes 9, no. 10: 492. https://doi.org/10.3390/genes9100492
APA StyleStrážnická, M., Marková, S., Searle, J. B., & Kotlík, P. (2018). Playing Hide-and-Seek in Beta-Globin Genes: Gene Conversion Transferring a Beneficial Mutation between Differentially Expressed Gene Duplicates. Genes, 9(10), 492. https://doi.org/10.3390/genes9100492