Minimizing Contamination from Plastic Labware in the Quantification of C16 and C18 Fatty Acids in Filter Samples of Atmospheric Particulate Matter and Their Utility in Apportioning Cooking Source Contribution to Urban PM2.5


Abstract
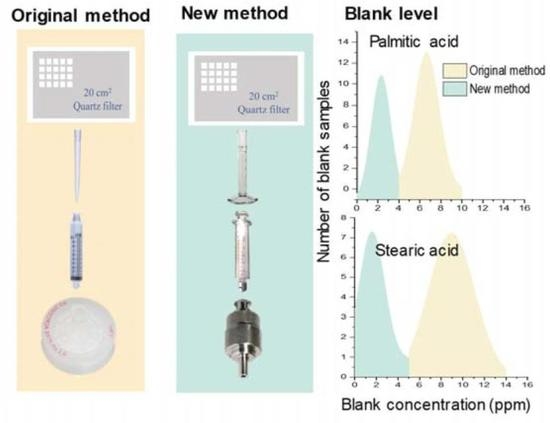
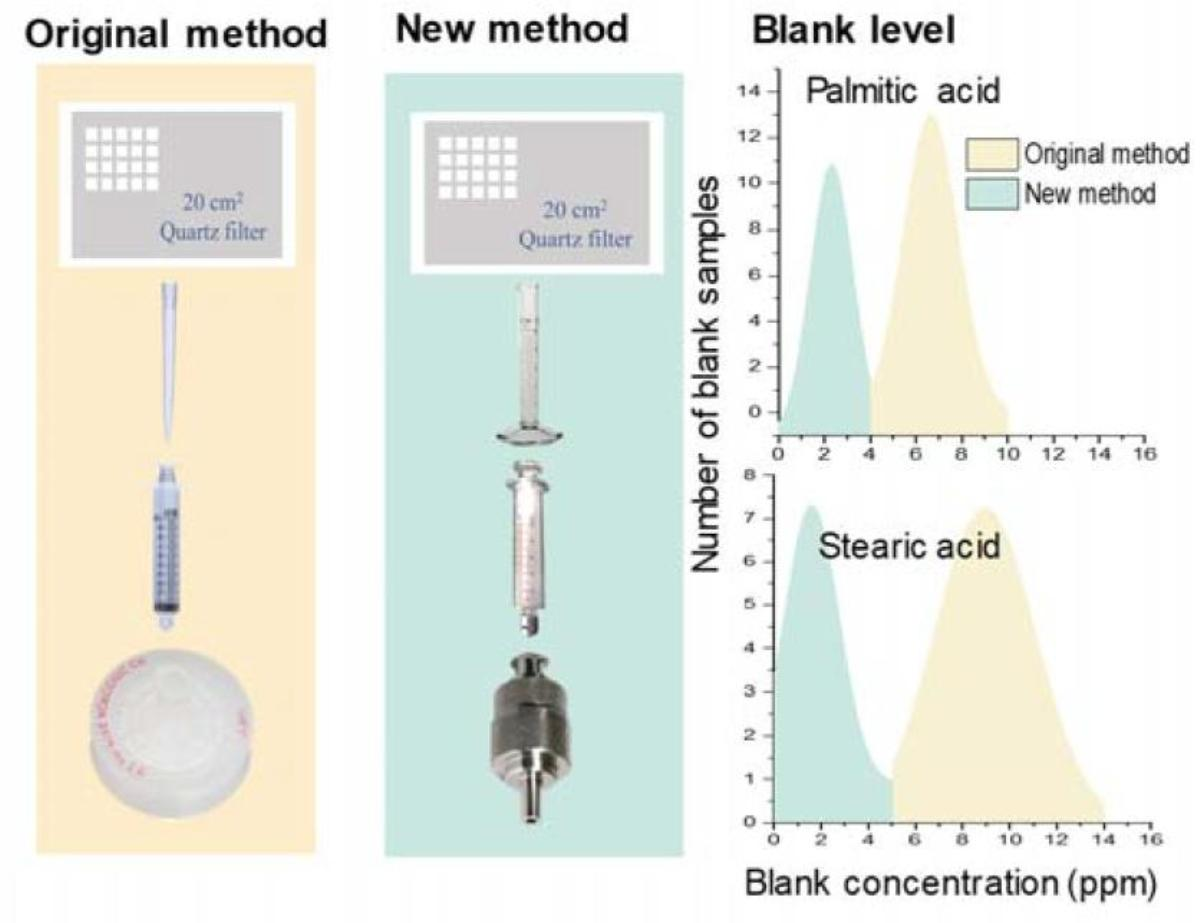
:
1. Introduction
2. Experiments
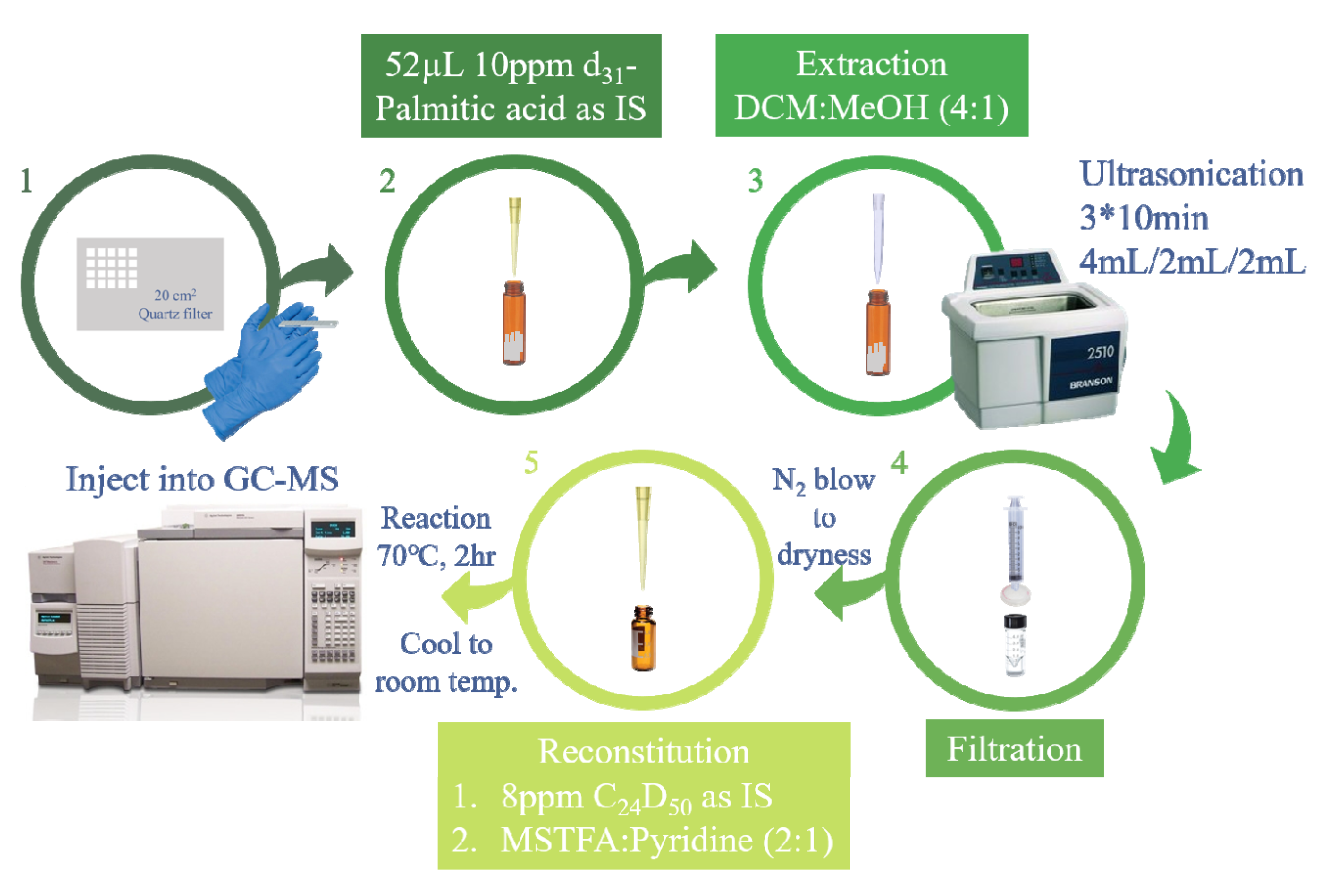
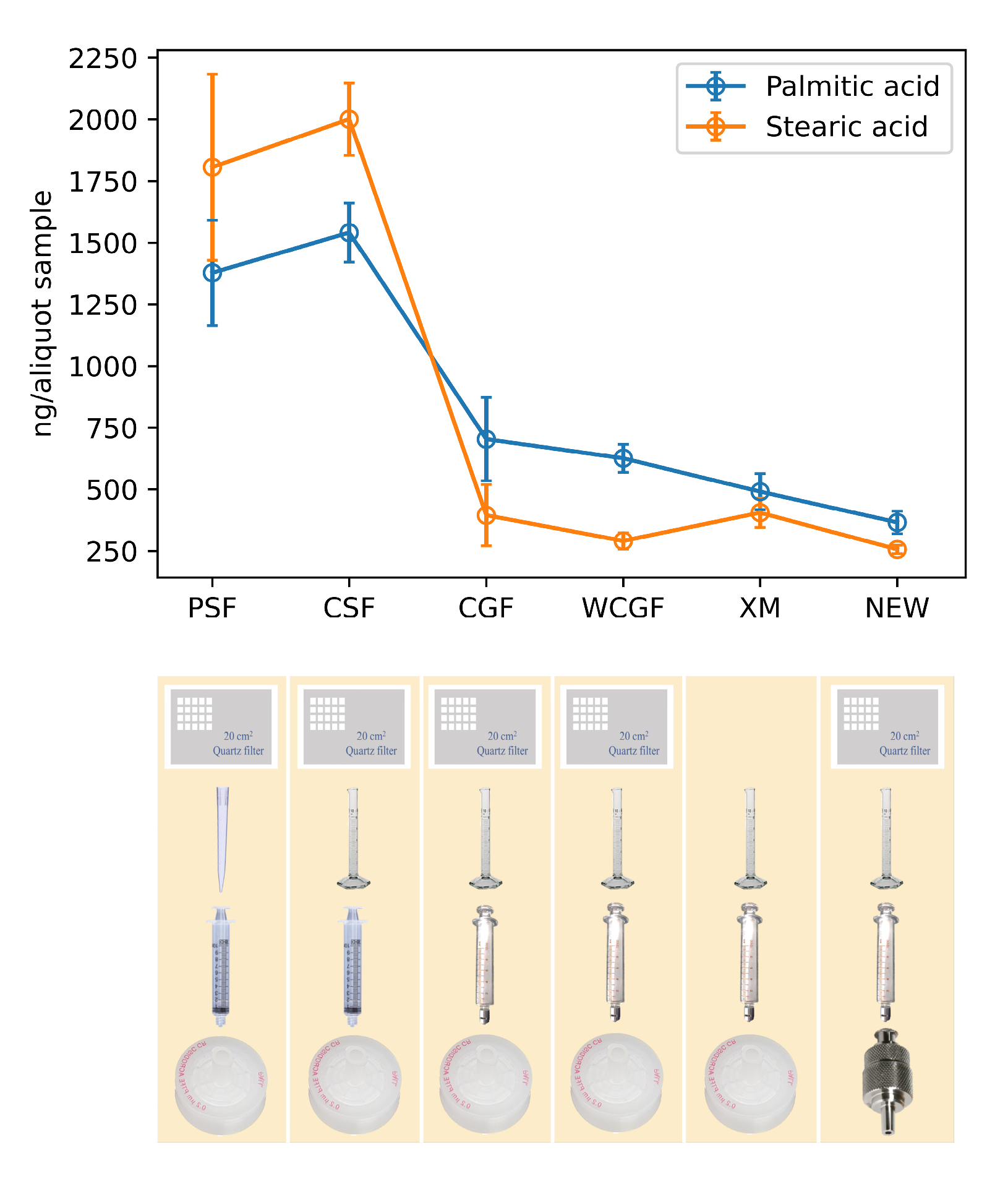
2.1. Original Analytical Method
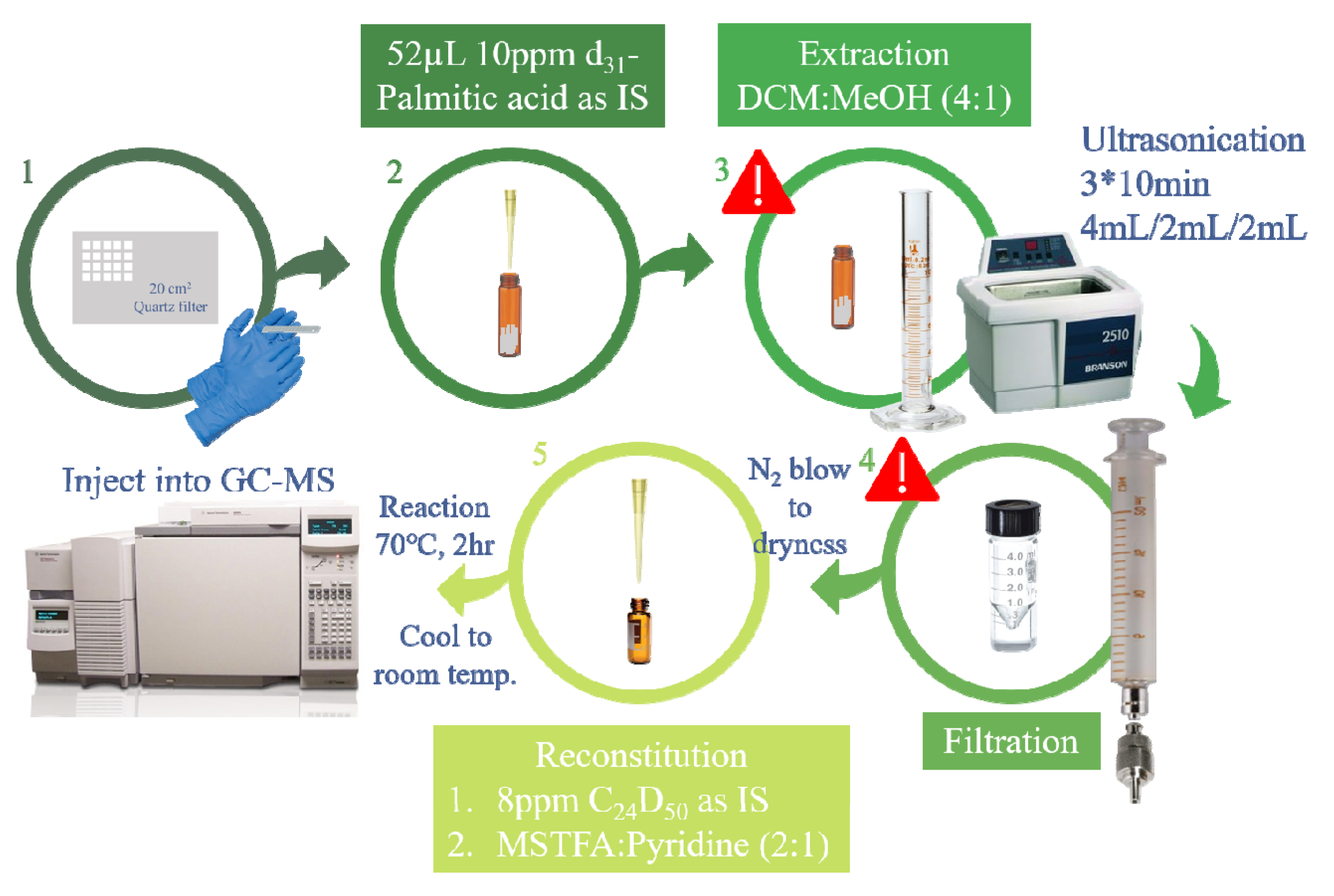
2.2. Revised Analytical Method
2.3. Ambient Filter Samples and Their Analysis
2.4. Source Apportionment by PMF Model with Cooking Related Tracers
3. Results and Discussions
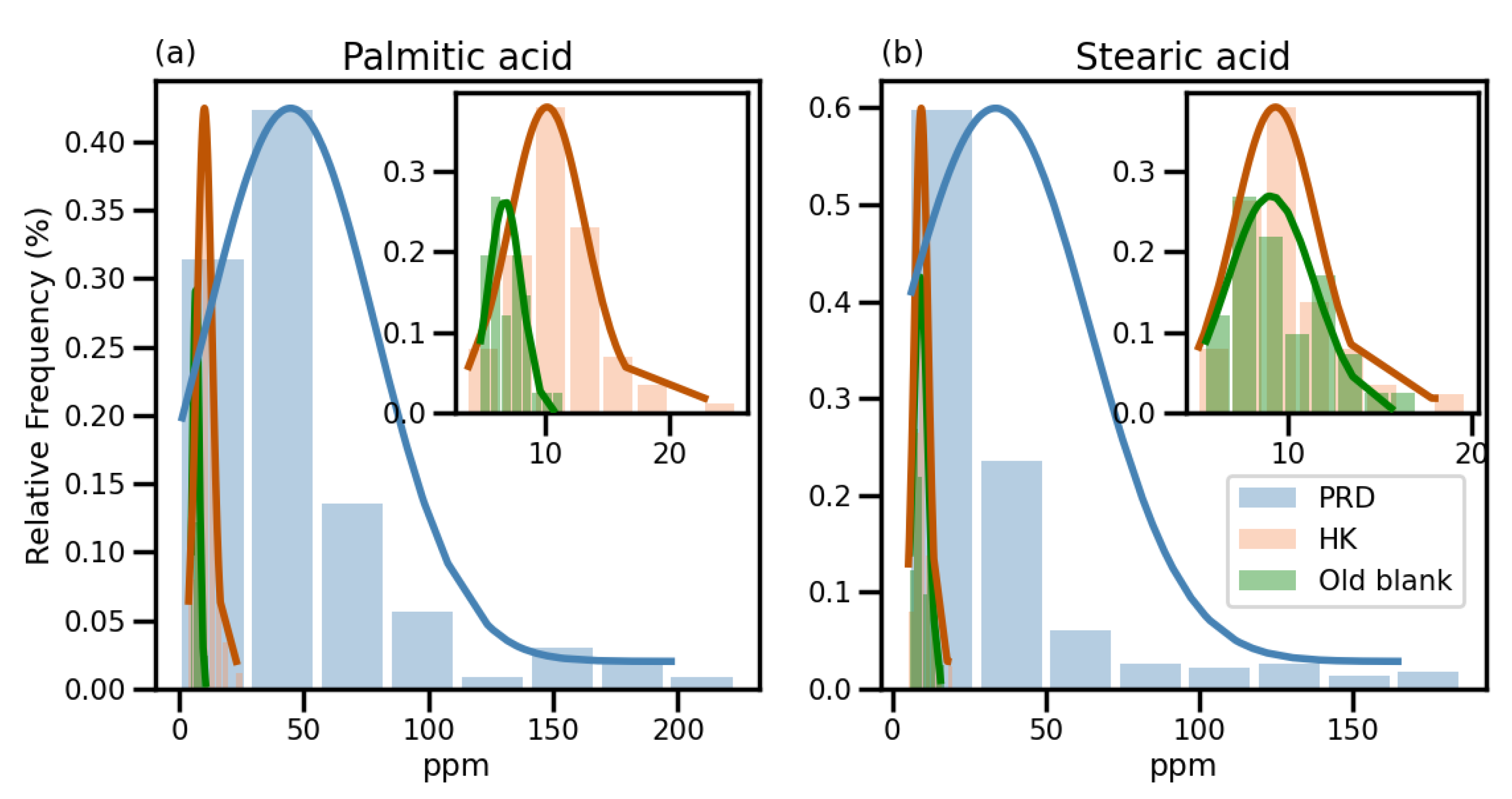
3.1. Characterization of Contamination of C16:0 and C18:0 Fatty Acids in the Original Method
3.2. Identification of Contamination Sources
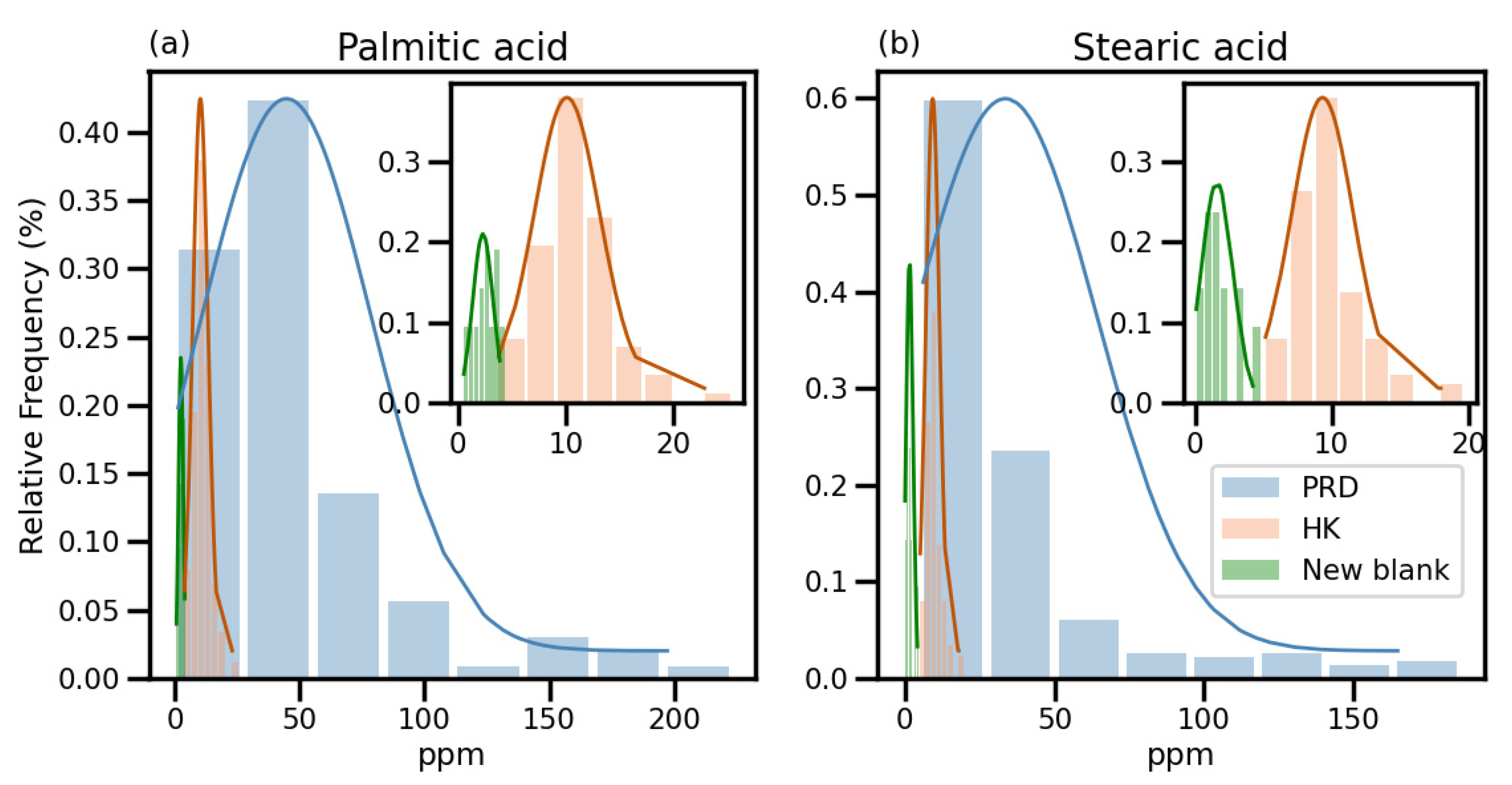
3.3. Evaluation of Revised Method
3.3.1. Contamination Levels
3.3.2. Limit of Detection (LOD) and Recoveries
3.3.3. Paired t-Tests
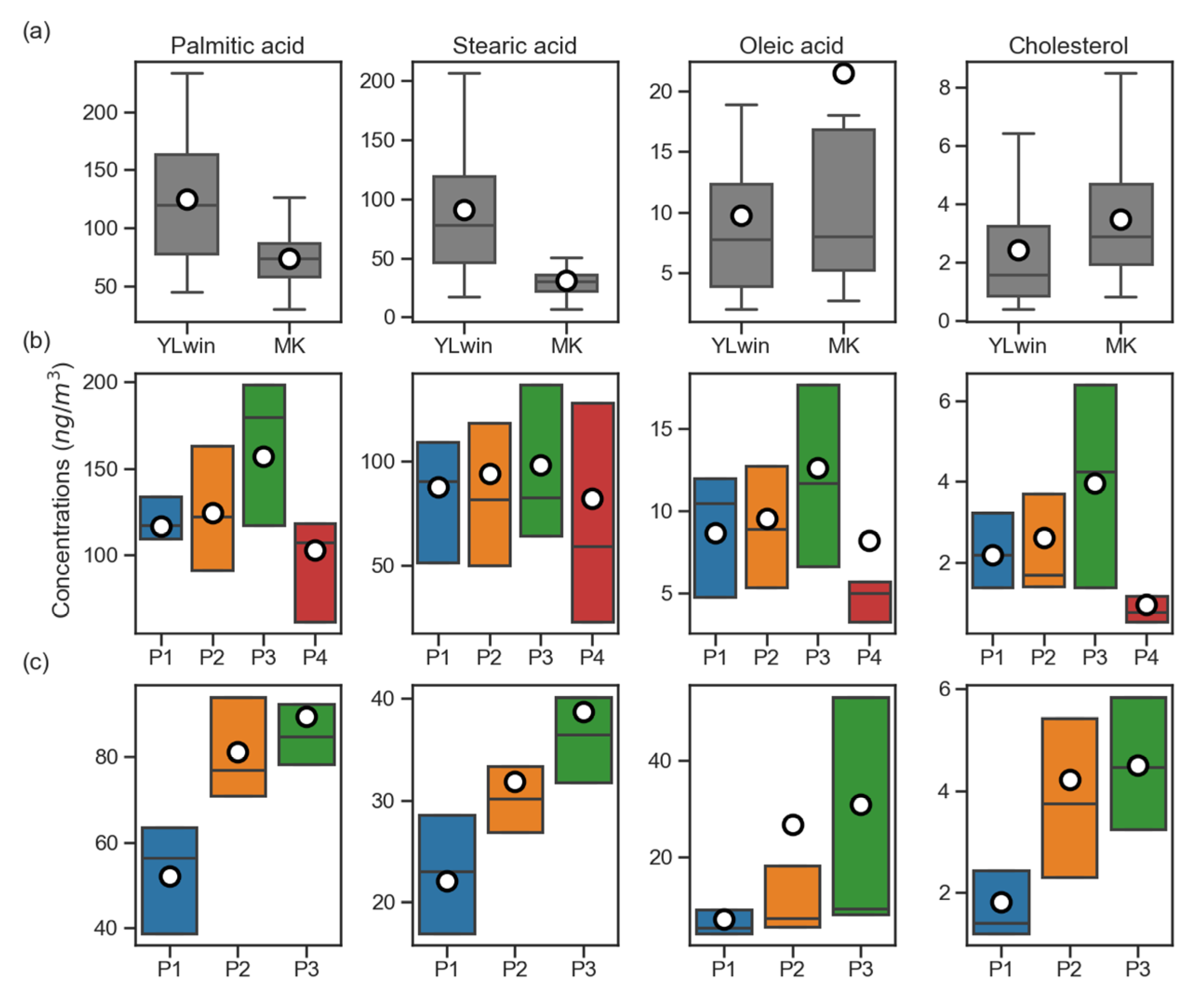
3.4. Source Analysis Using PMF Model
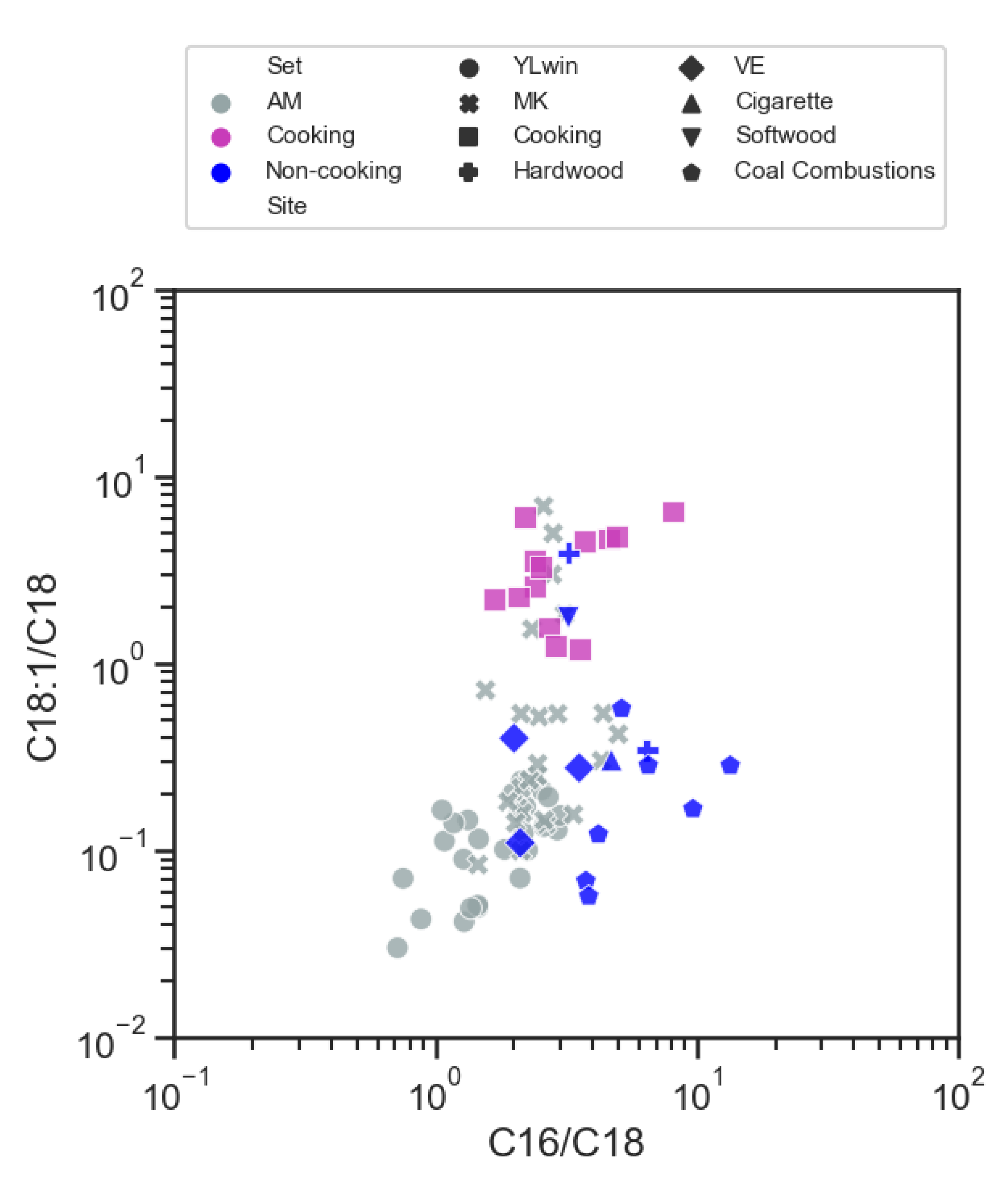
3.4.1. Abundances and Diurnal Variations of Cooking Related Tracers
3.4.2. Selection of PMF Input Species
3.4.3. PMF Input of Uncertainties
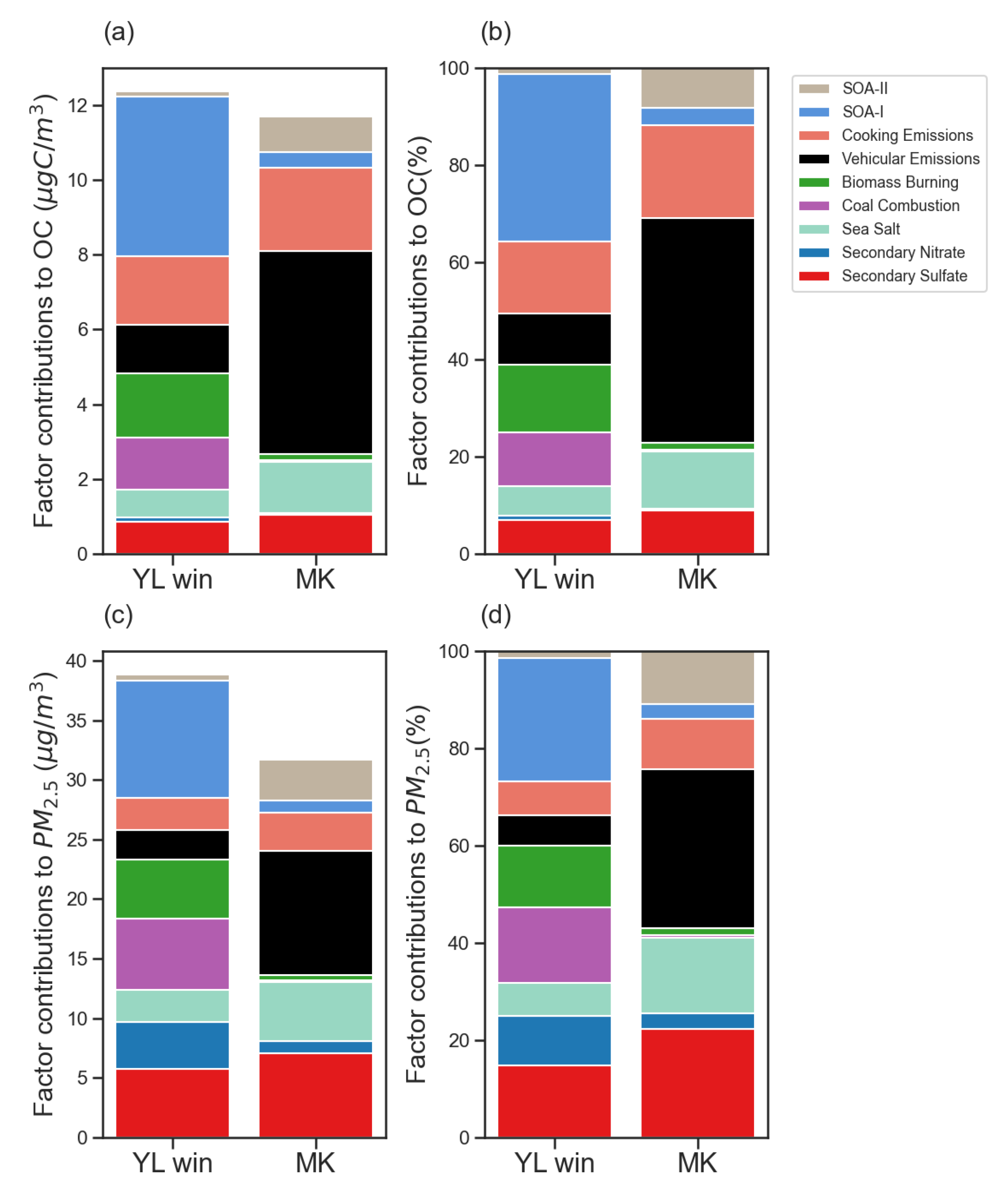
3.4.4. PMFCholC16C18 Solution
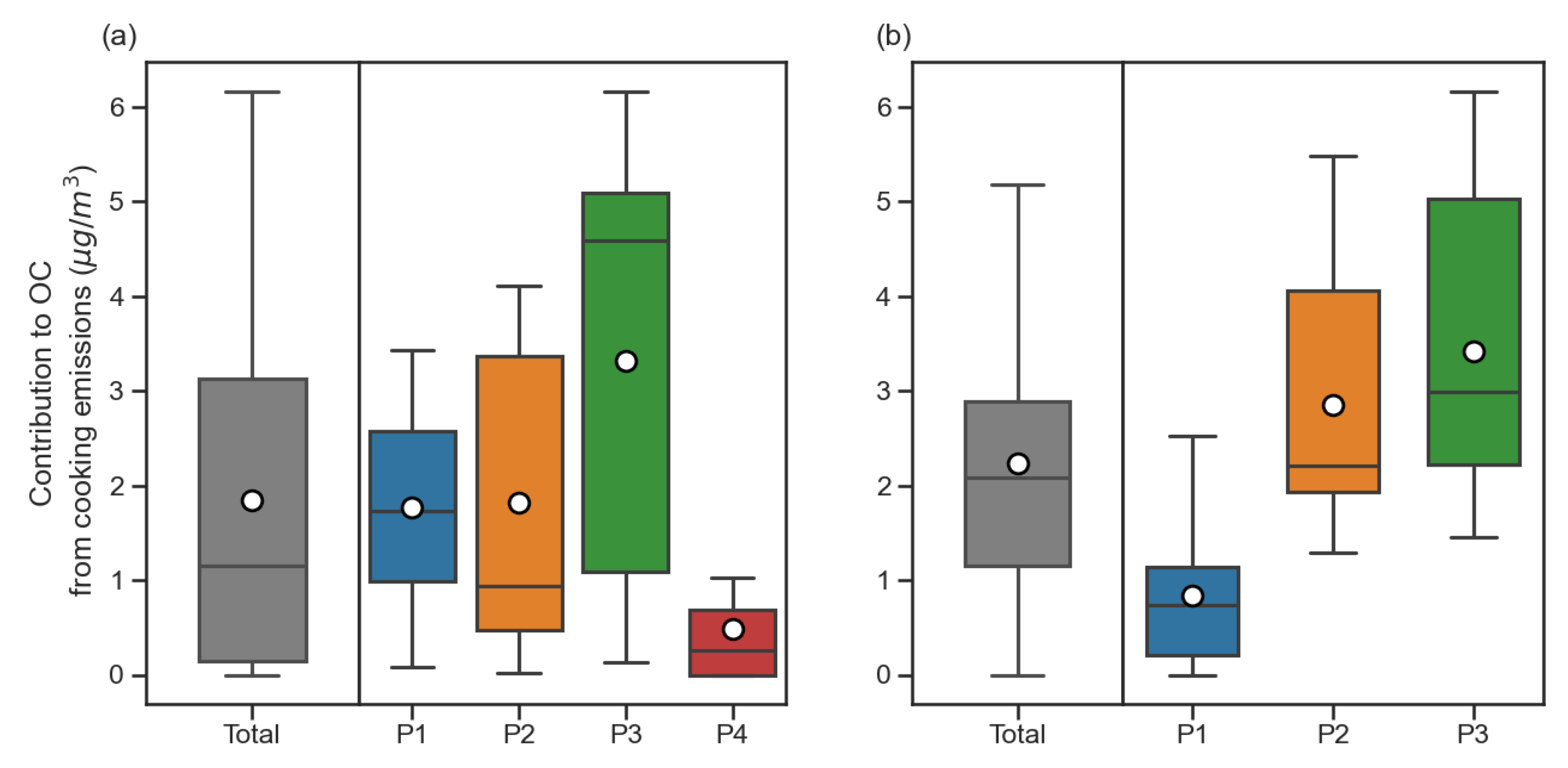
3.4.5. PM2.5 and OC Contribution from Cooking Emissions in PMFCholC16C18
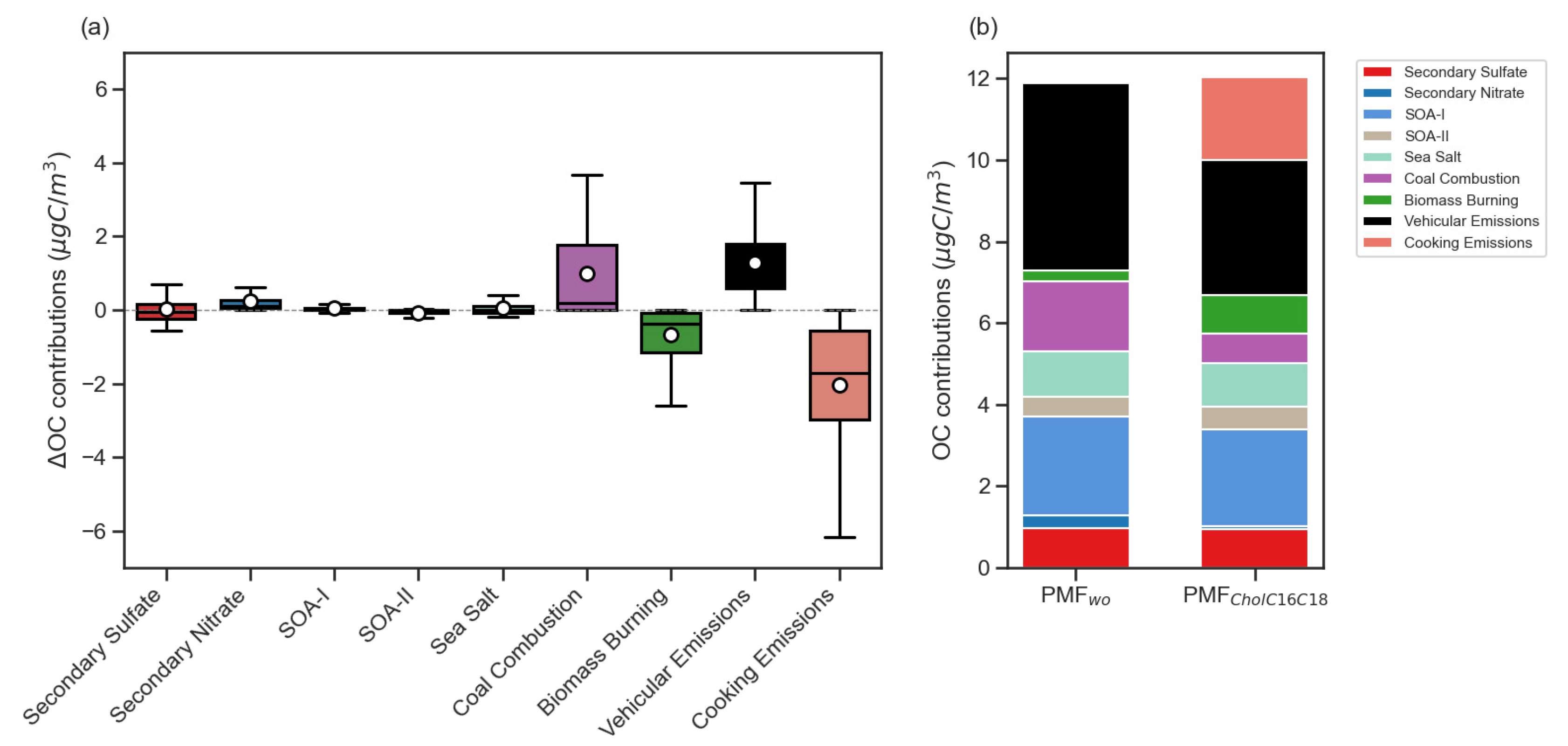
3.4.6. PMF Runs with Cholesterol or Fatty Acids Excluded
3.4.7. Impact of Cooking Related Tracers in PMF
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nolte, C.G.; Schauer, J.J.; Cass, G.R.; Simoneit, B.R.T. Highly polar organic compounds present in meat smoke. Environ. Sci. Technol. 1999, 33, 3313–3316. [Google Scholar] [CrossRef]
- Rogge, W.F.; Cass, G.R.; Hlldemann, L.M.; Mazurek, M.A.; Slmoneit, B.R.T. Sources of Fine Organic Aerosol. 1. Charbroilers and Meat Cooking Operations. Environ. Sci. Technol. 1991. [Google Scholar] [CrossRef]
- Schauer, J.J.; Kleeman, M.J.; Cass, G.R.; Simoneit, B.R.T. Measurement of emissions from air pollution sources. 4. C1–C27 organic compounds from cooking with seed oils. Environ. Sci. Technol. 2002, 36, 567–575. [Google Scholar] [CrossRef] [PubMed]
- He, L.Y.; Hu, M.; Huang, X.F.; Yu, B.D.; Zhang, Y.H.; Liu, D.Q. Measurement of emissions of fine particulate organic matter from Chinese cooking. Atmos. Environ. 2004, 38, 6557–6564. [Google Scholar] [CrossRef]
- McDonald, J.D.; Zielinska, B.; Fujita, E.M.; Sagebiel, J.C.; Chow, J.C.; Watson, J.G. Emissions from charbroiling and grilling of chicken and beef. J. Air Waste Manag. Assoc. 2003, 53, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.F.; He, L.Y.; Hu, M.; Canagaratna, M.R.; Sun, Y.; Zhang, Q.; Zhu, T.; Xue, L.; Zeng, L.W.; Liu, X.G.; et al. Highly time-resolved chemical characterization of atmospheric submicron particles during 2008 Beijing Olympic games using an aerodyne high-resolution aerosol mass spectrometer. Atmos. Chem. Phys. 2010, 10, 8933–8945. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.P.; Li, Y.J.; Yu, J.Z.; Louie, P.K.K.; Chan, C.K. Characteristics of submicron particulate matter at the Urban roadside in downtown Hong Kong-Overview of 4 months of continuous high-Resolution aerosol mass spectrometer measurements. J. Geophys. Res. 2015, 120, 7040–7058. [Google Scholar] [CrossRef]
- Sun, C.; Lee, B.P.; Huang, D.; Li, Y.J.; Schurman, M.I.; Louie, P.K.K.; Luk, C.; Chan, C.K. Continuous measurements at the urban roadside in an Asian megacity by Aerosol Chemical Speciation Monitor (ACSM): Particulate matter characteristics during fall and winter seasons in Hong Kong. Atmos. Chem. Phys. 2016, 16, 1713–1728. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Hopke, P.K.; Norris, G.; Williams, R.; Paatero, P. Source apportionment and analysis on ambient and personal exposure samples with a combined receptor model and an adaptive blank estimation strategy. Atmos. Environ. 2006, 40, 3788–3801. [Google Scholar] [CrossRef]
- Abdullahi, K.L.; Delgado-Saborit, J.M.; Harrison, R.M. Emissions and indoor concentrations of particulate matter and its specific chemical components from cooking: A review. Atmos. Environ. 2013, 71, 260–294. [Google Scholar] [CrossRef]
- Hou, X.; Zhuang, G.; Lin, Y.; Li, J.; Jiang, Y.; Fu, J.S. Emission of fine organic aerosol from traditional charcoal broiling in China. J. Atmos. Chem. 2008. [Google Scholar] [CrossRef]
- Schauer, J.J.; Kleeman, M.J.; Cass, G.R.; Simoneit, B.R.T. Measurement of emissions from air pollution sources. 1. C1 through C29 organic compounds from meat charbroiling. Environ. Sci. Technol. 1999. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, M.; Slanina, S.; Zhang, Y. Chemical compositions of fine particulate organic matter emitted from Chinese cooking. Environ. Sci. Technol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wan, T.S.M.; Fang, M.; Wang, F. Characterization of the non-volatile organic compounds in the aerosols of Hong Kong—Identification, abundance and origin. Atmos. Environ. 1997. [Google Scholar] [CrossRef]
- Fraser, M.P.; Yue, Z.W.; Buzcu, B. Source apportionment of fine particulate matter in Houston, TX, using organic molecular markers. Atmos. Environ. 2003. [Google Scholar] [CrossRef]
- Robinson, A.L.; Subramanian, R.; Donahue, N.M.; Bernardo-Bricker, A.; Rogge, W.F. Source apportionment of molecular markers and organic aerosol. 3. Food cooking emissions. Environ. Sci. Technol. 2006. [Google Scholar] [CrossRef]
- Schauer, J.J.; Cass, G.R. Source apportionment of wintertime gas-phase and particle-phase air pollutants using organic compounds as tracers. Environ. Sci. Technol. 2000. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Cass, G.R.; Schauer, J.J.; Edgerton, E.S. Source apportionment of PM2.5 in the southeastern United States using solvent-extractable organic compounds as tracers. Environ. Sci. Technol. 2002. [Google Scholar] [CrossRef]
- Ding, X.; Wang, X.M.; Zheng, M. The influence of temperature and aerosol acidity on biogenic secondary organic aerosol tracers: Observations at a rural site in the central Pearl River Delta region, South China. Atmos. Environ. 2011. [Google Scholar] [CrossRef]
- Sin, D.W.M.; Fung, W.H.; Choi, Y.Y.; Lam, C.H.; Louie, P.K.K.; Chow, J.C.; Watson, J.G. Seasonal and spatial variation of solvent extractable organic compounds in fine suspended particulate matter in Hong Kong. J. Air Waste Manag. Assoc. 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Fang, M.; Wang, F.; To, K.L. Characterization of the solvent extractable organic compounds in PM2.5 aerosols in Hong Kong. Atmos. Environ. 2000. [Google Scholar] [CrossRef]
- Cavonius, L.R.; Albers, E.; Undeland, I. In vitro bioaccessibility of proteins and lipids of pH-shift processed Nannochloropsis oculata microalga. Food Funct. 2016. [Google Scholar] [CrossRef] [Green Version]
- Cavonius, L.R.; Carlsson, N. Fatty acid contaminations originating from commercially available solid phase-extraction columns. Chem. Sci. Rev. Lett. 2015, 4, 1206–1209. [Google Scholar]
- Notter, S.J.; Stuart, B.H.; Dent, B.B.; Keegan, J. Solid-phase extraction in combination with GC/MS for the quantification of free fatty acids in adipocere. Eur. J. Lipid Sci. Technol. 2008. [Google Scholar] [CrossRef]
- Prasad, M.R.; Jones, R.M.; Young, H.S.; Kaplinsky, L.B.; Das, D.K. Analysis of tissue free fatty acids isolated by aminopropyl bonded-phase columns. J. Chromatogr. B Biomed. Sci. Appl. 1988, 428, 221–228. [Google Scholar] [CrossRef]
- Gunash, J.; Henao, J.J.A.; Stark, K.D. Palmitic and Stearic Free Fatty Acids Are Consistently Found in Materials used for Dried Blood Spot Collection. FASEB J. 2017, 31, 955-13. [Google Scholar] [CrossRef]
- Liu, G.; Mühlhäusler, B.S.; Gibson, R.A. Evaluation of contamination associated with current blood spot technology for determining the fatty acid status of individuals. Eur. J. Lipid Sci. Technol. 2015. [Google Scholar] [CrossRef]
- Yao, C.H.; Liu, G.Y.; Yang, K.; Gross, R.W.; Patti, G.J. Inaccurate quantitation of palmitate in metabolomics and isotope tracer studies due to plastics. Metabolomics 2016. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Huang, X.H.H.; Chow, K.S.; Wang, Q.; Zhang, T.; Wu, D.; Yu, J.Z. Abundance and Sources of Phthalic Acids, Benzene-Tricarboxylic Acids, and Phenolic Acids in PM2.5 at Urban and Suburban Sites in Southern China. ACS Earth Space Chem. 2018, 2, 147–158. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, X.H.H.; Tam, F.C.V.; Zhang, X.; Liu, K.M.; Yeung, C.; Feng, Y.; Cheng, Y.Y.; Wong, Y.K.; Ng, W.M.; et al. Source apportionment of fine particulate matter in Macao, China with and without organic tracers: A comparative study using positive matrix factorization. Atmos. Environ. 2019, 198, 183–193. [Google Scholar] [CrossRef]
- Wong, Y.K.; Huang, X.H.H.; Cheng, Y.Y.; Louie, P.K.K.; Yu, A.L.C.; Tang, A.W.Y.; Chan, D.H.L.; Yu, J.Z. Estimating contributions of vehicular emissions to PM2.5 in a roadside environment: A multiple approach study. Sci. Total Environ. 2019, 672, 776–788. [Google Scholar] [CrossRef]
- Paatero, P.; Tapper, U. Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values. Environmetrics 1994, 5, 111–126. [Google Scholar] [CrossRef]
- Paatero, P. Least squares formulation of robust non-negative factor analysis. Chemom. Intell. Lab. Syst. 1997, 37, 23–35. [Google Scholar] [CrossRef]
- Zhang, Y.; Schauer, J.J.; Zhang, Y.; Zeng, L.; Wei, Y.; Liu, Y.; Shao, M. Characteristics of particulate carbon emissions from real-world Chinese coal combustion. Environ. Sci. Technol. 2008, 42, 5068–5073. [Google Scholar] [CrossRef] [PubMed]
- Schauer, J.J.; Kleeman, M.J.; Cass, G.R.; Simoneit, B.R.T. Measurement of emissions from air pollution sources. 3. C1–C29 organic compounds from fireplace combustion of wood. Environ. Sci. Technol. 2001, 35, 1716–1728. [Google Scholar] [CrossRef]
- Xie, M.; Hannigan, M.P.; Barsanti, K.C. Impact of gas/particle partitioning of semivolatile organic compounds on source apportionment with positive matrix factorization. Environ. Sci. Technol. 2014, 48, 9053–9060. [Google Scholar] [CrossRef]
- Wang, Q.; He, X.; Hilda Huang, X.H.; Griffith, S.M.; Feng, Y.; Zhang, T.; Zhang, Q.; Wu, D.; Yu, J.Z. Impact of secondary organic aerosol tracers on tracer-based source apportionment of organic carbon and PM2.5: A case study in the pearl river delta, China. ACS Earth Space Chem. 2017, 1, 562–571. [Google Scholar] [CrossRef]
- Kaltsonoudis, C.; Kostenidou, E.; Louvaris, E.; Psichoudaki, M.; Tsiligiannis, E.; Florou, K.; Liangou, A.; Pandis, S.N. Characterization of fresh and aged organic aerosol emissions from meat charbroiling. Atmos. Chem. Phys. 2017, 17, 7143–7155. [Google Scholar] [CrossRef] [Green Version]
- Schauer, J.J.; Kleeman, M.J.; Cass, G.R.; Simoneit, B.R.T. Measurement of emissions from air pollution sources. 5. C1–C32 organic compounds from gasoline-powered motor vehicles. Environ. Sci. Technol. 2002, 36, 1169–1180. [Google Scholar] [CrossRef]
- Polissar, A.V.; Hopke, P.K.; Paatero, P.; Malm, W.C.; Sisler, J.F. Atmospheric aerosol over Alaska 2. Elemental composition and sources. J. Geophys. Res. Atmos. 1998, 103, 19045–19057. [Google Scholar] [CrossRef]
- Reff, A.; Eberly, S.I.; Bhave, P.V. Receptor modeling of ambient particulate matter data using positive matrix factorization: Review of existing methods. J. Air Waste Manag. Assoc. 2007, 57, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.G.; Eberly, S.; Paatero, P.; Norris, G.A. Methods for estimating uncertainty in PMF solutions: Examples with ambient air and water quality data and guidance on reporting PMF results. Sci. Total Environ. 2015, 518–519, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.F.; Yu, J.Z.; Yuan, Z.; Lau, A.K.H.; Louie, P.K.K. Source analysis of high particulate matter days in Hong Kong. Atmos. Environ. 2009, 43, 1196–1203. [Google Scholar] [CrossRef]
- Louie, P.K.K.; Watson, J.G.; Chow, J.C.; Chen, A.; Sin, D.W.M.; Lau, A.K.H. Seasonal characteristics and regional transport of PM2.5 in Hong Kong. Atmos. Environ. 2005, 39, 1695–1710. [Google Scholar] [CrossRef]
- Li, Y.C.; Yu, J.Z.; Ho, S.S.H.; Schauer, J.J.; Yuan, Z.; Lau, A.K.H.; Louie, P.K.K. Chemical characteristics and source apportionment of fine particulate organic carbon in Hong Kong during high particulate matter episodes in winter 2003. Atmos. Res. 2013, 120–121, 88–98. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Old | New | |
|---|---|---|---|
| LOD | LOD | Recovery | |
| Keto/hydroxy carboxylic acids | |||
| Glyceric acid | 0.51 ± 0.17 | 0.83 ± 0.24 | 93.4 ± 21.9 |
| Pinonic acid | 0.96 ± 0.21 | 0.90 ± 0.32 | 87.7 ± 6.7 |
| Dicarboxylic acids | |||
| Succinic acid | 1.22 ± 0.29 | 1.29 ± 0.50 | 133.8 ± 26.9 |
| Glutaric acid | 0.54 ± 0.25 | 1.12 ± 0.70 | 73.9 ± 7.2 |
| Adipic acid | 0.59 ± 0.34 | 0.74 ± 0.16 | 90.1 ± 15.6 |
| Pimelic acid | 0.78 ± 0.20 | 0.66 ± 0.11 | 96.1 ± 13.1 |
| Suberic acid | 0.48 ± 0.16 | 0.78 ± 0.20 | 97.2 ± 13.3 |
| Azelaic acid | 0.51 ± 0.23 | 1.01 ± 0.18 | 101.8 ± 11.5 |
| Phthalic acid | 0.68 ± 0.20 | 0.74 ± 0.19 | 102.4 ± 14.3 |
| Isophthalic acid | 0.43 ± 0.16 | 0.74 ± 0.15 | 110.0 ± 14.9 |
| Terephthalic acid | 1.02 ± 0.32 | 1.26 ± 0.33 | 112.9 ± 17.6 |
| Hydroxycarboxylic acids | |||
| Malic acid | 0.56 ± 0.16 | 1.15 ± 0.60 | 61.6 ± 14.7 |
| Tartaric acid | 0.75 ± 0.18 | 2.04 ± 0.50 | 34.5 ± 18.2 |
| Citramalic acid | 0.53 ± 0.15 | 1.31 ± 0.27 | 80.1 ± 11.9 |
| Citric acid | 1.04 ± 0.57 | 2.76 ± 0.60 | 23.5 ± 10.9 |
| Fatty acids | |||
| Palmitic acid | 1.36 ± 0.70 | 0.88 ± 0.45 | 86.2 ± 41.2 1 |
| Stearic acid | 2.42 ± 1.62 | 1.06 ± 0.17 | 115.9 ± 45.1 1 |
| Sterol | |||
| Cholesterol | 0.61 ± 0.50 | 0.98 ± 0.31 | 109.5 ± 7.6 |
| Hydroxybenzoic acids | |||
| 3-hydroxybenzoic acid | 0.49 ± 0.28 | 0.70 ± 0.23 | 118.5 ± 19.7 |
| 4-hydroxybenzoic acid | 0.43 ± 0.25 | 0.60 ± 0.15 | 110.5 ± 30.3 |
| Vanillic acid | 0.53 ± 0.03 | 0.58 ± 0.17 | 114.0 ± 18.6 |
| Syringic acid | 0.44 ± 0.17 | 0.82 ± 0.32 | 94.7 ± 14.2 |
| Benzene tricarboxylic acids | |||
| 1,2,4-benzenetricarboxylic acid | 0.76 ± 0.34 | 1.18 ± 0.41 | 80.8 ± 19.2 |
| 1,3,5-benzenetricarboxylic acid | 0.60 ± 0.26 | 0.64 ± 0.26 | 97.5 ± 9.8 |
| Saccharides | |||
| Galactosan | 0.55 ± 0.48 | 0.75 ± 0.25 | 113.9 ± 31.0 |
| Mannosan | 0.47 ± 0.15 | 0.62 ± 0.14 | 106.3 ± 18.5 |
| Levoglucosan | 0.45 ± 0.19 | 0.66 ± 0.15 | 111.9 ± 19.3 |
| Xylitol | 0.42 ± 0.14 | 0.99 ± 0.83 | 117.8 ± 18.6 |
| Arabitol | 0.40 ± 0.12 | 0.86 ± 0.57 | 120.0 ± 20.2 |
| Mannitol | 0.46 ± 0.28 | 0.75 ± 0.25 | 110.5 ± 14.4 |
| Sorbitol | 0.41 ± 0.24 | 0.72 ± 0.18 | 111.8 ± 16.3 |
| Glycerol | 0.50 ± 0.38 | 0.89 ± 0.16 | 119.9 ± 25.9 |
| Erythritol | 0.53 ± 0.11 | 1.13 ± 0.47 | 120.6 ± 24.1 |
| Fructose | 0.44 ± 0.15 | 1.02 ± 0.41 | 132.4 ± 28.0 |
| Xylose | 0.37 ± 0.27 | 1.0 ± 0.4 | 108.6 ± 12.7 |
| Mannose | 0.62 ± 0.12 | 1.43 ± 0.30 | 115.5 ± 11.9 |
| Glucose | 1.19 ± 0.65 | 1.88 ± 0.51 | 104.9 ± 6.9 |
| Maltose | 0.81 ± 0.59 | 1.23 ± 0.24 | 94.5 ± 4.9 |
| Sucrose | 0.56 ± 0.18 | 0.43 ± 0.32 | 108.6 ± 2.2 |
| Trehalose | 0.36 ± 0.11 | 0.47 ± 0.12 | 115.3 ± 5.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.Y.; Yu, J.Z. Minimizing Contamination from Plastic Labware in the Quantification of C16 and C18 Fatty Acids in Filter Samples of Atmospheric Particulate Matter and Their Utility in Apportioning Cooking Source Contribution to Urban PM2.5. Atmosphere 2020, 11, 1120. https://doi.org/10.3390/atmos11101120
Cheng YY, Yu JZ. Minimizing Contamination from Plastic Labware in the Quantification of C16 and C18 Fatty Acids in Filter Samples of Atmospheric Particulate Matter and Their Utility in Apportioning Cooking Source Contribution to Urban PM2.5. Atmosphere. 2020; 11(10):1120. https://doi.org/10.3390/atmos11101120
Chicago/Turabian StyleCheng, Yuk Ying, and Jian Zhen Yu. 2020. "Minimizing Contamination from Plastic Labware in the Quantification of C16 and C18 Fatty Acids in Filter Samples of Atmospheric Particulate Matter and Their Utility in Apportioning Cooking Source Contribution to Urban PM2.5" Atmosphere 11, no. 10: 1120. https://doi.org/10.3390/atmos11101120
APA StyleCheng, Y. Y., & Yu, J. Z. (2020). Minimizing Contamination from Plastic Labware in the Quantification of C16 and C18 Fatty Acids in Filter Samples of Atmospheric Particulate Matter and Their Utility in Apportioning Cooking Source Contribution to Urban PM2.5. Atmosphere, 11(10), 1120. https://doi.org/10.3390/atmos11101120