The Global Hepatitis B Virus Genotype Distribution Approximated from Available Genotyping Data



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
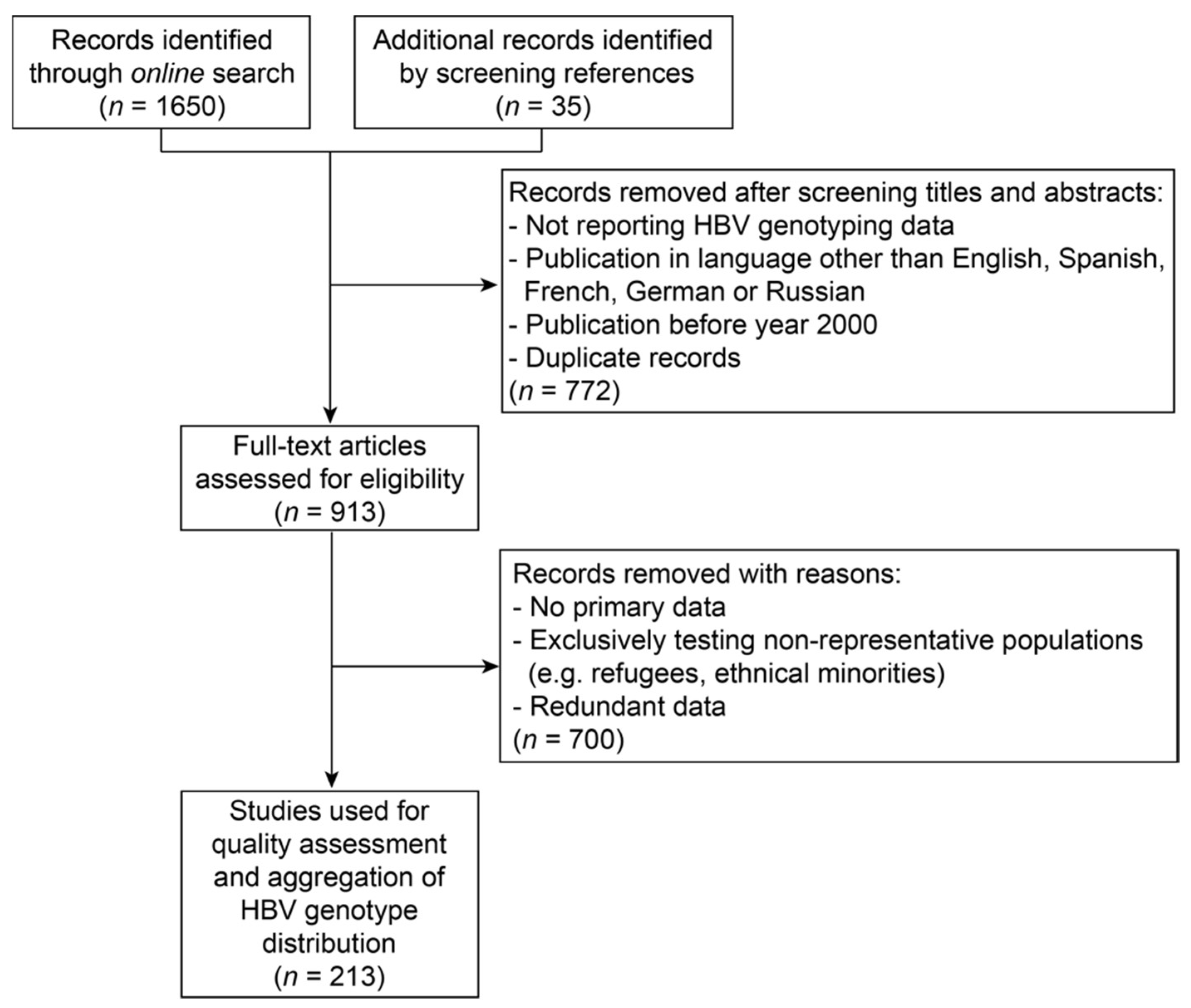
2.1. Literature Search for Hepatitis B Virus Genotyping Data
- Study population was based exclusively on individuals of foreign origin (e.g., refugees) or an ethnical minority. If information on country of origin was available, data were allocated to the respective country;
- Secondary data;
- Redundant data which were also described in another record.
2.2. Extracted Variables and Assumptions
- Year of publication;
- Publication type;
- Year of sample collection (if no year was available, two years prior to publication was assumed);
- Date of analysis (if no information was available, one year prior to publication was assumed);
- Location of sample collection;
- Selection criteria of study participants;
- Sex and age of tested population;
- Method of genotyping;
- Number of samples and result of genotyping.
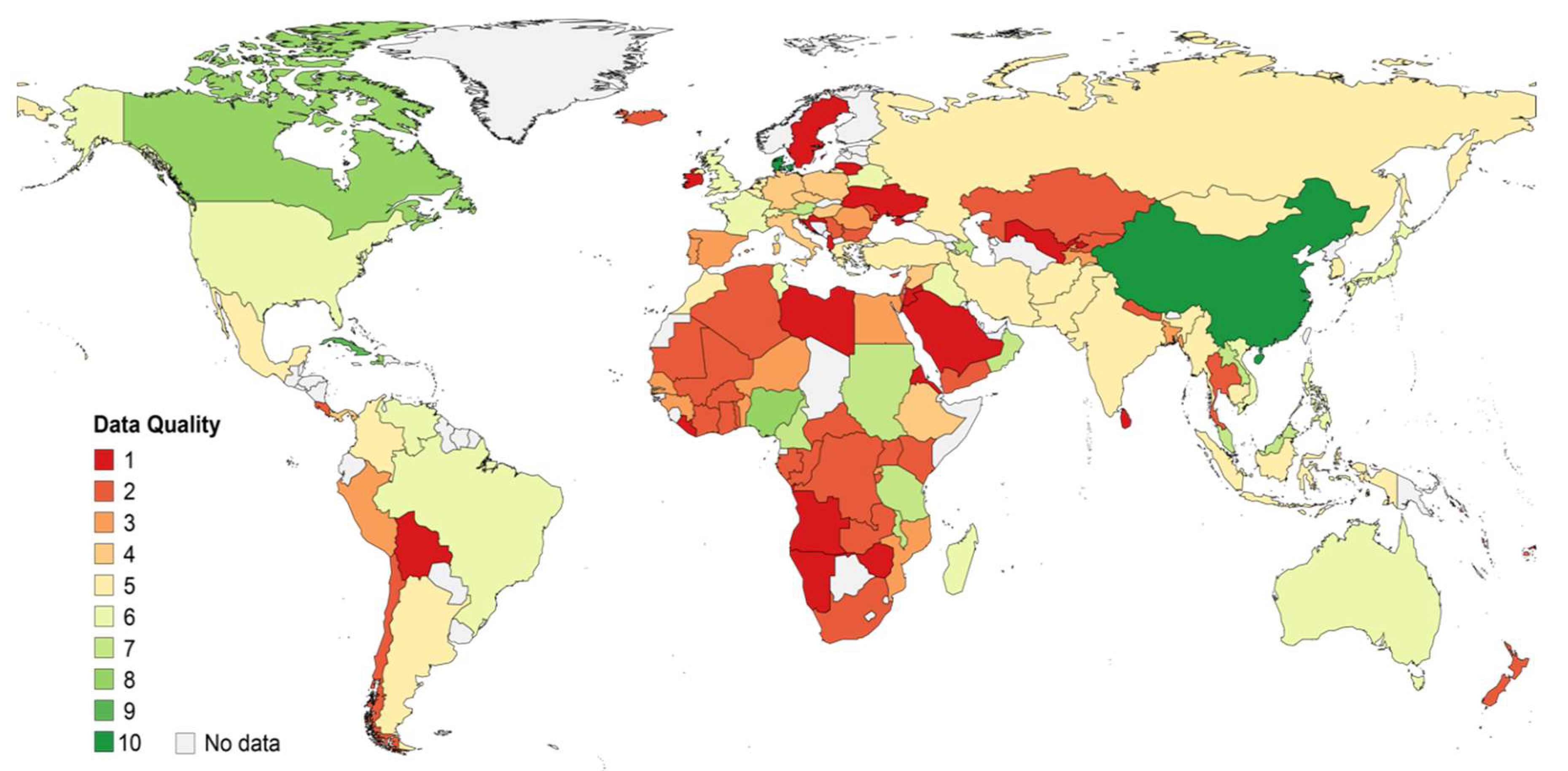
2.3. Qualitative Assessment of Data
- A study quality score was calculated with equal weighting from two different scores, the genotyping and the generalizability score (lowest score implying the lowest quality):
- (a)
- Genotyping score: Reliability of genotype information. A weighted average was calculated from scores for:
- ○
- Year of sample analysis in studies (30% weighting) (median was used in case of year range provided):
- ▪
- Before 2010 (before genotype I was described): Score 1;
- ▪
- 2010 or later: Score 2.
- ○
- Ability of the method to correctly identify genotype, including recombinant viruses (e.g., genotype I) (70% weighting):
- ▪
- Non-sequencing-based methods like probe-/PCR-/restriction fragment length polymorphism (RFLP), or enzyme immunoassay (EIA) (partly not capable to detect all genotypes/high risk to misclassify recombinant viruses): Score 1;
- ▪
- Region of viral genome sequenced (capable to identify all genotypes but with medium risk to misclassify recombinant viruses): Score 3;
- ▪
- Whole viral genome sequenced (capable to identify all genotypes including recombinant viruses): Score 5.
- (b)
- Generalizability score: Potential for generalizability of genotype information to chronic HBV infections in a country in the year 2015. A weighted average was calculated from scores for:
- ○
- Representation of country by the study location (40% weighting):
- ▪
- Samples derived from a single town or region: Score 1;
- ▪
- Samples collected from several regions or nationwide: Score 2.
- ○
- Representation of HBV-infected population by the study population (40% weighting):
- ▪
- Favored selection (e.g., individuals of specific age group, with a specific risk factor to acquire HBV infection or showing a specific sequala): Score 1;
- ▪
- Non-favored selection (e.g., all HBV-DNA positive individuals): Score 2.
- ○
- Year of sample collection (20% weighting). Median was used in case of year range provided:
- ▪
- Before 2000: Score 1;
- ▪
- 2000–2010: Score 2;
- ▪
- 2011 or later: Score 3.
- A country quality score was calculated as the weighted average of study quality scores of all studies providing results for a certain country. Each study was weighted by the proportion of samples it contributed. The result was multiplied by a score for the sum of genotyped samples from all studies for the country:
- ▪
- <100 samples: Score 1;
- ▪
- 100–999 samples: Score 2;
- ▪
- 1000 or more samples: Score 3.
2.4. Aggregation of Genotyping Data
2.5. Approximation of Number of Infections with Each Hepatitis B Virus Genotype
2.6. Creation of Maps and Additional Software
3. Results
3.1. Description of Hepatitis B Virus Genotyping Data
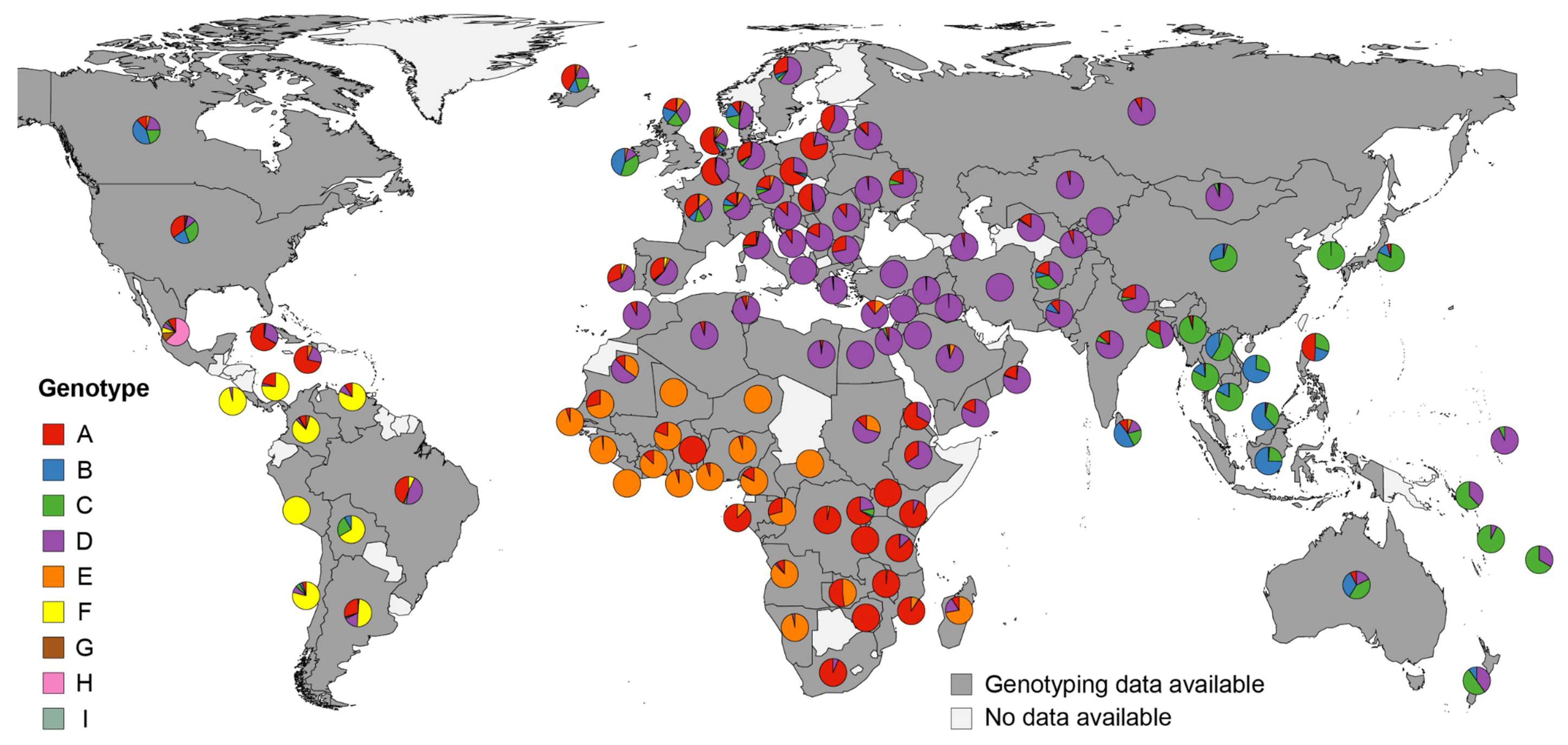
3.2. World-Wide Hepatitis B Virus Genotype Appearance
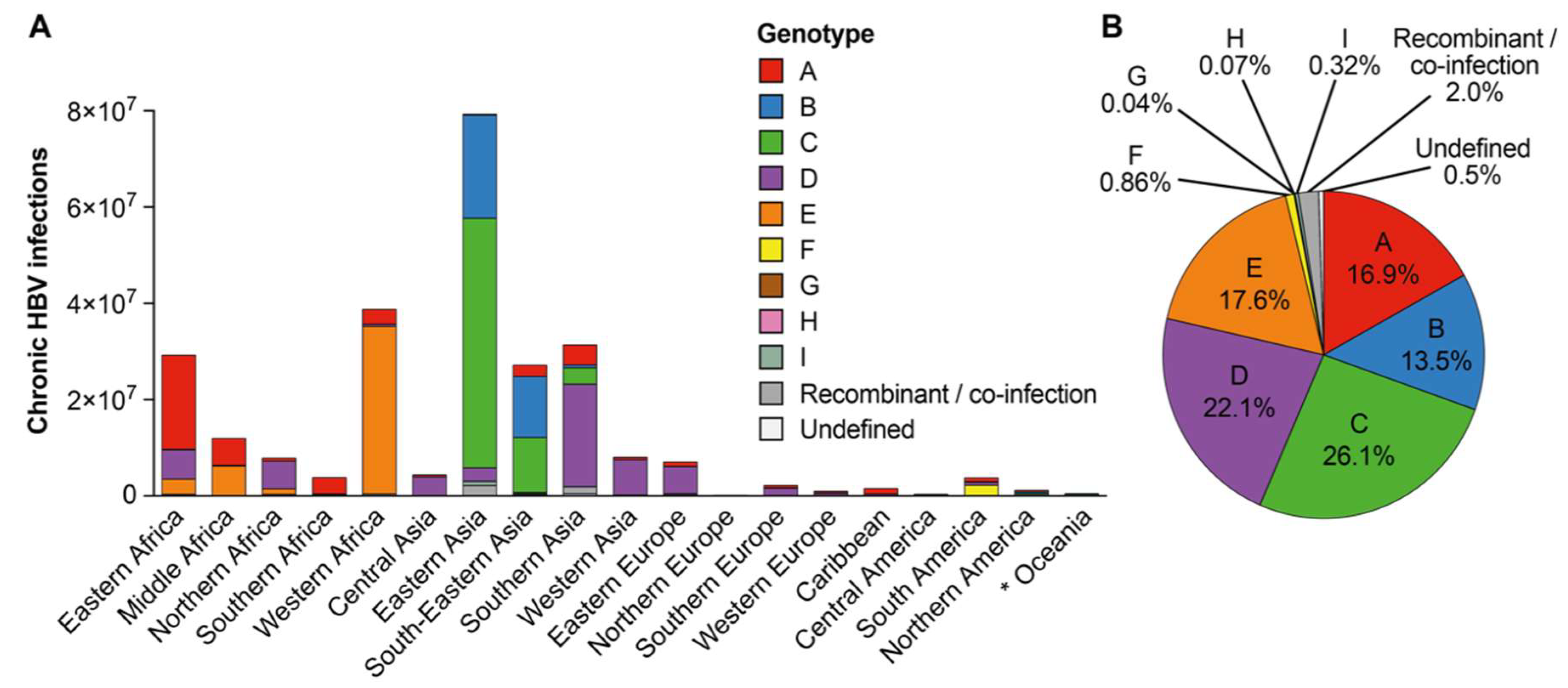
3.3. Approximation of Number of Chronic Hepatitis B Virus Infections with Each Hepatitis B Virus Genotype
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015, 386, 1546–1555. [Google Scholar] [CrossRef]
- Word Health Organisation. Global Hepatitis Report 2017; World Health Organisation: Geneva, Switzerland, 2017. [Google Scholar]
- Kramvis, A. Genotypes and genetic variability of hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Pourkarim, M.R.; Amini-Bavil-Olyaee, S.; Kurbanov, F.; Van Ranst, M.; Tacke, F. Molecular identification of hepatitis B virus genotypes/subgenotypes: Revised classification hurdles and updated resolutions. World J. Gastroenterol. 2014, 20, 7152–7168. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Trinh, T.N.; Abe, K. New complex recombinant genotype of hepatitis B virus identified in Vietnam. J. Virol. 2008, 82, 5657–5663. [Google Scholar] [PubMed]
- Tatematsu, K.; Tanaka, Y.; Kurbanov, F.; Sugauchi, F.; Mano, S.; Maeshiro, T.; Nakayoshi, T.; Wakuta, M.; Miyakawa, Y.; Mizokami, M. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a japanese patient and provisionally assigned to new genotype J. J. Virol. 2009, 83, 10538–10547. [Google Scholar] [CrossRef] [PubMed]
- Kurbanov, F.; Tanaka, Y.; Kramvis, A.; Simmonds, P.; Mizokami, M. When should “I” consider a new hepatitis B virus genotype? J. Virol. 2008, 82, 8241–8242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Yuan, Q.; Ge, S.X.; Wang, H.Y.; Zhang, Y.L.; Chen, Q.R.; Zhang, J.; Chen, P.J.; Xia, N.S. Molecular and phylogenetic analyses suggest an additional hepatitis B virus genotype “I”. PLoS ONE 2010, 5, e9297. [Google Scholar] [CrossRef] [PubMed]
- Arankalle, V.A.; Gandhe, S.S.; Borkakoty, B.J.; Walimbe, A.M.; Biswas, D.; Mahanta, J. A novel HBV recombinant (genotype I) similar to Vietnam/Laos in a primitive tribe in Eastern India. J. Viral Hepat. 2010, 17, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Osiowy, C.; Kaita, K.; Solar, K.; Mendoza, K. Molecular characterization of hepatitis B virus and a 9-year clinical profile in a patient infected with genotype I. J. Med. Virol. 2010, 82, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Locarnini, S.; Littlejohn, M.; Aziz, M.N.; Yuen, L. Possible origins and evolution of the hepatitis B virus (HBV). Semin. Cancer Biol. 2013, 23, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Sunbul, M. Hepatitis B virus genotypes: Global distribution and clinical importance. World J. Gastroenterol. 2014, 20, 5427–5434. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A. The clinical implications of hepatitis B virus genotypes and HBeAg in pediatrics. Rev. Med. Virol. 2016, 26, 285–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.J.; Kao, J.H. Global perspective on the natural history of chronic hepatitis B: Role of hepatitis B virus genotypes A to J. Semin. Liver Dis. 2013, 33, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Krekulova, L.; Rehak, V.; da Silva Filho, H.P.; Zavoral, M.; Riley, L.W. Genotypic distribution of hepatitis B virus in the Czech Republic: A possible association with modes of transmission and clinical outcome. Eur. J. Gastroenterol. Hepatol. 2003, 15, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Inui, A.; Fujisawa, T.; Takano, T.; Tajiri, H.; Murakami, J.; Suzuki, M. Transmission route and genotype of chronic hepatitis B virus infection in children in Japan between 1976 and 2010: A retrospective, multicenter study. Hepatol. Res. 2015, 45, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Kao, J.H. Hepatitis B virus genotypes and variants. Cold Spring Harb. Perspect. Med. 2015, 5, a021436. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Borold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the origin and evolution of hepatitis B viruses by means of a family of non-enveloped fish viruses. Cell Host Microbe 2017, 22, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Paraskevis, D.; Magiorkinis, G.; Magiorkinis, E.; Ho, S.Y.; Belshaw, R.; Allain, J.P.; Hatzakis, A. Dating the origin and dispersal of hepatitis B virus infection in humans and primates. Hepatology 2013, 57, 908–916. [Google Scholar] [CrossRef] [PubMed]
- United Nations, Department of Economic and Social Affairs Population Division. World Population Prospects: The 2017 Revision, Dvd ed.; United Nations: New York, NY, USA, 2017. [Google Scholar]
- Reed, F.A.; Tishkoff, S.A. African human diversity, origins and migrations. Curr. Opin. Genet. Dev. 2006, 16, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.M.; Goel, A.; Kannangai, R.; Abraham, P. Further evidence of hepatitis B virus genotype I circulation in Northeast India. Infect. Genet. Evol. 2013, 18, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhu, C.; Zheng, W.; Carr, M.J.; Higgins, D.G.; Zhang, Z. Subgenotype reclassification of genotype B hepatitis B virus. BMC Gastroenterol. 2012, 12, 116. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhu, C.; Zheng, W.; Zheng, W.; Ling, C.; Carr, M.J.; Higgins, D.G.; Zhang, Z. Subgenotyping of genotype C hepatitis B virus: Correcting misclassifications and identifying a novel subgenotype. PLoS ONE 2012, 7, e47271. [Google Scholar] [CrossRef] [PubMed]
- Yousif, M.; Kramvis, A. Genotype D of hepatitis B virus and its subgenotypes: An update. Hepatol. Res. 2013, 43, 355–364. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velkov, S.; Ott, J.J.; Protzer, U.; Michler, T. The Global Hepatitis B Virus Genotype Distribution Approximated from Available Genotyping Data. Genes 2018, 9, 495. https://doi.org/10.3390/genes9100495
Velkov S, Ott JJ, Protzer U, Michler T. The Global Hepatitis B Virus Genotype Distribution Approximated from Available Genotyping Data. Genes. 2018; 9(10):495. https://doi.org/10.3390/genes9100495
Chicago/Turabian StyleVelkov, Stoyan, Jördis J. Ott, Ulrike Protzer, and Thomas Michler. 2018. "The Global Hepatitis B Virus Genotype Distribution Approximated from Available Genotyping Data" Genes 9, no. 10: 495. https://doi.org/10.3390/genes9100495
APA StyleVelkov, S., Ott, J. J., Protzer, U., & Michler, T. (2018). The Global Hepatitis B Virus Genotype Distribution Approximated from Available Genotyping Data. Genes, 9(10), 495. https://doi.org/10.3390/genes9100495