Comparative Analysis of Surface Layer Glycoproteins and Genes Involved in Protein Glycosylation in the Genus Haloferax

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and 16S rRNA/polB Gene Sequencing
2.2. Genomic DNA Extraction by Spooling
2.3. Phylogenetic Analysis
2.4. Estimation of the Significance of a Protein Sequence Alignment Using PRSS
2.5. Protein Function Prediction
2.6. Recombination Analysis
2.7. Average Nucleotide Identity Calculation
2.8. Culture Conditions
2.9. Protein Sample Preparation
2.10. SDS-PAGE and Protein Detection
2.11. Mass Spectrometry
3. Results
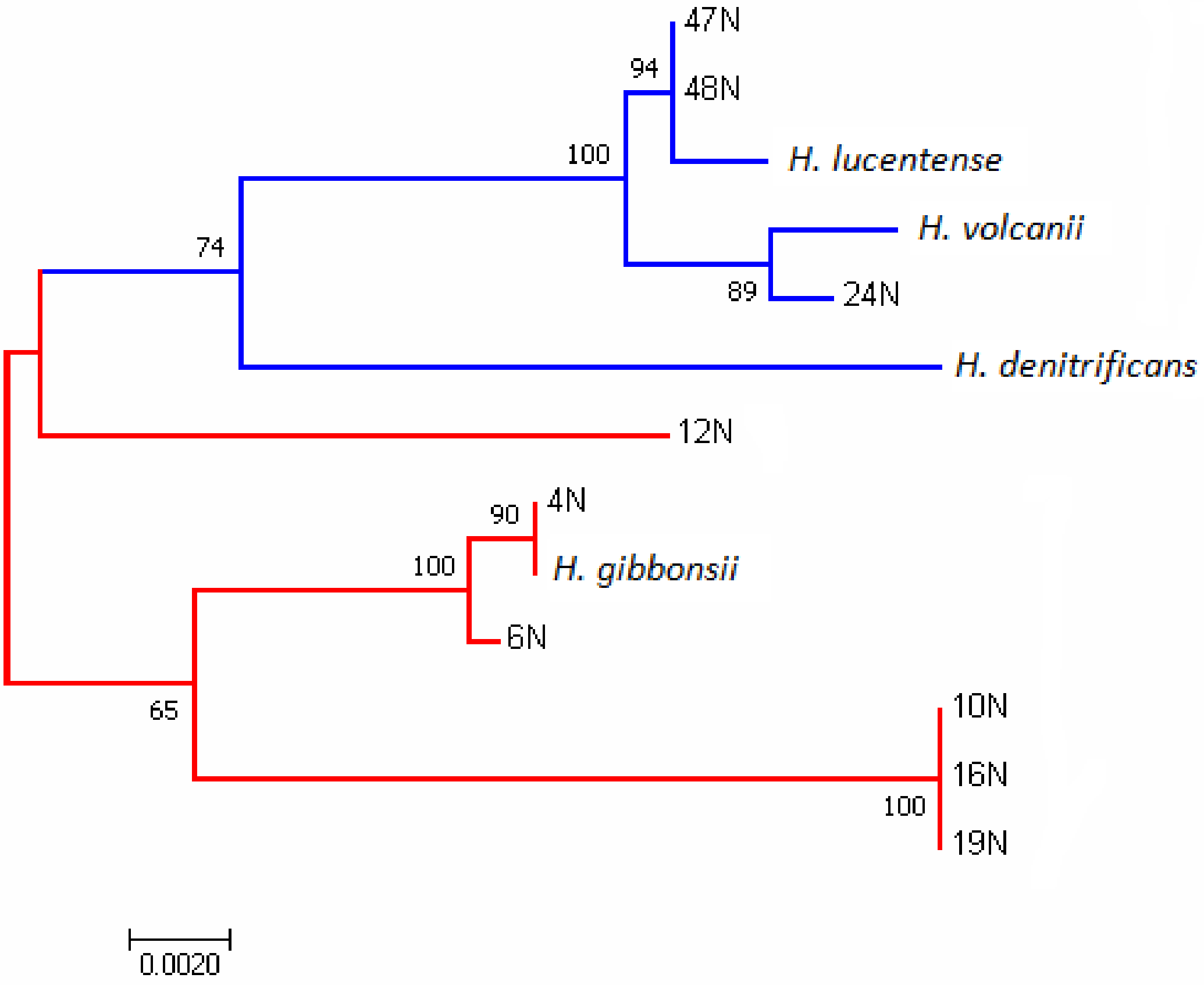
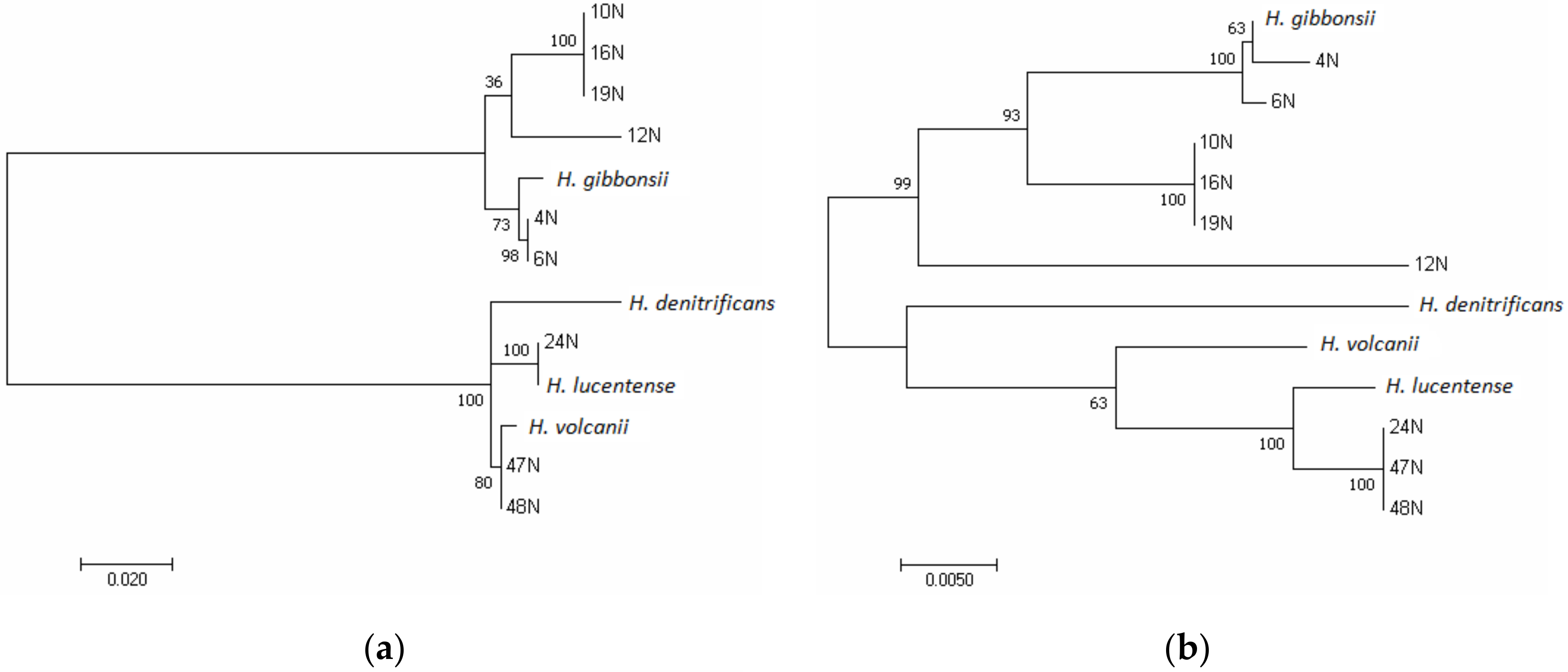
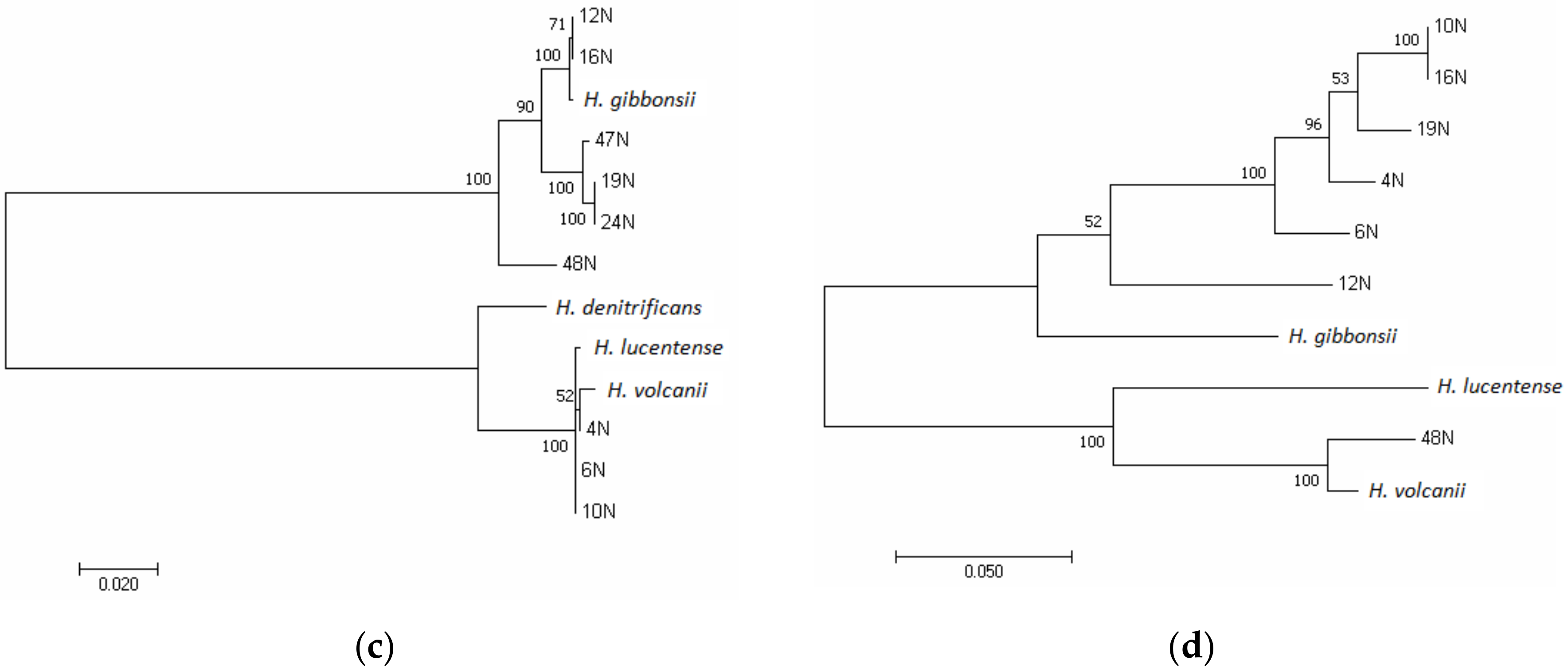
3.1. Phylogenetic Analysis of the Isolates Using the rpoB1 Gene and ANI
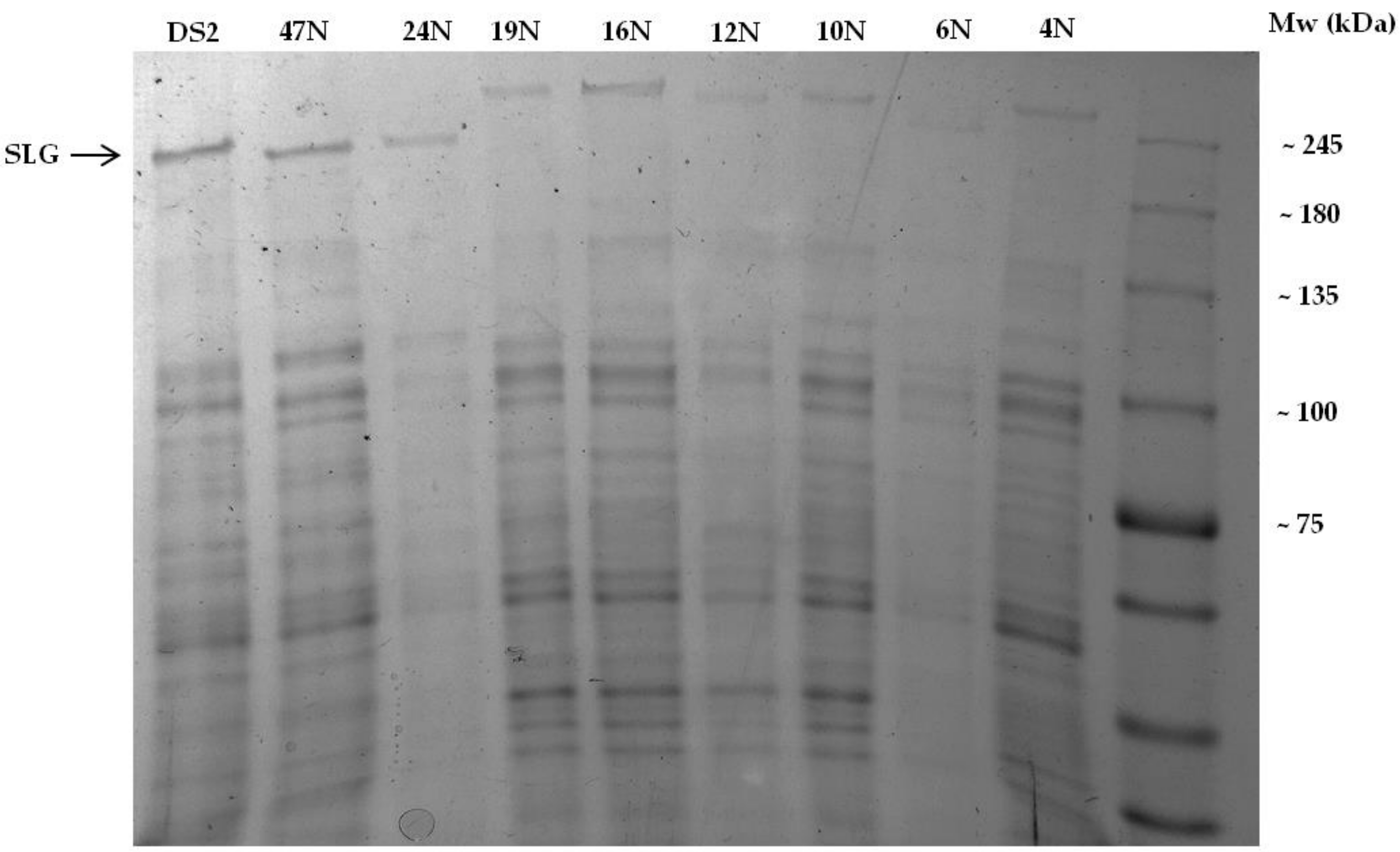
3.2. Two Distinct S-layer Glycoproteins Exist within the Closely-Related Haloferax Isolates
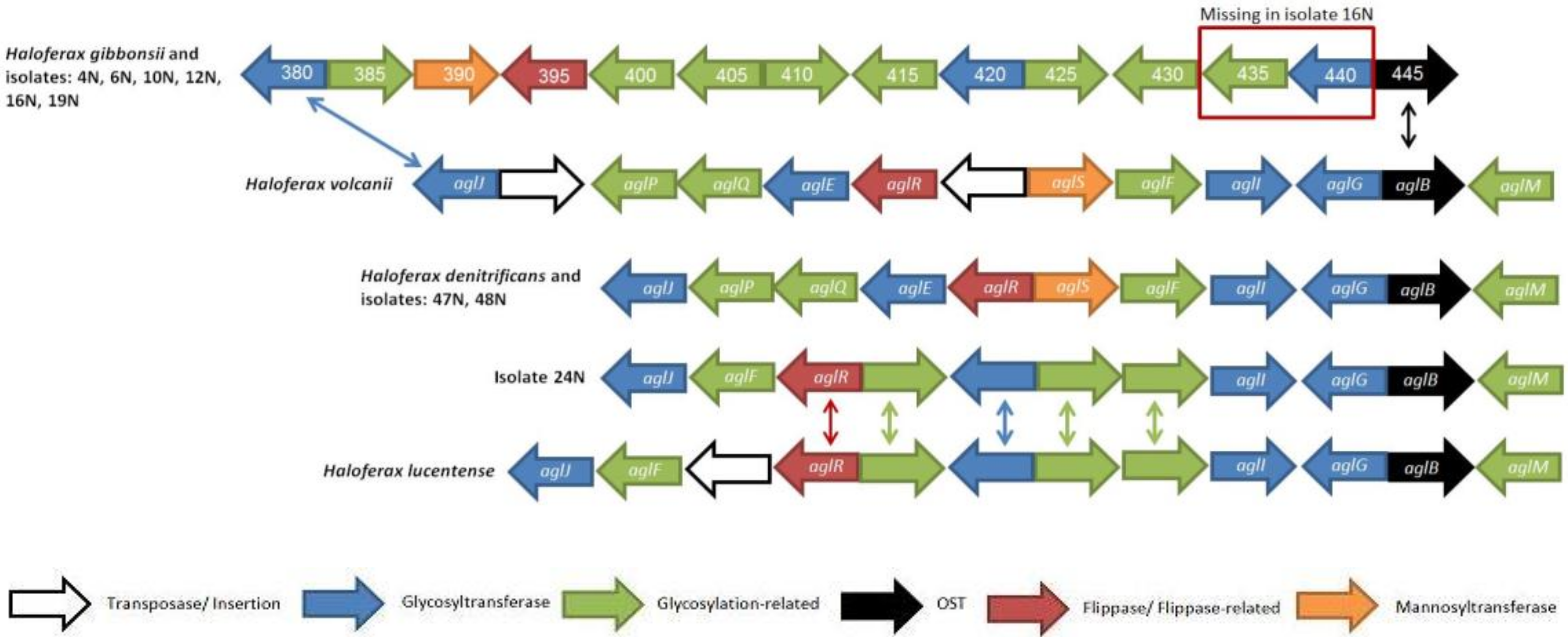
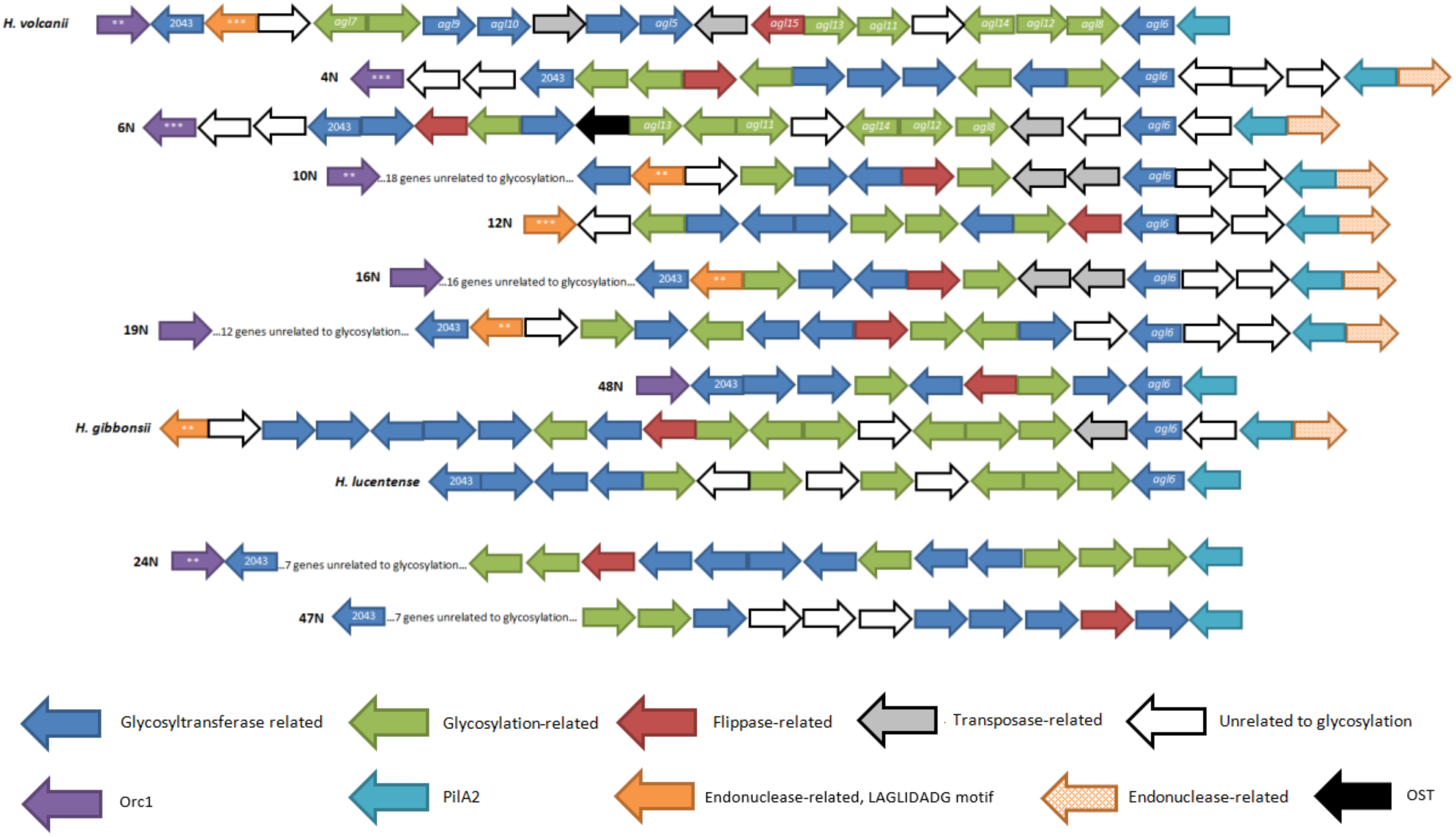
3.3. The Composition of Genes in N-Glycosylation Pathway Clusters Varies in the Isolates
3.4. The Glycosyltransferase Gene aglJ Is Relatively Conserved
3.5. Phylogeny of the Mannosyltransferase-Encoding aglD Gene Is Similar to aglJ in the H. gibbonsii Clade but Not in the H. volcanii-Related Isolates
3.6. The Gene Encoding AglB Most Likely Underwent Lateral Gene Transfer in Haloferax Species
3.7. Alternative Glycosylation Clusters in Isolates and Related Haloferax Species
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Akca, E.; Claus, H.; Schultz, N.; Karbach, G.; Schlott, B.; Debaerdemaeker, T.; Declercq, J.-P.; König, H. Genes and derived amino acid sequences of S-layer proteins from mesophilic, thermophilic, and extremely thermophilic methanococci. Extremophiles 2002, 6, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Tripepi, M.; You, J.; Temel, S.; Önder, Ö.; Brisson, D.; Pohlschröder, M. N-glycosylation of Haloferax volcanii flagellins requires known Agl proteins and is essential for biosynthesis of stable flagella. J. Bacteriol. 2012, 194, 4876–4887. [Google Scholar] [CrossRef] [PubMed]
- Esquivel, R.N.; Schulze, S.; Xu, R.; Hippler, M.; Pohlschroder, M. Identification of Haloferax volcanii Pilin N-glycans with diverse roles in pilus biosynthesis, adhesion, and microcolony formation. J. Biol. Chem. 2016, 291, 10602–10614. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, F.; Aebi, M. Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Eichler, J.; Arbiv, A.; Cohen-Rosenzweig, C.; Kaminski, L.; Kandiba, L.; Konrad, Z. N-glycosylation in Haloferax volcanii: Adjusting the sweetness. Front. Microbiol. 2013, 4, 403. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Naparstek, S.; Calo, D.; Eichler, J. Protein glycosylation as an adaptive response in Archaea: growth at different salt concentrations leads to alterations in Haloferax volcanii S-layer glycoprotein N-glycosylation. Environ. Microbiol. 2012, 14, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Abu-Qarn, M.; Giordano, A.; Battaglia, F.; Trauner, A.; Hitchen, P.G.; Morris, H.R.; Dell, A.; Eichler, J. Identification of AglE, a second glycosyltransferase involved in N-glycosylation of the Haloferax volcanii S-layer glycoprotein. J. Bacteriol. 2008, 190, 3140–3146. [Google Scholar] [CrossRef] [PubMed]
- Yurist-Doutsch, S.; Abu-Qarn, M.; Battaglia, F.; Morris, H.R.; Hitchen, P.G.; Dell, A.; Eichler, J. aglF, aglG and aglI, novel members of a gene island involved in the N-glycosylation of the Haloferax volcanii S-layer glycoprotein. Mol. Microbiol. 2008, 69, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Naparstek, S.; Kaminski, L.; Konrad, Z.; Eichler, J. Distinct glycan-charged phosphodolichol carriers are required for the assembly of the pentasaccharide N-linked to the Haloferax volcanii S-layer glycoprotein. Mol. Microbiol. 2010, 78, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, L.; Eichler, J. Identification of residues important for the activity of Haloferax volcanii AglD, a component of the archaeal N-glycosylation pathway. Archaea 2010, 2010, 315108. [Google Scholar] [CrossRef] [PubMed]
- Abu-Qarn, M.; Yurist-Doutsch, S.; Giordano, A.; Trauner, A.; Morris, H.R.; Hitchen, P.; Medalia, O.; Dell, A.; Eichler, J. Haloferax volcanii AglB and AglD are involved in N-glycosylation of the S-layer glycoprotein and proper assembly of the surface Layer. J. Mol. Biol. 2007, 374, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Kandiba, L.; Lin, C.-W.; Aebi, M.; Eichler, J.; Guerardel, Y. Structural characterization of the N-linked pentasaccharide decorating glycoproteins of the halophilic archaeon Haloferax volcanii. Glycobiology 2016, 26, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Plavner, N.; Eichler, J. Defining the topology of the N-glycosylation pathway in the halophilic archaeon Haloferax volcanii. J. Bacteriol. 2008, 190, 8045–8052. [Google Scholar] [CrossRef] [PubMed]
- Calo, D.; Guan, Z.; Naparstek, S.; Eichler, J. Different routes to the same ending: Comparing the N-glycosylation processes of Haloferax volcanii and Haloarcula marismortui, two halophilic archaea from the Dead Sea. Mol. Microbiol. 2011, 81, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Rosenzweig, C.; Yurist-Doutsch, S.; Eichler, J. AglS, a novel component of the Haloferax volcanii N-glycosylation pathway, is a dolichol phosphate-mannose mannosyltransferase. J. Bacteriol. 2012, 194, 6909–6916. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, L.; Guan, Z.; Abu-Qarn, M.; Konrad, Z.; Eichler, J. AglR is required for addition of the final mannose residue of the N-linked glycan decorating the Haloferax volcanii S-layer glycoprotein. Biochim. Biophys. Acta-Gen. Subj. 2012, 1820, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Yurist-Doutsch, S.; Magidovich, H.; Ventura, V.V.; Hitchen, P.G.; Dell, A.; Eichler, J. N-glycosylation in Archaea: On the coordinated actions of Haloferax volcanii AglF and AglM. Mol. Microbiol. 2010, 75, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Arbiv, A.; Yurist-Doutsch, S.; Guan, Z.; Eichler, J.; Hashimoto, W. AglQ is a novel component of the Haloferax volcanii N-glycosylation pathway. PLoS ONE 2013, 8, e81782. [Google Scholar] [CrossRef] [PubMed]
- Magidovich, H.; Yurist-Doutsch, S.; Konrad, Z.; Ventura, V.V.; Dell, A.; Hitchen, P.G.; Eichler, J. AglP is a S-adenosyl-l-methionine-dependent methyltransferase that participates in the N-glycosylation pathway of Haloferax volcanii. Mol. Microbiol. 2010, 76, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Yurist-Doutsch, S.; Eichler, J. Manual annotation, transcriptional analysis, and protein expression studies reveal novel genes in the agl cluster responsible for N-glycosylation in the halophilic archaeon Haloferax volcanii. J. Bacteriol. 2009, 191, 3068–3075. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, L.; Guan, Z.; Yurist-Doutsch, S.; Eichler, J. Two distinct N-glycosylation pathways process the Haloferax volcanii S-layer glycoprotein upon changes in environmental salinity. MBio 2013, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, L.; Lurie-Weinberger, M.N.; Allers, T.; Gophna, U.; Eichler, J. Phylogenetic- and genome-derived insight into the evolution of N-glycosylation in Archaea. Mol. Phylogenet. Evol. 2013, 68, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Youssef, N.H.; Ashlock-Savage, K.N.; Elshahed, M.S. Phylogenetic diversities and community structure of members of the extremely halophilic Archaea (order Halobacteriales) in multiple saline sediment habitats. Appl. Environ. Microbiol. 2012, 78, 1332–1344. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R. Effective protein sequence comparison. Methods Enzymol. 1996, 266, 227–258. [Google Scholar] [PubMed]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred Server at its core. J. Mol. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Smith, J. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.M.; Konstantinidis, K.T. Bypassing cultivation to identify bacterial species culture-independent genomic approaches identify credibly distinct clusters, avoid cultivation bias, and provide true insights into microbial species. Microbe 2014, 9, 111–118. [Google Scholar]
- Klappenbach, J.A.; Goris, J.; Vandamme, P.; Coenye, T.; Konstantinidis, K.T.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Allers, T.; Barak, S.; Liddell, S.; Wardell, K.; Mevarech, M. Improved strains and plasmid vectors for conditional overexpression of His-tagged proteins in Haloferax volcanii. Appl. Environ. Microbiol. 2010, 76, 1759–1769. [Google Scholar] [CrossRef] [PubMed]
- He, F. Laemmli-SDS-PAGE. Bio-Protocol 2011, 1, 16. [Google Scholar] [CrossRef]
- Walsh, D.A.; Bapteste, E.; Kamekura, M.; Doolittle, W.F. Evolution of the RNA Polymerase B’ Subunit Gene (rpoB’) in Halobacteriales: a complementary molecular marker to the SSU rRNA Gene. Mol. Biol. Evol. 2004, 21, 2340–2351. [Google Scholar] [CrossRef] [PubMed]
- Papke, R.T.; Koenig, J.E.; Rodríguez-Valera, F.; Doolittle, W.F. Frequent recombination in a saltern population of Halorubrum. Science 2004, 306, 1928–1929. [Google Scholar] [PubMed]
- Papke, R.T.; Zhaxybayeva, O.; Feil, E.J.; Sommerfeld, K.; Muise, D.; Doolittle, W.F. Searching for species in haloarchaea. Proc. Natl. Acad. Sci. USA 2007, 104, 14092–14097. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Gogarten, J.P.; Papke, R.T. Quantifying homologous replacement of loci between haloarchaeal species. Genome Biol. Evol. 2012, 4, 1223–1244. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.M.; Konstantinidis, K.T. The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. Peer J. Prepr. 2016. [Google Scholar] [CrossRef]
- Sumper, M.; Berg, E.; Mengele, R.; Strobel, I. Primary structure and glycosylation of the S-layer protein of Haloferax volcanii. J. Bacteriol. 1990, 172, 7111–7118. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S. F.; Gish, W.; Miller, W.; Myers, E. W.; Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [PubMed]
- Gish, W.; States, D. J. Identification of protein coding regions by database similarity search. Nat. Genet. 1993, 3, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, L.; Abu-Qarn, M.; Guan, Z.; Naparstek, S.; Ventura, V.V.; Raetz, C.R.H.; Hitchen, P.G.; Dell, A.; Eichler, J. AglJ adds the first sugar of the N-linked pentasaccharide decorating the Haloferax volcanii S-layer glycoprotein. J. Bacteriol. 2010, 192, 5572–5579. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Rosenzweig, C.; Guan, Z.; Shaanan, B.; Eichler, J. Substrate promiscuity: AglB, the archaeal oligosaccharyltransferase, can process a variety of lipid-linked glycans. Appl. Environ. Microbiol. 2014, 80, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Vrionis, H.A.; Schneider, J.; Berezuk, A.; Khursigara, C.M.; Jarrell, K.F. Complementation of an AglB Mutant of Methanococcus maripaludis with heterologous oligosaccharyltransferases. PLoS ONE 2016, 11, e0167611. [Google Scholar] [CrossRef] [PubMed]
- Omelchenko, M.V.; Makarova, K.S.; Wolf, Y.I.; Rogozin, I.B.; Koonin, E.v. Evolution of mosaic operons by horizontal gene transfer and gene displacement in situ. Genome Biol. 2003, 4, R55. [Google Scholar] [CrossRef] [PubMed]
- Eichler, J.; Koomey, M. Sweet new roles for protein glycosylation in prokaryotes. Trends Microbiol. 2017, 25, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Dassa, B.; London, N.; Stoddard, B.L.; Schueler-Furman, O.; Pietrokovski, S. Fractured genes: A novel genomic arrangement involving new split inteins and a new homing endonuclease family. Nucleic Acids Res. 2009, 37, 2560–2573. [Google Scholar] [CrossRef] [PubMed]
- Belle, A.; Landthaler, M.; Shub, D.A. Intronless homing: Site-specific endonuclease SegF of bacteriophage T4 mediates localized marker exclusion analogous to homing endonucleases of group I introns. Genes Dev. 2002, 16, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Thane Papke, R.; Naor, A.; Gophna, U. Speciation in the shadow of recombination and lateral gene transfer. In Lateral Gene Transfer in Evolution; Springer: New York, NY, USA, 2013; pp. 275–289. [Google Scholar]
- Andam, C.P.; Williams, D.; Gogarten, J.P. Biased gene transfer mimics patterns created through shared ancestry. Proc. Natl. Acad. Sci. USA 2010, 107, 10679–10684. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.J.; Friedman, J.; Cordero, O.X.; Preheim, S.P.; Timberlake, S.C.; Szabo, G.; Polz, M.F.; Alm, E.J. population genomics of early events in the ecological differentiation of bacteria. Science 2012, 336, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Valera, F.; Martin-Cuadrado, A.-B.; Rodriguez-Brito, B.; Pašić, L.; Thingstad, T.F.; Rohwer, F.; Mira, A. Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 2009, 7, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Avrani, S.; Wurtzel, O.; Sharon, I.; Sorek, R.; Lindell, D. Genomic island variability facilitates Prochlorococcus–virus coexistence. Nature 2011, 474, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Cuadros-Orellana, S.; Martin-Cuadrado, A.-B.; Legault, B.; D’Auria, G.; Zhaxybayeva, O.; Papke, R.T.; Rodriguez-Valera, F. Genomic plasticity in prokaryotes: The case of the square haloarchaeon. ISME J. 2007, 1, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Croucher, N.J.; Kagedan, L.; Thompson, C.M.; Parkhill, J.; Bentley, S.D.; Finkelstein, J.A.; Lipsitch, M.; Hanage, W.P. Selective and genetic constraints on pneumococcal serotype switching. PLoS Genet. 2015, 11, e1005095. [Google Scholar] [CrossRef] [PubMed]
- Taketani, M.; Donia, M.S.; Jacobson, A.N.; Lambris, J.D.; Fischbach, M.A. A phase-variable surface layer from the gut symbiont Bacteroides thetaiotaomicron. MBio 2015, 6, e01339-15. [Google Scholar] [CrossRef] [PubMed]
- Daubin, V.; Lerat, E.; Perrière, G. The source of laterally transferred genes in bacterial genomes. Genome Biol. 2003, 4, R57. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Juez, G.; Rodriguez-Valera, F. Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites. Mol. Microbiol. 1993, 9, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rodriguez, R.; Haitjema, C.; Huang, Q.; Nam, K.H.; Bernardis, S.; Ke, A.; DeLisa, M.P. Envelope stress is a trigger of CRISPR RNA-mediated DNA silencing in Escherichia coli. Mol. Microbiol. 2011, 79, 584–599. [Google Scholar] [CrossRef] [PubMed]
- Barzel, A.; Naor, A.; Privman, E.; Kupiec, M.; Gophna, U. Homing endonucleases residing within inteins: Evolutionary puzzles awaiting genetic solutions. Biochem. Soc. Trans. 2011, 39, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Naor, A.; Altman-Price, N.; Soucy, S.M.; Green, A.G.; Mitiagin, Y.; Turgeman-Grott, I.; Davidovich, N.; Gogarten, J.P.; Gophna, U. Impact of a homing intein on recombination frequency and organismal fitness. Proc. Natl. Acad. Sci. USA 2016, 113, E4654–E4661. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate Number | Place of Isolation | Number of Contigs over 1 Kbp | GC Content (%) | Total Length (bp) * |
|---|---|---|---|---|
| 4N | Michmoret pool1 | 32 | 65.31 | 4,196,082 |
| 6N | Michmoret pool2 | 23 | 65.12 | 4,414,718 |
| 10N | Michmoret pool2 | 54 | 65.2 | 4,278,547 |
| 12N | Michmoret pool2 | 46 | 65.32 | 4,031,013 |
| 16N | Michmoret pool2 | 10 | 65.74 | 3,971,935 |
| 19N | Atlit pool2 | 29 | 64.29 | 4,185,404 |
| 24N | Atlit pool4 | 9 | 65.74 | 3,914,077 |
| 47N | Atlit pool8 | 11 | 65.84 | 3,845,131 |
| 48N | Atlit pool8 | 11 | 65.82 | 3,925,572 |
| Haloferax volcanii DS2 | Dead Sea | 65 | 4,012,900 | |
| Haloferax gibbonsii ATCC 33959 | Saltern, Rio de Janeiro | 62.8 | 3,918,454 | |
| Haloferax denitrificans ATCC 35960 | Saltern, California | 66.3 | 3,825,970 | |
| Haloferax lucentense DSM 14919 | Saltern, Spain | 66.39 | 3,619,064 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalev, Y.; Soucy, S.M.; Papke, R.T.; Gogarten, J.P.; Eichler, J.; Gophna, U. Comparative Analysis of Surface Layer Glycoproteins and Genes Involved in Protein Glycosylation in the Genus Haloferax. Genes 2018, 9, 172. https://doi.org/10.3390/genes9030172
Shalev Y, Soucy SM, Papke RT, Gogarten JP, Eichler J, Gophna U. Comparative Analysis of Surface Layer Glycoproteins and Genes Involved in Protein Glycosylation in the Genus Haloferax. Genes. 2018; 9(3):172. https://doi.org/10.3390/genes9030172
Chicago/Turabian StyleShalev, Yarden, Shannon M. Soucy, R. Thane Papke, J. Peter Gogarten, Jerry Eichler, and Uri Gophna. 2018. "Comparative Analysis of Surface Layer Glycoproteins and Genes Involved in Protein Glycosylation in the Genus Haloferax" Genes 9, no. 3: 172. https://doi.org/10.3390/genes9030172
APA StyleShalev, Y., Soucy, S. M., Papke, R. T., Gogarten, J. P., Eichler, J., & Gophna, U. (2018). Comparative Analysis of Surface Layer Glycoproteins and Genes Involved in Protein Glycosylation in the Genus Haloferax. Genes, 9(3), 172. https://doi.org/10.3390/genes9030172