Complete Genome Sequence of the Model Halovirus PhiH1 (ΦH1)

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus DNA and Sequencing Methods
2.2. CRISPR Spacer Searches
2.3. Bioinformatic Methods
2.4. Data Availability
3. Results and Discussion
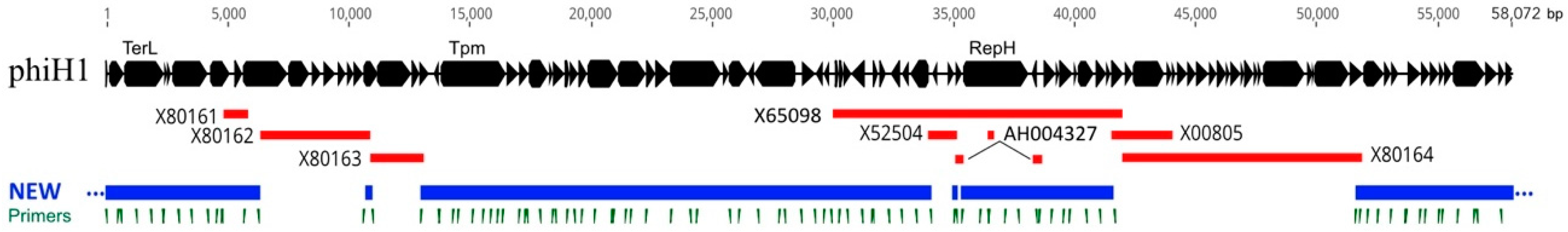
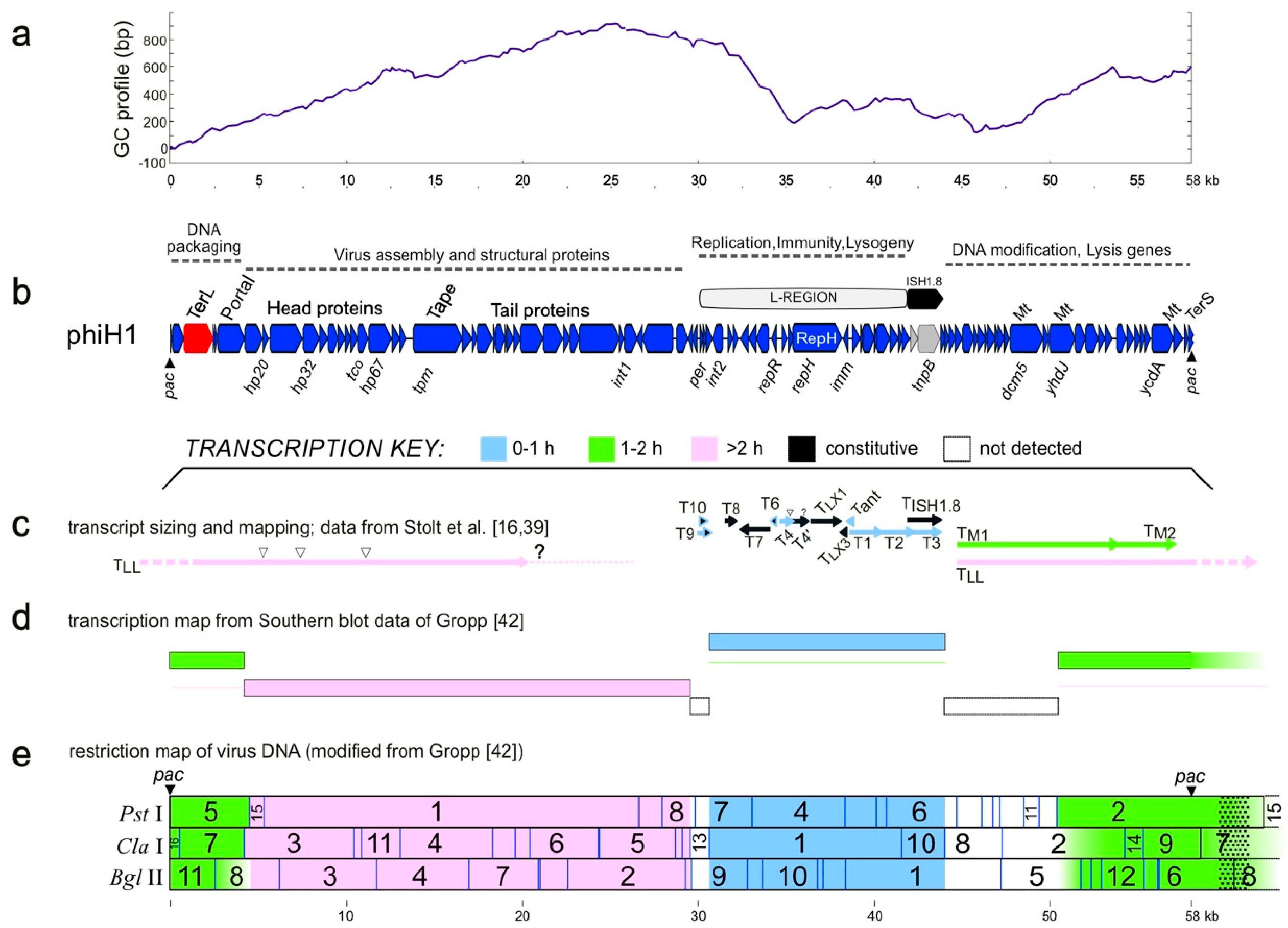
3.1. Sequence and Annotation of PhiH1
3.2. Matches to CRISPR Spacers
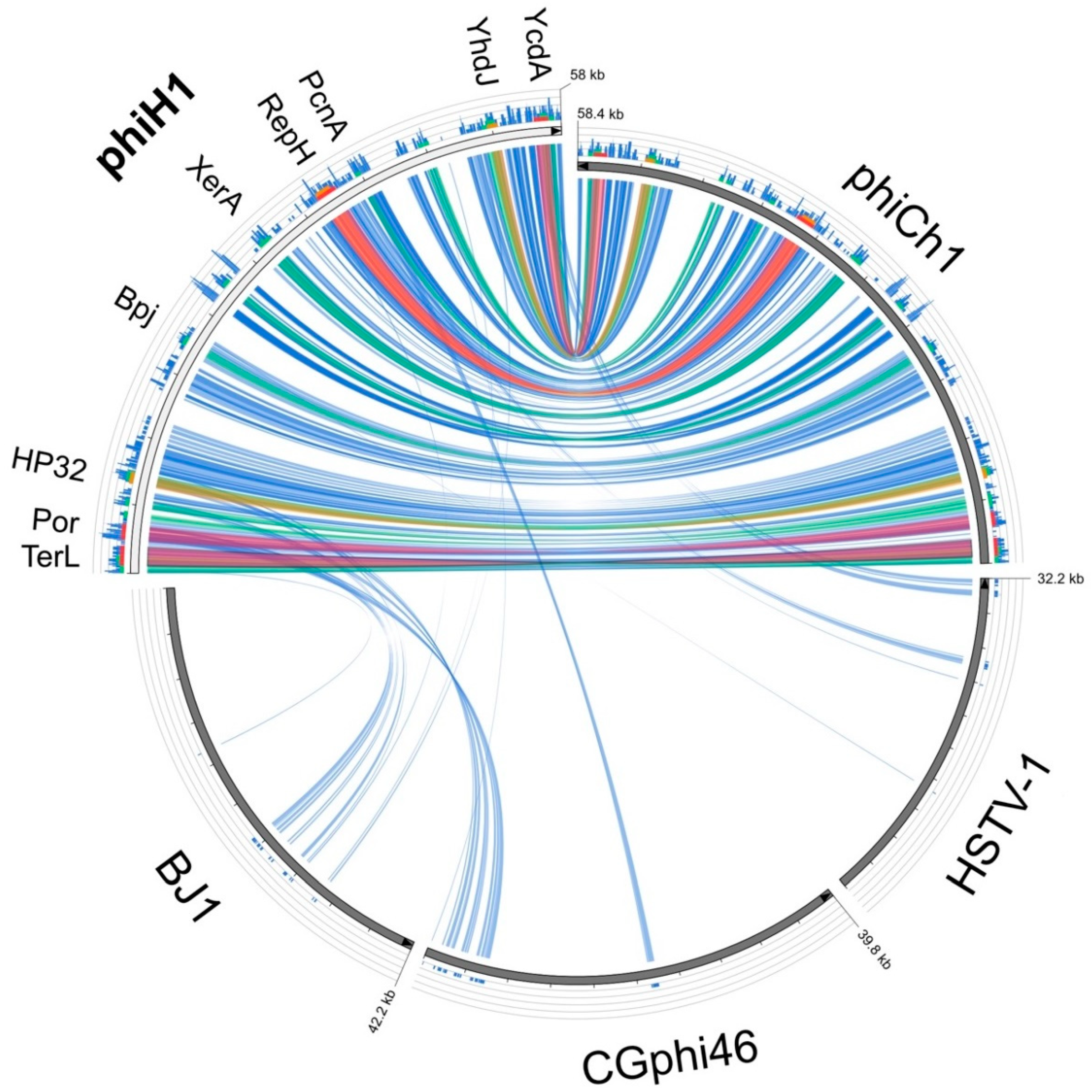
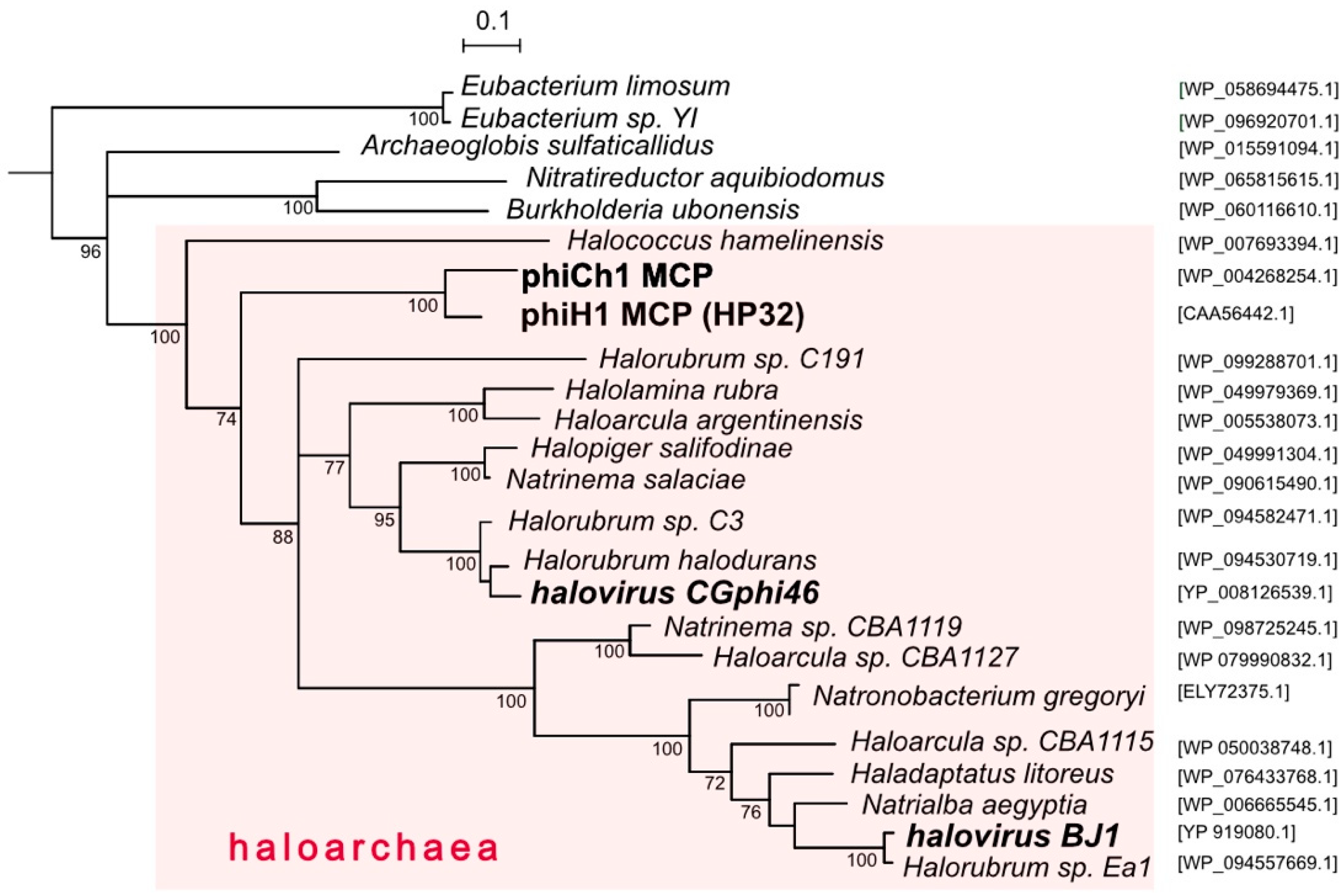
3.3. Relatives and Phylogeny of PhiH1
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schnabel, H.; Zillig, W.; Pfaffle, M.; Schnabel, R.; Michel, H.; Delius, H. Halobacterium halobium phage ΦH. EMBO J. 1982, 1, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Zillig, W.; Gropp, F.; Henschen, A.; Neumann, H.; Palm, P.; Reiter, W.D.; Rettenberger, M.; Schnabel, H.; Yeats, S. Archaebacteria virus host systems. Syst. Appl. Microbiol. 1986, 7, 58–66. [Google Scholar] [CrossRef]
- Zillig, W.; Reiter, W.-D.; Palm, P.; Gropp, F.; Neumann, H.; Rettenberger, M. Viruses of Archaebacteria. In The Bacteriophages; Calendar, R., Ed.; Plenum Publishing Corpn: New York, NY, USA, 1988. [Google Scholar]
- Schnabel, H.; Zillig, W. Circular structure of the genome of phage ΦH in a lysogenic Halobacterium halobium. Mol. Gen. Genet. 1984, 193, 422–426. [Google Scholar] [CrossRef]
- Schnabel, H.; Schramm, E.; Schnabel, R.; Zillig, W. Structural variability in the genome of phage ΦH of Halobacterium halobium. Mol. Gen. Genet. 1982, 188, 370–377. [Google Scholar] [CrossRef]
- ICTV Report, C. ICTV Online (10th) Report on Virus Taxonomy. Available online: https://talk.ictvonline.org/taxonomy/p/taxonomy-history?taxnode_id=20170459 (accessed on 19 March 2018).
- Krupovic, M.; Dutilh, B.E.; Adriaenssens, E.M.; Wittmann, J.; Vogensen, F.K.; Sullivan, M.B.; Rumnieks, J.; Prangishvili, D.; Lavigne, R.; Kropinski, A.M.; et al. Taxonomy of prokaryotic viruses: Update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 2016, 161, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Cline, S.W.; Doolittle, W.F. Efficient transfection of the archaebacterium Halobacterium halobium. J. Bacteriol. 1987, 169, 1341–1344. [Google Scholar] [CrossRef] [PubMed]
- Blaseio, U.; Pfeifer, F. Transformation of Halobacterium halobium: Development of vectors and investigation of gas vesicle synthesis. Proc. Natl. Acad. Sci. USA 1990, 87, 6772–6776. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. In vivo studies on the effects of immunity genes on early lytic transcription in the Halobacterium salinarium phage ϕH. Mol. Gen. Genet. 1992, 235, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Ken, R.; Hackett, N.R. Halobacterium halobium strains lysogenic for phage phiH contain a protein resembling coliphage repressors. J. Bacteriol. 1991, 173, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. In vivo and in vitro analysis of transcription of the L region from the Halobacterium salinarium phage ϕH: Definition of a repressor-enhancing gene. Virology 1993, 195, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. Antisense RNA mediates transcriptional processing in an archaebacterium, indicating a novel kind of RNase activity. Mol. Microbiol. 1993, 7, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, H. An immune strain of Halobacterium halobium carries the invertible L segment of phage ΦH as a plasmid. Proc. Natl. Acad. Sci. USA 1984, 81, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. Transcription of the halophage ΦH repressor gene is abolished by transcription from an inversely oriented lytic promoter. FEBS Lett. 1994, 344, 125–128. [Google Scholar] [CrossRef] [Green Version]
- Stolt, P.; Zillig, W. Gene regulation in halophage ΦH; more than promoters. Syst. Appl. Microbiol. 1993, 16, 591–596. [Google Scholar] [CrossRef]
- Witte, A.; Baranyi, U.; Klein, R.; Sulzner, M.; Luo, C.; Wanner, G.; Krüger, D.H.; Lubitz, W. Characterization of Natronobacterium magadii phage ϕCh1, a unique archaeal phage containing DNA and RNA. Mol. Microbiol. 1997, 23, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Baranyi, U.; Rössler, N.; Greineder, B.; Scholz, H.; Witte, A. Natrialba magadii virus ϕCh1: First complete nucleotide sequence and functional organization of a virus infecting a haloalkaliphilic archaeon. Mol. Microbiol. 2002, 45, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Selb, R.; Derntl, C.; Klein, R.; Alte, B.; Hofbauer, C.; Kaufmann, M.; Beraha, J.; Schoner, L.; Witte, A. The viral gene ORF79 encodes a repressor regulating induction of the lytic life cycle in the haloalkaliphilic virus phiCh1. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Siddaramappa, S.; Challacombe, J.F.; Decastro, R.E.; Pfeiffer, F.; Sastre, D.E.; Gimenez, M.I.; Paggi, R.A.; Detter, J.C.; Davenport, K.W.; Goodwin, L.A.; et al. A comparative genomics perspective on the genetic content of the alkaliphilic haloarchaeon Natrialba magadii ATCC 43099T. BMC Genom. 2012, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Zillig, W.; Palm, P.; Reiter, W.D.; Gropp, F.; Puhler, G.; Klenk, H.P. Comparative evaluation of gene expression in Archaebacteria. Eur. J. Biochem. 1988, 173, 473–482. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 11 October 2018).
- Gordon, D. Viewing and editing assembled sequences using Consed. Curr. Protoc. Bioinform. 2003, 2. [Google Scholar] [CrossRef]
- Skennerton, C.T.; Imelfort, M.; Tyson, G.W. Crass: Identification and reconstruction of CRISPR from unassembled metagenomic data. Nucleic Acids Res. 2013, 41, e105. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M. The Sequence Read Archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyall-Smith, M.; Pfeiffer, F. The PL6-family plasmids of Haloquadratum are virus-related. Front. Microbiol. 2018, 9, 1070. [Google Scholar] [CrossRef] [PubMed]
- CRISPRs Web Server. Available online: http://crispr.i2bc.paris-saclay.fr/ (accessed on 11 October 2018).
- Lomsadze, A.; Gemayel, K.; Tang, S.; Borodovsky, M. Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res. 2018, 28, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information BLAST. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 11 October 2018).
- YASS Genomic Similarity Search Tool. Available online: http://bioinfo.lifl.fr/yass/index.php (accessed on 11 October 2018).
- Geneious. Available online: https://www.geneious.com/geneious/ (accessed on 11 October 2018).
- Circoletto. Available online: http://tools.bat.infspire.org/circoletto/ (accessed on 11 October 2018).
- I-Tasser, Protein Structure & Function Predictions. Available online: https://zhanglab.ccmb.med.umich.edu/I-TASSER (accessed on 11 October 2018).
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef] [PubMed]
- CPT Phage Galaxy. Available online: https://cpt.tamu.edu/galaxy-pub/ (accessed on 11 October 2018).
- VIRFAM, Remote Homology Detection of Viral Protein Families. Available online: http://biodev.cea.fr/virfam/ (accessed on 11 October 2018).
- Schnabel, H.; Schnabel, R.; Yeats, S.; Tu, J.; Gierl, A.; Neumann, H.; Zillig, W. Genome organization and transcription in Archaebacteria. Folia Biol. (Praha) 1984, 30, 2–6. [Google Scholar] [PubMed]
- Pfeiffer, F.; Schuster, S.C.; Broicher, A.; Falb, M.; Palm, P.; Rodewald, K.; Ruepp, A.; Soppa, J.; Tittor, J.; Oesterhelt, D. Evolution in the laboratory: The genome of Halobacterium salinarum strain R1 compared to that of strain NRC-1. Genomics 2008, 91, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Grampp, B.; Zillig, W. Genes for DNA cytosine methyltransferases and structural proteins, expressed during lytic growth by the phage ΦH of the archaebacterium Halobacterium salinarium. Biol. Chem. Hoppe Seyler 1994, 375, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Pavelka, M.S., Jr.; Butler, J.S. Structure-function analysis of VapB4 antitoxin identifies critical features of a minimal VapC4 toxin-binding module. J. Bacteriol. 2015, 197, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhang, C.T. GC-Profile: A web-based tool for visualizing and analyzing the variation of GC content in genomic sequences. Nucleic Acids Res. 2006, 34, W686–W691. [Google Scholar] [CrossRef] [PubMed]
- Gropp, F. Genexpression im Archaebakterium Halobacterium halobium: Der Phage ΦH und die DNA-abhängige RNA-Polymerase. Ph.D. Thesis, Ludwig-Maximilians-Universitaet Muenchen, Munich, Germany, 26 July 1989. [Google Scholar]
- Gropp, F.; Grampp, B.; Stolt, P.; Palm, P.; Zillig, W. The immunity-conferring plasmid pϕHL from the Halobacterium salinarium phage ϕH: Nucleotide sequence and transcription. Virology 1992, 190, 45–54. [Google Scholar] [CrossRef]
- ISfinder. Available online: https://isfinder.biotoul.fr/ (accessed on 11 October 2018).
- Vogelsang-Wenke, H.; Oesterhelt, D. Isolation of a halobacterial phage with a fully cytosine-methylated genome. MGG Mol. Gen. Genet. 1988, 211, 407–414. [Google Scholar] [CrossRef]
- Baranyi, U.; Klein, R.; Lubitz, W.; Kruger, D.H.; Witte, A. The archaeal halophilic virus-encoded Dam-like methyltransferase M. ϕCh1-I methylates adenine residues and complements dam mutants in the low salt environment of Escherichia coli. Mol. Microbiol. 2000, 35, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Pagaling, E.; Haigh, R.D.; Grant, W.D.; Cowan, D.A.; Jones, B.E.; Ma, Y.; Ventosa, A.; Heaphy, S. Sequence analysis of an archaeal virus isolated from a hypersaline lake in Inner Mongolia, China. BMC Genom. 2007, 8, 410. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, M.K.; Laurinmäki, P.; Russell, D.A.; Ko, C.C.; Jacobs-Sera, D.; Hendrix, R.W.; Bamford, D.H.; Butcher, S.J. Structure of the archaeal head-tailed virus HSTV-1 completes the HK97 fold story. Proc. Natl. Acad. Sci. USA 2013, 110, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Forterre, P.; Bamford, D.H. Comparative analysis of the mosaic genomes of tailed archaeal viruses and proviruses suggests common themes for virion architecture and assembly with tailed viruses of bacteria. J. Mol. Biol. 2010, 397, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.L.; Jiang, W.; Rixon, F.J.; Chiu, W. Common ancestry of herpesviruses and tailed DNA bacteriophages. J. Virol. 2005, 79, 14967–14970. [Google Scholar] [CrossRef] [PubMed]
- Kamekura, M.; Dyall-Smith, M. Taxonomy of the family Halobacteriaceae and the description of two new genera Halorubrobacterium and Natrialba. J. Gen. Appl. Microbiol. 1995, 41, 333–350. [Google Scholar] [CrossRef]
- Tindall, B.J.; Ross, H.N.M.; Grant, W.D. Natronobacterium gen. nov. and Natronococcus gen. nov. Two new genera of haloalkaliphilic archaebacteria. Syst. Appl. Microbiol. 1984, 5, 41–57. [Google Scholar] [CrossRef]
- Kalenov, S.V.; Baurina, M.M.; Skladnev, D.A.; Kuznetsov, A.Y. High-effective cultivation of Halobacterium salinarum providing with bacteriorhodopsin production under controlled stress. J. Biotechnol. 2016, 233, 211–218. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Start (nt) | Stop (nt) | Locus_tag | Length (bp) | Direction | Gene | Product | Homologs1: phiCh1, ORF pNMAG03 [Other] |
|---|---|---|---|---|---|---|---|
| 115 | 717 | PhiH1_005 | 603 | + | - | uncharacterized protein | PhiCh1p02, ORF1 Nmag_4251 |
| 710 | 2371 | PhiH1_010 | 1662 | + | terL | terminase large subunit TerL | PhiCh1p03, ORF2 Nmag_4252 |
| 2377 | 2505 | PhiH1_015 | 129 | + | - | uncharacterized protein | Nmag_4253 |
| 2498 | 2689 | PhiH1_020 | 192 | + | - | uncharacterized protein | PhiCh1p05, ORF4 Nmag_4255 |
| 2686 | 4242 | PhiH1_025 | 1557 | + | por | portal protein Por | PhiCh1p07, ORF6 Nmag_4257 |
| 4246 | 5187 | PhiH1_030 | 942 | + | - | head morphogenesis protein | PhiCh1p08, ORF7 Nmag_4258 |
| 5261 | 5587 | PhiH1_035 | 327 | + | hp20 | capsid protein HP20 | [AJF28118.1] |
| 5667 | 7466 | PhiH1_040 | 1800 | + | - | prohead protease | 4 PhiCh1p09, ORF8 4 PhiCh1p10, ORF9 Nmag_4259 |
| 7506 | 8468 | PhiH1_045 | 963 | + | hp32 | major capsid protein HP32 | PhiCh1p12, ORF11 Nmag_4260 |
| 8481 | 8933 | PhiH1_050 | 453 | + | - | uncharacterized protein | PhiCh1p13, ORF12 Nmag_4261 |
| 8940 | 9542 | PhiH1_055 | 603 | + | ada | head-tail adaptor protein Ada | PhiCh1p14, ORF13 Nmag_4262 |
| 9539 | 9919 | PhiH1_060 | 381 | + | hco | head closure protein type 1 Hco | PhiCh1p15, ORF14 Nmag_4263 |
| 9921 | 10,202 | PhiH1_065 | 282 | + | - | uncharacterized protein | PhiCh1p16, ORF15 Nmag_4264 |
| 10,202 | 10,636 | PhiH1_070 | 435 | + | nep | probable neck protein type 1 Nep | PhiCh1p17, ORF16 Nmag_4265 |
| 10,643 | 11,239 | PhiH1_075 | 597 | + | tco | tail completion protein type 1 Tco | PhiCh1p18, ORF17 Nmag_4266 |
| 11,259 | 12,557 | PhiH1_080 | 1299 | + | hp67 | tail sheath protein HP67 | PhiCh1p19, ORF18 Nmag_4267 |
| 12,607 | 13,002 | PhiH1_085 | 396 | + | - | probable structural protein | PhiCh1p20, ORF19 Nmag_4268 |
| 13,006 | 13,407 | PhiH1_090 | 402 | + | - | uncharacterized protein | PhiCh1p21, ORF20 Nmag_4269 |
| 13,572 | 13,745 | PhiH1_095 | 174 | − | - | DUF4177 domain protein | [SEH60446.1] |
| 13,792 | 16,581 | PhiH1_100 | 2790 | + | tpm | tape-measure tail protein Tpm | 4 PhiCh1p23, ORF22 4 PhiCh1p24, ORF23 Nmag_4272 |
| 16,583 | 17,104 | PhiH1_105 | 522 | + | - | uncharacterized protein | PhiCh1p25, ORF24 Nmag_4273 |
| 17,108 | 17,446 | PhiH1_110 | 339 | + | - | uncharacterized protein | PhiCh1p26, ORF25 Nmag_4274 |
| 17,450 | 18,298 | PhiH1_115 | 849 | + | - | uncharacterized protein | PhiCh1p27, ORF26 Nmag_4275 |
| 18,306 | 18,446 | PhiH1_120 | 141 | + | - | CxxC motif protein | [SEH61109.1] |
| 18,443 | 18,988 | PhiH1_125 | 546 | + | - | uncharacterized protein | PhiCh1p29, ORF28 Nmag_4276 |
| 18,988 | 19,146 | PhiH1_130 | 159 | + | - | uncharacterized protein | - |
| 19,143 | 19,508 | PhiH1_135 | 366 | + | - | virus-related protein | [AGM10900.1] |
| 19,505 | 19,867 | PhiH1_140 | 363 | + | - | uncharacterized protein | PhiCh1p30, ORF29 Nmag_4277 |
| 19,874 | 21,148 | PhiH1_145 | 1275 | + | bpj | baseplate J family protein Bpj | PhiCh1p31, ORF30 Nmag_4278 |
| 21,135 | 22,277 | PhiH1_150 | 1143 | + | - | uncharacterized protein | PhiCh1p32, ORF31 Nmag_4279 |
| 22,295 | 22,678 | PhiH1_155 | 384 | + | - | virus-related protein | [AFH21897.1] |
| 22,683 | 23,249 | PhiH1_160 | 567 | + | - | virus-related protein | [AFH21653.1] |
| 23,252 | 25,504 | PhiH1_165 | 2253 | + | - | repeat-containing tail fibre protein | PhiCh1p37, ORF36 Nmag_4282 PhiCh1p35, ORF34 Nmag_4286 |
| 25,506 | 25,787 | PhiH1_170 | 282 | + | - | uncharacterized protein | Nmag_4285 |
| 25,825 | 26,499 | PhiH1_175 | 675 | + | int1 | tyrosine integrase/recombinase Int1 | PhiCh1p36, ORF35 Nmag_4284 |
| 26,490 | 26,792 | PhiH1_180 | 303 | − | - | uncharacterized protein | Nmag_4283 |
| 26,798 | 27,766 | PhiH1_185 | 969 | − | - | repeat-containing tail fibre protein 2 | PhiCh1p37, ORF36 Nmag_4282 PhiCh1p35, ORF34 Nmag_4286 |
| 27,803 | 28,150 | PhiH1_190 | 348 | + | - | YncB-like endonuclease | [AGM11801.1] |
| 28,153 | 28,386 | PhiH1_195 | 234 | + | - | virus-related protein | [AGC34510.1] |
| 28,379 | 28,675 | PhiH1_200 | 297 | + | - | uncharacterized protein | [EMA49173.1] |
| 28,682 | 28,783 | PhiH1_205 | 102 | + | - | uncharacterized protein | - |
| 28,788 | 29,357 | PhiH1_210 | 570 | + | - | transmembrane domain protein | - |
| 29,394 | 29,642 | PhiH1_215 | 249 | − | - | uncharacterized protein | - |
| 29,651 | 29,941 | PhiH1_220 | 291 | − | - | uncharacterized protein | PhiCh1p40, ORF39 Nmag_4289 |
| 30,104 | 30,244 | PhiH1_225 | 144 | + | - | uncharacterized protein | - |
| 30,250 | 30,414 | PhiH1_230 | 165 | + | - | uncharacterized protein | PhiCh1p44, ORF43 Nmag_4292 |
| 30,411 | 30,806 | PhiH1_235 | 396 | + | - | VapC family toxin | PhiCh1p45, ORF44 Nmag_4293 |
| 30,803 | 31,465 | PhiH1_240 | 663 | − | int2 | tyrosine integrase/recombinase Int2 | PhiCh1p46, ORF45 Nmag_4294 |
| 31,680 | 31,934 | PhiH1_245 | 255 | + | - | uncharacterized protein | - |
| 31,939 | 32,271 | PhiH1_250 | 333 | + | - | uncharacterized protein | Nmag_4297 |
| 32,420 | 32,857 | PhiH1_255 | 438 | − | - | HNH-type endonuclease | PhiCh1p48, ORF47 Nmag_4296 |
| 32,854 | 33,255 | PhiH1_260 | 402 | − | - | uncharacterized protein | [ELY96531.1] |
| 33,248 | 34,024 | PhiH1_265 | 777 | − | - | parA domain protein | PhiCh1p47, ORF46 Nmag_4295 |
| 34,161 | 34,430 | PhiH1_270 | 270 | − | repR | repressor protein RepR | 5 PhiCh1p49, ORF48 5 Nmag_4298 [ELZ06324.1] |
| 34,730 | 35,071 | PhiH1_275 | 342 | + | - | uncharacterized protein | - |
| 35,068 | 35,424 | PhiH1_280 | 357 | + | - | uncharacterized protein | PhiCh1p50, ORF49 |
| 35,381 | 38,167 | PhiH1_285 | 2787 | + | repH | plasmid replication protein RepH | 4 PhiCh1p54, ORF53 4 PhiCh1p55, ORF54 Nmag_4299 |
| 38,262 | 38,489 | PhiH1_290 | 228 | − | imm | probable immunity protein Imm | PhiCh1p56, ORF55 Nmag_4300 |
| 38,733 | 39,263 | PhiH1_295 | 531 | + | - | transcriptional regulator, PadR-like family | PhiCh1p57, ORF56 Nmag_4301 |
| 39,260 | 39,385 | PhiH1_300 | 126 | + | - | CxxC motif protein | - |
| 39,382 | 39,978 | PhiH1_305 | 597 | + | - | uncharacterized protein | PhiCh1p59, ORF58 Nmag_4303 |
| 39,975 | 40,133 | PhiH1_310 | 159 | + | - | uncharacterized protein | - |
| 40,153 | 40,902 | PhiH1_315 | 750 | + | pcnA | DNA polymerase sliding clamp PcnA | PhiCh1p60, ORF59 Nmag_4211 |
| 40,908 | 41,339 | PhiH1_320 | 432 | + | - | uncharacterized protein | PhiCh1p61, ORF60 Nmag_4212 |
| 41,339 | 41,554 | PhiH1_325 | 216 | + | - | uncharacterized protein | PhiCh1p62, ORF61 Nmag_4213 |
| 41,547 | 42,041 | PhiH1_330 | 495 | + | - | uncharacterized protein | - |
| 42,098 | 42,490 | PhiH1_335 | 393 | + | tnpA | IS200-type transposase TnpA | [CAP12925.1] |
| 42,492 | 43,748 | PhiH1_340 | 1257 | + | tnpB | IS1341-type transposase TnpB | [CAP12926.1] |
| 43,808 | 44,014 | PhiH1_345 | 207 | + | - | uncharacterized protein | - |
| 44,007 | 44,234 | PhiH1_350 | 228 | + | - | uncharacterized protein | PhiCh1p66, ORF65 Nmag_4217 |
| 44,231 | 44,656 | PhiH1_355 | 426 | + | - | CxxC motif protein | PhiCh1p68, ORF67 Nmag_4219 |
| 44,646 | 45,026 | PhiH1_360 | 381 | + | - | uncharacterized protein | PhiCh1p69, ORF68 Nmag_4220 |
| 45,023 | 45,646 | PhiH1_365 | 624 | + | - | HNH-type endonuclease | [KYG11427.1] |
| 45,639 | 45,926 | PhiH1_370 | 288 | + | - | uncharacterized protein | PhiCh1p71, ORF70 Nmag_4222 |
| 45,919 | 46,350 | PhiH1_375 | 432 | + | - | DUF4326 domain protein | PhiCh1p72, ORF71 Nmag_4223 |
| 46,343 | 46,441 | PhiH1_380 | 99 | + | - | uncharacterized protein | - |
| 46,438 | 46,884 | PhiH1_385 | 447 | + | - | CxxC motif protein | PhiCh1p74, ORF73 Nmag_4225 |
| 46,865 | 47,038 | PhiH1_390 | 174 | + | - | uncharacterized protein | 5 PhiCh1p73, ORF72 5 Nmag_4224 |
| 47,031 | 47,447 | PhiH1_395 | 417 | + | - | uncharacterized protein | - |
| 47,440 | 47,739 | PhiH1_400 | 300 | + | - | NTPase protein | [PLX87675.1] |
| 47,732 | 49,618 | PhiH1_405 | 1887 | + | dcm5 | C-5 cytosine-specific DNA methylase Dcm5 | 5 PhiCh1p81, ORF80 [PCR88664.1] |
| 49,611 | 49,931 | PhiH1_410 | 321 | + | - | uncharacterized protein | PhiCh1p82, ORF81 Nmag_4234 |
| 49,918 | 50,037 | PhiH1_415 | 120 | + | - | CxxC motif protein | - |
| 50,091 | 51,452 | PhiH1_420 | 1362 | + | yhdJ | DNA methylase N-4/N-6 domain protein YhdJ | PhiCh1p83, ORF82 Nmag_4235 |
| 51,449 | 52,024 | PhiH1_425 | 576 | + | - | uncharacterized protein | PhiCh1p84, ORF83 Nmag_4236 |
| 52,021 | 52,791 | PhiH1_430 | 771 | + | - | uncharacterized protein | PhiCh1p85, ORF84 Nmag_4237 |
| 52,784 | 53,152 | PhiH1_435 | 369 | + | - | uncharacterized protein | PhiCh1p88, ORF87 Nmag_4240 |
| 53,145 | 53,504 | PhiH1_440 | 360 | + | - | uncharacterized protein | PhiCh1p89, ORF88 Nmag_4241 |
| 53,788 | 54,369 | PhiH1_445 | 582 | + | - | CxxC motif protein | PhiCh1p90, ORF89 Nmag_4242 |
| 54,403 | 54,771 | PhiH1_450 | 369 | + | - | uncharacterized protein | PhiCh1p91, ORF90 Nmag_4243 |
| 54,794 | 55,147 | PhiH1_455 | 354 | + | - | uncharacterized protein | - |
| 55,144 | 55,401 | PhiH1_460 | 258 | + | - | transmembrane domain protein | PhiCh1p93, ORF92 Nmag_4244 |
| 55,394 | 55,729 | PhiH1_465 | 336 | + | - | transmembrane domain protein 3 | PhiCh1p94, ORF93 Nmag_4245 |
| 55,794 | 57,053 | PhiH1_470 | 1260 | + | ycdA | DNA methylase N-4/N-6 domain protein YcdA | PhiCh1p95, ORF94 Nmag_4246 |
| 57,046 | 57,564 | PhiH1_475 | 519 | + | - | uncharacterized protein | PhiCh1p96, ORF95 Nmag_4247 |
| 57,621 | 57,830 | PhiH1_480 | 210 | + | - | CxxC motif protein | PhiCh1p98, ORF97 Nmag_4249 |
| 57,827> | <63 | PhiH1_485 | 309 | + | terS | terminase small subunit TerS | PhiCh1p01, ORF98 Nmag_4250 |
| No. | CRISPR Spacer Matches to phiH1 1 | Translation 2 |
|---|---|---|
 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyall-Smith, M.; Pfeifer, F.; Witte, A.; Oesterhelt, D.; Pfeiffer, F. Complete Genome Sequence of the Model Halovirus PhiH1 (ΦH1). Genes 2018, 9, 493. https://doi.org/10.3390/genes9100493
Dyall-Smith M, Pfeifer F, Witte A, Oesterhelt D, Pfeiffer F. Complete Genome Sequence of the Model Halovirus PhiH1 (ΦH1). Genes. 2018; 9(10):493. https://doi.org/10.3390/genes9100493
Chicago/Turabian StyleDyall-Smith, Mike, Felicitas Pfeifer, Angela Witte, Dieter Oesterhelt, and Friedhelm Pfeiffer. 2018. "Complete Genome Sequence of the Model Halovirus PhiH1 (ΦH1)" Genes 9, no. 10: 493. https://doi.org/10.3390/genes9100493
APA StyleDyall-Smith, M., Pfeifer, F., Witte, A., Oesterhelt, D., & Pfeiffer, F. (2018). Complete Genome Sequence of the Model Halovirus PhiH1 (ΦH1). Genes, 9(10), 493. https://doi.org/10.3390/genes9100493