1. Introduction
The release of large quantities of trace gas pollutants into the tropical troposphere from human burning practices and wildfires in Africa exerts a significant influence on global atmospheric composition, most importantly on the relatively pristine Southern Hemisphere (SH), tropical Atlantic [
1,
2,
3] and around the burning regions. The resulting emissions from biomass burning (BB) impact directly on air quality by reducing visibility with concurrent enhancement in aerosol concentrations and associated reduction in photochemical activity. It has also been shown that the long range transport of pollutants emitted from BB can affect air quality thousands of kilometers away from the source [
4]. It is estimated that open burning in Africa accounts for more than 50% of the total global BB emissions during any typical year [
5,
6]. It is therefore important to quantify the sensitivity of SH atmospheric composition to the uncertainty associated with African BB emission estimates.
The large interannual variability in the timing, distribution and extent of burning events can presently be mapped by making use of satellite observations and using the total burned area products [
6,
7,
8,
9]. To quantify the strength of the emission sources methodologies have been developed which combine such satellite observations with, e.g., terrestrial ecosystem models that account for differences in both land type and the availability of biomass for burning. This has lead to the development of a number of bottom-up emission inventories for use in three-dimensional global chemistry-transport models (3D global CTMs) [
6,
8,
9]. A recent intercomparison of BB emission inventories for the year 2003 has highlighted large differences in both the seasonality and the total flux of gases emitted from tropical burning regions [
10]. This study concluded that no preference can be given towards which methodology captures BB events most accurately, where there are both seasonal and regional differences in the performance of each inventory. This introduces significant uncertainty in the associated emissions of CO, nitrogen oxides (NO
x), non-methane hydrocarbons (NMHCs) and carbonaceous aerosols from BB emission sources. Thus, the most advanced bottom-up BB inventories that are currently available for use in 3D global CTMs exhibit significant differences in the integrated seasonal fluxes.
The strong convective uplift of BB emissions due to the increased air buoyancy above fires means that BB emissions exhibit a vertical injection profile that is commonly accounted for in 3D global CTMs [
11]. The vertical distribution can be constrained using statistical composites derived using satellite measurements [
12], with injection heights being dependant on the cumulative effects of fire intensity, land type, season and the local meteorological parameters. Although BB emissions in the tropics are rarely injected directly into the free troposphere (FT) [
13,
14], the long range transport out of the main burning regions can be influenced by the method with which BB emissions are introduced into large-scale atmospheric models [
3]. Therefore, the description of BB emissions in global CTMs may have consequences concerning the fraction of the emitted species transported away from the source regions and the subsequent conclusions drawn as to the importance of African BB for the chemical composition of the troposphere in the tropics and SH.
Typical applications of 3D global CTMs include: (i) the quantification of the long range transport of pollutants between continents and more remote locations; (ii) the investigation of chemical source-receptor relationships; and (iii) the provision of estimates of the atmospheric lifetimes of key trace gases such as CO (τCO), O
3 (τO
3) and CH
4 (τCH
4). Recent multi-model intercomparison studies estimate that in large-scale global models τCO, τO
3 and τCH
4 have typical values of ~2 months, ~22 days and ~8.7 years, respectively [
15,
16]. These chemical lifetimes control the impact of emission sources on air quality far away from the source area by changing the resident surface concentrations [
17]. Moreover, any increases in the global burdens of the greenhouse gases CH
4 and O
3 impact on climate due to their warming potential. Such warming has the potential to induce a positive feedback on wildfire incidence via more frequent drought conditions [
18].
The removal of CO and CH
4 is principally governed by the resident concentration of the OH radical formed via the photolysis of O
3 in the presence of H
2O, where O
3 is efficiently regenerated in the presence of NO
x and sunlight. A chemical budget analysis from a typical CTM simulation reveals that CO and CH
4 scavenge ~40% and ~16% of the available global OH in the troposphere, respectively, with a large fraction of the oxidation occurring in the tropics (30°S–30°N). The impacts of BB emissions on τ
CH4 are manifold, with the net impact being rather uncertain. For instance, in response to the widespread 1997 Indonesian wildfires, Duncan
et al. [
19] have shown that τ
CH4 increases due to large-scale reductions in OH (>20%). This reduction was ascribed to enhanced CO emissions released from the fires, enhanced heterogeneous loss of HO
x radicals and reductions in UV light caused by increases in BB aerosols. However other more recent studies have suggested that the observed increase in CH
4 during 1997 measured by the NOAA Earth System Research Laboratory (ESRL) surface monitoring network was due to enhanced CH
4 emissions directly from increased BB rather than due to increased competition for OH [
20]. Other studies suggest a combination of both CH
4 emissions and lifetime changes to explain the variable annual growth rate [
21]. Also the anomalous climatic conditions that favour intensive BB events result in a dry, arid environment with reduced cloudiness, which impact OH, O
3 (via lower biogenic and BB NO
x), τCO and τCH
4.
For anthropogenic emissions it has been shown that the global OH distribution, and thus τCH
4, is significantly affected by the ratio of CO and N (as nitrogen oxides) emitted into the atmosphere [
22,
23]. For BB the CO/N ratio is typically much higher than anthropogenic emissions due to incomplete combustion processes [
23]. This has the potential to impact the global CO burden, and thus τCH
4, due to a lower fraction of the emitted CO being oxidized in/around the source region. To date to our knowledge there has been no similar investigation as to how the potential variability in the CO/N ratio from BB emission inventories impacts on the atmospheric lifetimes of abundant trace gases when applied in a global CTM, which is a major focus of this study.
In this paper we investigate the uncertainties introduced towards simulating global tropospheric composition associated with differences between bottom-up BB emission estimates when applied in a global 3D CTM. For this purpose we examine the influence on global air quality, the long range transport of pollutants out of BB source regions and the perturbations introduced in estimating global atmospheric lifetimes of dominant trace species. We also investigate the impact of increasing the update frequency with respect to the temporal distribution of BB emissions and the parameterization used for introducing tropical BB emission into the boundary layer on both tropical and SH composition. Finally we show that there is a compensating effect towards the perturbations introduced towards the oxidative capacity of the troposphere due to the CO/N ratio defined in different BB emission inventories, which constrains the effects on the global lifetimes for dominant trace gases.
2. Model Simulations and Emissions
The global 3D CTM used for this study is TM4, where the version applied here has been comprehensively described elsewhere [
3]. The model is run at a horizontal resolution of 3° × 2° using 34 vertical levels with a model top of 0.1 hPa and is driven by meteorological fields from the European Centre for Medium-Range Weather Forecasting (ECMWF) operational analysis. The model uses the modified CBM4 chemical mechanism [
24] updated to include recent recommendations to the reaction rate data [
25]. Photolysis rates are calculated using the parameterization of Landgraf and Crutzen [
26], modified to account for attenuation effects by clouds and ground albedo [
27]. The chosen simulation year is 2006, for which the distribution of both CO and O
3 in TM4 have recently been evaluated against a host of different measurements taken in and around the African continent [
3,
28,
29] and where burning intensity in Africa is representative of a typical year [
6]. For global anthropogenic and biogenic emissions we use the emission inventories provided by the EU-RETRO project for the year 2000 (
http://retro.enes.org).
In this study we principally focus on differences between BB inventories provided for the African domain (here defined as the region 34°S–34°N, 20°W–40°E). BB emission estimates are taken from bottom-up BB inventories, namely: GFEDv2 [
8], GFEDv3 [
6], and AMMABB [
9], for which large differences have been shown to exist in the emission estimates for both trace gases and aerosols [
9,
10]. For regions outside the African domain we adopt the GFEDv2 emission inventory for BB in all simulations. The update frequency of these inventories is monthly, where the original inventories are provided on either a 1° × 1° or 0.5° × 0.5° grid resolution and subsequently coarsened for application in TM4. To account for typical burning practises a daily burning cycle is applied to all BB emissions over the latitudinal range 20°S–20°N peaking at 2 pm local time [
30]. For the majority of simulations BB emissions between 20°S–20°N are introduced into the first 2 km of the troposphere, with ~50% being placed between 0 and 1 km and ~50% being placed between 1 and 2 km. For the mid- and high latitudes, burning heights are adopted from the literature [
11]. To quantify the global impact of African BB emissions on both long range transport and the atmospheric burdens and lifetimes of dominant trace species we perform a simulation where African BB emissions are turned off (referred to as NONE). In all simulations the CH
4 surface concentrations are constrained by relaxation towards observations made at pristine locations as part of the NOAA surface network. This allows TM4 to capture the latitudinal gradient and seasonal cycles which are present, as well as the variability in the CH
4 growth rate over recent decades [
20]. Therefore differences in the estimates of direct CH
4 emissions from BB are not accounted for in this study, although these are rather small typically ranging between 17 and 20 Tg CH
4 yr
−1 [
6,
8]. In these simulations a CH
4 surface gradient exists of ~1,770 ppb at 35°S and ~1,850 ppb at 35°N. A one year spin-up period is adopted for all simulations using the relevant BB emission inventory available for the year 2005.
Table 1.
A definition of the sensitivity simulations conducted in this study. These different emission inventories are applied for the African domain only. All other regions use GFEDv2 monthly estimates with latitudinally dependant injection heights from Dentener
et al. [
11].
Table 1.
A definition of the sensitivity simulations conducted in this study. These different emission inventories are applied for the African domain only. All other regions use GFEDv2 monthly estimates with latitudinally dependant injection heights from Dentener et al. [11].
| Definition of Simulation | Biomass Burning Emission Inventory | Injection Height (km) | Temporal Update Frequency |
|---|
| GFEDv2 | GFEDv2 | 0–2 | Monthly |
| GFEDv3 | GFEDv3 | 0–2 | Monthly |
| AMMABB | AMMABB | 0–2 | Monthly |
| AMMABB_LOWNOX | AMMABB | 0–2 | Monthly |
| 8DAY | GFEDv2 | 0–2 | 8-day |
| HIGH-IH | GFEDv2 | 0–3 | Monthly |
Sensitivity studies are also defined to investigate the effect of increasing the update frequency of the temporal distribution of BB events and increasing the injection heights used for introducing tropical BB emissions (referred to as 8-DAY and HIGH-IH, respectively). For the 8-DAY simulation we adopted the GFEDv2 8-day emission inventory [
6] where a new temporal distribution and emission flux is defined every 8 days of simulation time. For HIGH-IH we change the vertical distribution between 30°S and 30°N from the default (see above) to that derived from a 5-year record of satellite observations [
12] which results in no emissions in the first 100 m, ~25% between 100 and 1,000 m, ~50% between 1 and 2 km and ~25% between 2 and 3 km. Finally, to investigate the effect that BB NO
x emissions have on mitigating the long range transport of CO we perform a simulation using the AMMABB inventory where the BB NO
x is reduced by 50% (AMMABB_LOWNOX). An overview of all the simulations used for this study and the BB inventories adopted for Africa in each simulation are given in
Table 1.
3. Comparison of Biomass Burning Emission Inventories
The three BB emission inventories analyzed here use a similar modeling approach for the derivation of emission estimates [
10], where differences exist primarily due to the burned area product which is utilized and the input data (e.g., the definition and distribution of land types) to the modeling process. All BB emission inventories used in this study are commonly known as bottom-up inventories.
For the GFED inventories daily burnt area maps are derived from the Moderate Resolution Imaging Spectrophotometer (MODIS) [
31,
32]. The differences between versions 2 and 3 of the inventory are derived from changes that have been made to the method for calculating the monthly burned area product and fire activity from MODIS and the parameterizations applied in the Carnegie-Ames-Stanford-Approach (CASA) biogeochemical model [
33] used for calculating plant productivity, tree mortality, leaf litterfall and combustion completeness. For the global burned area product this results in a ~10% difference between versions used for deriving the GFEDv2 and GFEDv3 inventories [
32], with a ~60% increase for southern Africa. In total 13 different vegetation classes are defined ranging from deciduous needleleaf to Savanna, including both an Urban and Barren classification. A fixed vegetation map including fractional tree cover for the year 2001 is adopted regardless of the year [
34]. The soil moisture component is calculated using precipitation values from the Global Precipitation Climatology Project (GPCP, [
35]) and the temperature taken from climatological values. The CASA model produces a monthly input file for the derivation of BB emission fluxes on a 0.5° × 0.5° grid resolution. The emission factors used for each of the seven different fire types are given in van der Werf
et al. [
6] and are based on the recommendations of Andreae and Merlet [
36], except for peat fires which are based on Christian
et al. [
37]. That the GFEDv3 emission inventory is more recent means that it supersedes the GFEDv2 emission inventory, and subsequently reduces the uncertainty associated with emissions due to BB. In spite of the increase in the burned area the annual emission fluxes in the GFEDv3 inventory are generally lower for many regions when compared to the corresponding GFEDv2 inventory [
6]. This leads to an associated decrease in the global CO emission flux for the GFEDv3 inventory, which has recently been questioned when comparing CO inversions performed using the 4DVAR version of TM5, optimized using surface measurements of CO [
38] and CO inversions performed using the MOZART model using satellite observations from the MOPITT (Measurements of Pollution in the Troposphere) satellite instrument [
39]. Details related to the uncertainties of the input parameters used for determining the GFEDv2 and GFEDv3 emission inventories are given in the literature [
6,
8].
Although two generations of the GFED inventory are applied in this study, no direct comparisons of CTM simulations using both versions of the inventory for African BB have appeared in the literature to our knowledge, thus warranting some investigation of the effects considering that GFEDv2 has been widely used during previous CTM studies [
3,
28]. Moreover, the 8-day product was only available for the GFEDv2 inventory at the time of this study. For the NMHC the GFED inventory provides a lumped total (given in Tg C yr
−1) which is segregated into the individual CBM4 aggregated species used for representing higher organics according to the ratios given in
Table 2. Here the organic species in the modified CBM4 mechanism for which BB contribute are thus: PAR represents paraffinic bonds, ETH represents ethene (CH
2=CH
2), OLE represents olefinic bonds, ALD2 represents aldehydes, MGLY represents methylgloxal (CH
3COCHO) and HCHO represents formaldehyde.
Table 2.
The ratios used for the segregation of the non-methane hydrocarbons (NMHC) emission fluxes provided by the GFED biomass burning emission inventory as an aggregated emission flux (Tg C) into transported trace species in the modified CBM4 chemical mechanism. For definitions of each species see the details provided in the text.
Table 2.
The ratios used for the segregation of the non-methane hydrocarbons (NMHC) emission fluxes provided by the GFED biomass burning emission inventory as an aggregated emission flux (Tg C) into transported trace species in the modified CBM4 chemical mechanism. For definitions of each species see the details provided in the text.
| Modified CBM4 Species | Fraction of Total Lumped NMHC | Modified CBM4 Species | Fraction of Total Lumped NMHC |
|---|
| PAR | 0.286 | ALD2 | 0.022 |
| ETH | 0.125 | MGLY | 0.006 |
| OLE | 0.068 | HCHO | 0.005 |
For the AMMABB inventory the L3JRC burned area product is used [
40] derived using daily images from the VEGETATION sensor on the Satellite Pour l’ Observation de la Terre (SPOT) instrument, with corrections applied for the GLC3 (open deciduous broadleaved tree cover) and GLC12 (deciduous closed-open shrubs) ecosystems using intercomparisons of the LANDSAT and L3JRC products [
9]. Also a fixed vegetation map for the year 2000 is adopted during the calculation of the emission fluxes [
10]. The total emission estimate for each trace gas is calculated using the designated biomass density, burning efficiency and emission factors for 14 separate vegetation types [
9]. The burning efficiency across vegetation types typical ranges from 0.25 (e.g., GLC1, broadleaf evergreen) to 0.9 (GLC11, Evergreen Shrubs). Again the emission factors used for the derivation of trace gas emissions are taken from Andreae and Merlet [
36]. Each contribution is then weighted by the fraction of each vegetation type contained in the 0.5° × 0.5° grid cell. The associated uncertainty related to each of the input parameters used to derive the emission fluxes are given in Liousse
et al. [
9].
For NO
x emissions separate estimates are given for NO and NO
2 to account for burning efficiency, although all NO
x emissions are introduced as NO in TM4 for computational stability. For the NMHC a total of 49 different emission fluxes are provided for a diverse range of Volatile Organic Compounds (VOC’s). These are partitioned into the CBM4 components following the recommendations given in Yarwood
et al. [
41].
Annual totals for the African BB emission estimates for CO, NO
x (as N), and the NMHC (as C) are given in
Table 3, along with the associated CO/N ratios. Also shown are the corresponding emission totals from anthropogenic and biogenic sources. The emissions from the AMMABB inventory are much higher than those provided in either GFEDv2 or GFEDv3 for all species, with differences of up to ~65% in the annual totals for Africa as found in previous studies for other years [
10]. Moreover, emissions due to BB dominate for the African continent regardless of the emission inventory which is employed showing the importance of African BB as a tropical emission source.
Table 3.
The annual emission totals of reactive trace gases released from Africa (34°S–34°N, 20°W–40°E) for 2006 as estimated by the various BB emission inventories adopted in this study. The numbers in parenthesis are the percentage contributions to the total annual global emissions emitted for each trace species. The corresponding CO/N emission ratios for Africa are provided in the column on the far right.
Table 3.
The annual emission totals of reactive trace gases released from Africa (34°S–34°N, 20°W–40°E) for 2006 as estimated by the various BB emission inventories adopted in this study. The numbers in parenthesis are the percentage contributions to the total annual global emissions emitted for each trace species. The corresponding CO/N emission ratios for Africa are provided in the column on the far right.
| | CO(Tg CO yr−1) | NOx(Tg N yr−1) | NMHC(Tg C yr−1) | CO/N Ratio |
|---|
| GFEDv2 | 149.29 (14.2) | 2.44 (5.3) | 4.45 (2.5) | 61.2 |
| GFEDv3 | 143.82 (13.8) | 2.10 (4.5) | 4.45 (2.5) | 68.5 |
| AMMABB | 243.64 (21.3) | 5.36 (10.4) | 16.35 (9.6) | 43.3 |
| AMMBB_LOWNOX | 243.64 (21.3) | 2.73 (5.5) | 16.35 (9.6) | 89.2 |
| Anthropogenic | 66.70 (6.4) | 3.85 (7.9) | 7.78 (12.5) | 17.8 |
| Biogenic | 28.58 (2.7) | 2.19 (4.5) | 9.84 (15.9) | 13.1 |
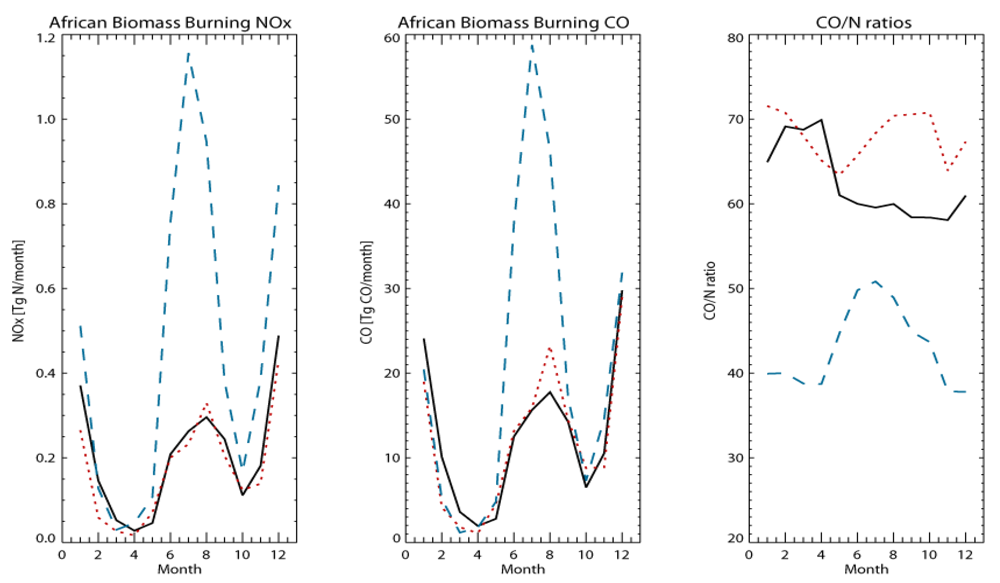
Figure 1 shows differences in the seasonal cycle of Africa BB emissions for both NO
x (left) and CO (middle), along with the corresponding seasonal cycle in the CO/N ratios (right). The two seasonal peaks in BB emissions occur from November to January (NDJ) in the Northern Hemisphere (NH) and from June to September (JJAS) in the SH. Largest differences occur between GFEDv2 and AMMABB during boreal summertime and are associated with the intense burning which occurs in southern Africa during this season. For NO
x, differences of ~50% also occur from NH burning during boreal wintertime. For both of the GFED inventories there are larger monthly emissions from NH burning than SH burning, whereas for AMMABB the emissions from SH burning dominate. Comparing GFEDv3 and AMMABB shows that the CO BB emission fluxes become quite similar from January to April compared to the corresponding NO
x emissions. Given that the emission factors for each trace species are similar between inventories this difference is related to the cumulative effect of differences in the burned area product, vegetation classifications and land use maps. The seasonal variation in the CO/N ratio shows that although the magnitude of the monthly CO/N ratios differ by ~30–50%, the seasonal variation becomes more similar between the GFEDv3 and the AMMABB inventories, although for the AMMABB inventory there is a maxima during August.
Figure 1.
Comparison of the monthly African (34°S–36°N, 20°W–40°E) emission totals for (left) NOx and (middle) CO in Tg N/month and Tg CO/month, respectively. The seasonal variation in the corresponding CO/N ratio is also shown (right). The inventories are: (__) GFEDv2, (.…) GFEDv3 and (---) AMMABB.
Figure 1.
Comparison of the monthly African (34°S–36°N, 20°W–40°E) emission totals for (left) NOx and (middle) CO in Tg N/month and Tg CO/month, respectively. The seasonal variation in the corresponding CO/N ratio is also shown (right). The inventories are: (__) GFEDv2, (.…) GFEDv3 and (---) AMMABB.
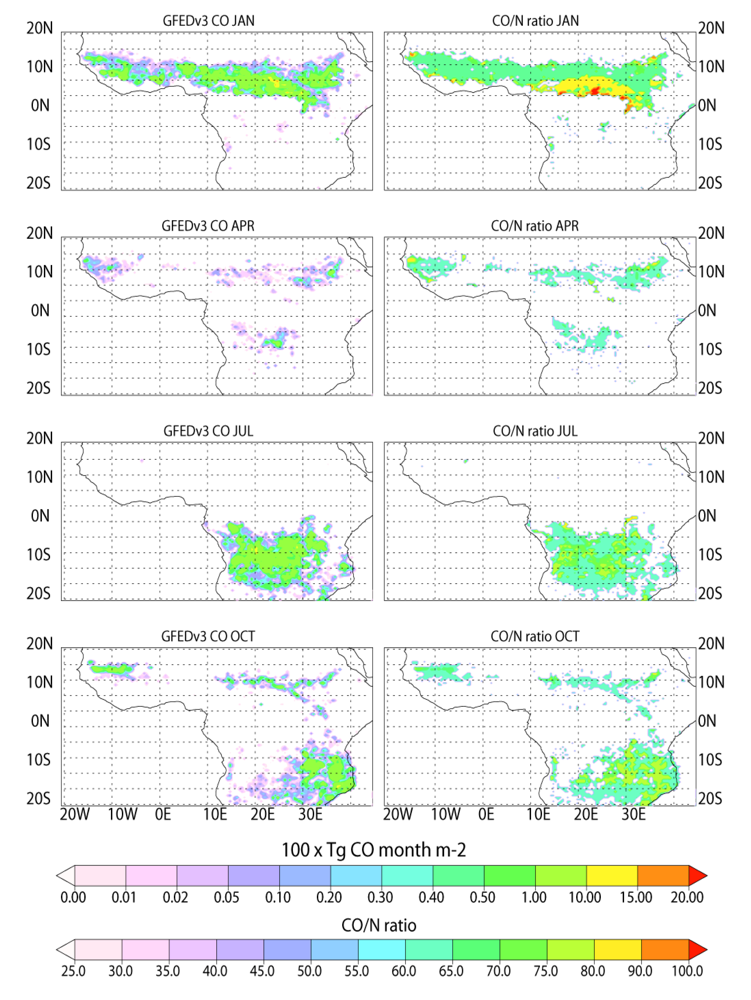
To highlight the differences in the temporal distribution of the emission estimates provided in each inventory,
Figure 2 and
Figure 3 show the monthly BB emission estimates for CO from Africa for January, April, July and October 2006 as provided in the GFEDv3 and AMMABB emission inventories, respectively. The corresponding CO/N ratios are also shown in all instances. Each emission inventory is shown on a 0.5° × 0.5° resolution. Both figures also show that there is significant variability with respect to the intensity of fires across any given latitude range for any given month. Comparing the total burned area in Africa for 2006 gives 195 × 10
4 km
2 for the L3JRC product [
42] and 237 × 10
4 km
2 for the GFEDv3 product [
32]. This results in the burned area in the L3JRC product being approximately half as large in northern Africa and two-thirds as large in southern Africa [
32]. Therefore, the amount of emissions released per unit area burnt is larger in the AMMABB inventory.
Figure 2.
The temporal distribution in African BB for (top to bottom) January, April, July and October during 2006 as provided in the GFEDv3 emission inventory. The corresponding CO/N ratio is also provided for all months.
Figure 2.
The temporal distribution in African BB for (top to bottom) January, April, July and October during 2006 as provided in the GFEDv3 emission inventory. The corresponding CO/N ratio is also provided for all months.
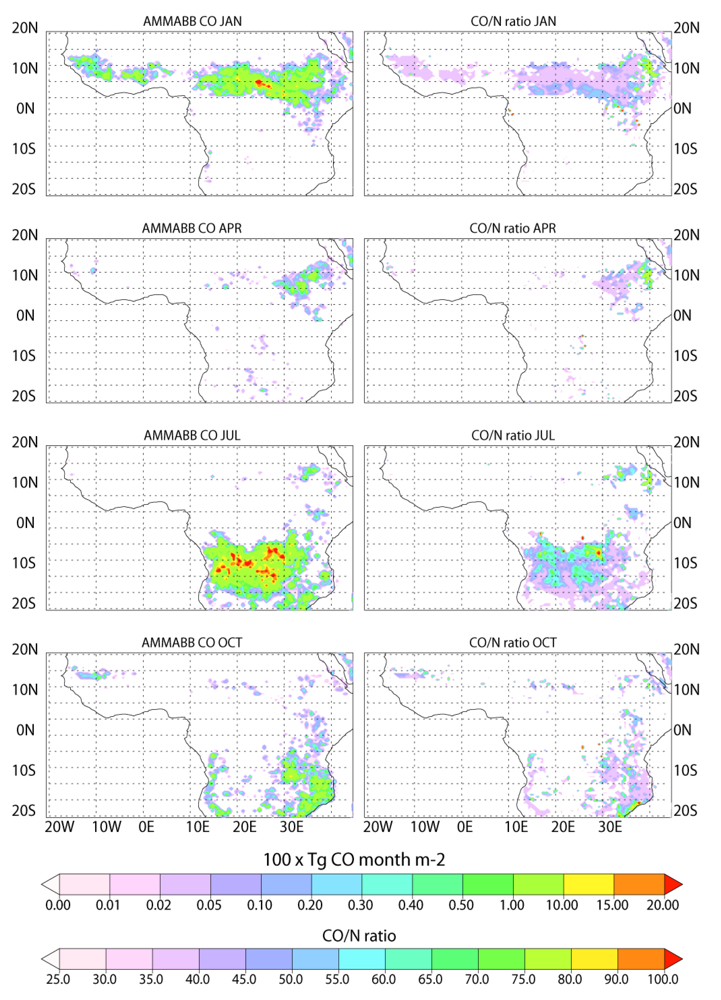
Figure 3.
As for
Figure 2 except showing the temporal distribution of the AMMABB emission inventory.
Figure 3.
As for
Figure 2 except showing the temporal distribution of the AMMABB emission inventory.
In general the seasonal hemispheric distribution is similar, although the extent of burning below (above) the equator is larger in the AMMABB inventory for January (July). The longitudinal distribution in the burning pattern is also very different, where the GFEDv3 inventory has more burning in West Africa throughout the year. Conversely the AMMABB inventory displays more burning activity towards the Horn of Africa. Previous studies have concluded that there is an underestimation in BB in Western Africa in the AMMABB inventory as a result of the L3JRC burned area product [
10,
40]. Stroppiana
et al. [
10] have performed direct comparisons of BB CO emissions derived for Africa during 2003 between the GFEDv3 and AMMABB inventories and found a correlation co-efficient (
R2) of 0.47, which agrees better than for other regions such as e.g., North America, where the reliability of the L3JRC burnt area product is thought to be low [
42]. They have also shown that for the three main land cover types (forest, savanna/grassland and agriculture) there are significant differences in the contribution of CO from each land type in Africa.
For much of the burning region the GFEDv3 inventory has CO/N ratios that range between 60 and 70, although there are regions in southern Africa which ratios can be as high as 80. The corresponding ratios in the AMMABB inventory are typically lower around 35 to 50, although the more intense burning events have ratios which are higher around 70 to 90. When applied in TM4, the spatial variability in these CO/N ratios becomes somewhat homogenized due to the coarsening step onto the horizontal grid of 3° × 2°. Thus using a model with higher resolution would introduce more significant regional variability into the African domain with respect to the CO/N ratio compared to the simulations presented here, which would subsequently affect tropospheric O
3 production [
43].
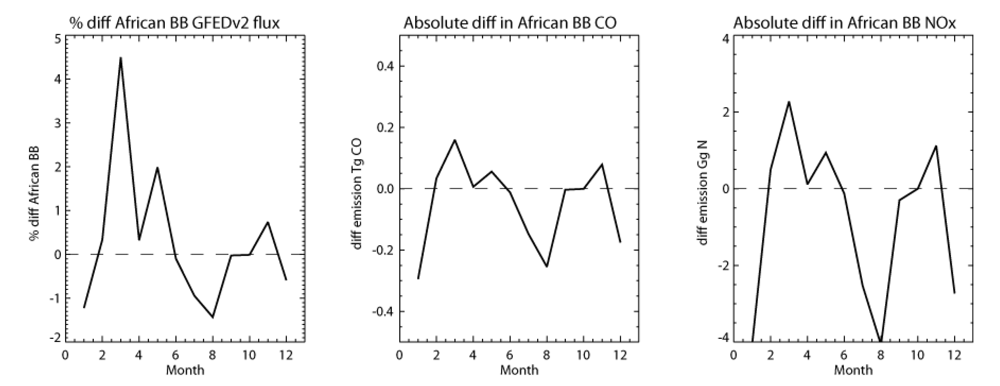
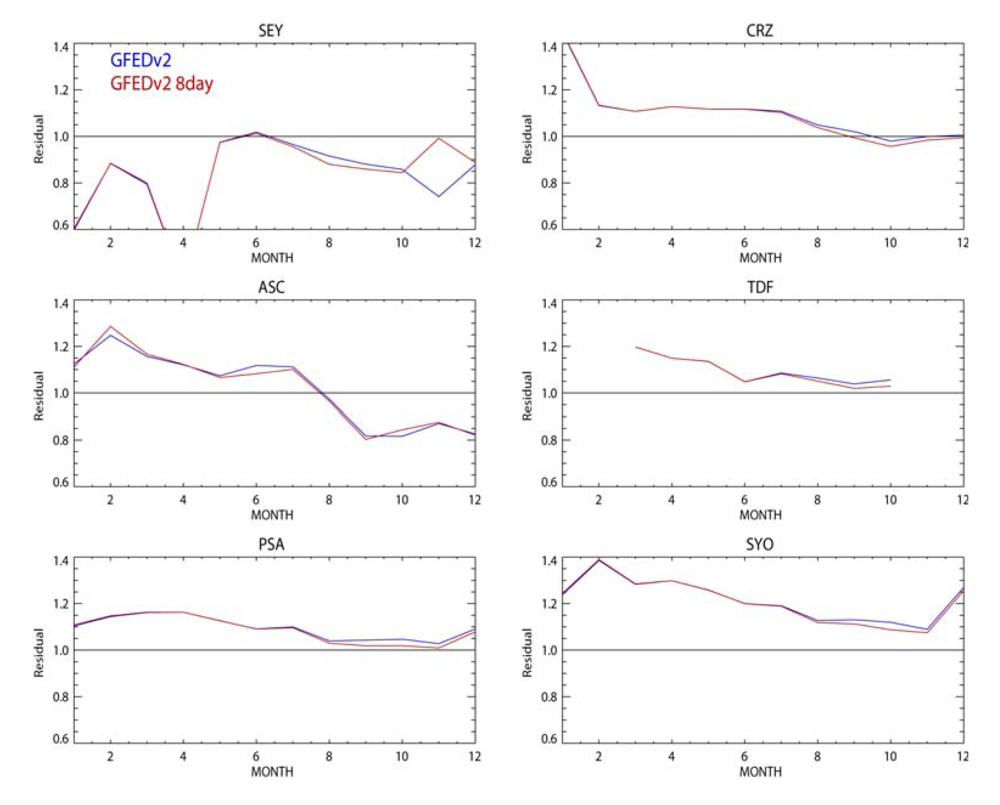
Finally we investigate the differences between the GFEDv2 and GFEDv2 8-day emission inventories for African (34°S–34°N, 20°W–40°E) CO emissions. Even if the annually integrated emission totals are approximately equal between the two different GFEDv2 inventories, the segregation of the emission fluxes into 8-day composites may result in different totals when the resulting emission is summed per calendar month.
Figure 4 shows the percentage differences in the monthly BB emissions fluxes from the African domain for 2006, as given in the 1° × 1° emission inventories used in this study. Also shown are the absolute differences in Tg CO and Gg N per month. It can be seen that there are generally decreases during the intensive BB seasons NDJ and JJAS. Integrating the absolute difference for the entire year results in a decrease of 0.5 Tg CO yr
−1 and 8.9 Gg N yr
−1 when applying the GFEDv2 8-day emission inventory for the African domain. These differences shift in location as governed by the location of the seasonal BB activity.
Figure 4.
The percentage difference in the monthly BB emission fluxes from Africa (34°S–34°N, 20°W–40°E) between the GFEDv2 and GFEDv2 8-day emission inventories. Also shown is the absolute difference in Tg CO and Gg N per month. The differences are calculated as (8-DAY-MONTHLY)/MONTHLY × 100.
Figure 4.
The percentage difference in the monthly BB emission fluxes from Africa (34°S–34°N, 20°W–40°E) between the GFEDv2 and GFEDv2 8-day emission inventories. Also shown is the absolute difference in Tg CO and Gg N per month. The differences are calculated as (8-DAY-MONTHLY)/MONTHLY × 100.
5. Changes to Global Burdens and Tropospheric Lifetimes
The large variability in the estimates of trace gas emissions contained in the different bottom-up BB emission inventories for Africa shown in
Table 2 has the potential to significantly impact the burdens and lifetimes of the ubiquitous trace gas species, O
3, CO and CH
4. In order to investigate the perturbations introduced we use a set of diagnostics which have been defined for assessing the performance of CTMs during multi-model intercomparison studies [
16]. For brevity we limit the discussion to the changes introduced by the different BB emission estimates, with the differences due to temporal variability and injection height being rather small in comparison and therefore of lesser interest.
For tropospheric O
3 the variability in the burden (BO
3), tropospheric lifetime (τO
3) and net annual production terms (pO
3) are given in
Table 4 for the different BB emission inventories. The percentage differences that are given are calculated with respect to the NONE simulation. Here we define a climatological tropopause [
60] throughout the analysis which ensures that the mass of the troposphere remains constant for all simulations therefore allowing a valid comparison of tropospheric burdens and lifetimes.
Table 4.
The variability in the global tropospheric burden and lifetime of O
3 during 2006 when adopting the different BB emission inventories for Africa. Also shown is the total global
in situ chemical production of O
3. The climatological tropopause of Lawrence
et al. [
60] is used to define the troposphere throughout. The relative percentage changes of tropospheric lifetimes and global burdens as compared to the NONE simulation are given in parenthesis.
Table 4.
The variability in the global tropospheric burden and lifetime of O3 during 2006 when adopting the different BB emission inventories for Africa. Also shown is the total global in situ chemical production of O3. The climatological tropopause of Lawrence et al. [60] is used to define the troposphere throughout. The relative percentage changes of tropospheric lifetimes and global burdens as compared to the NONE simulation are given in parenthesis.
| | BO3 (Tg O3) | O3 Lifetime (days) | pO3 (Tg O3) |
|---|
| NONE | 317.7 | 24.1 | 4328.1 |
| GFEDv2 | 323.0 (+1.7) | 23.6 (−1.9) | 4500.3 (+4.0) |
| GFEDv3 | 323.2 (+1.8) | 23.7 (−1.6) | 4492.5 (+3.8) |
| AMMABB | 332.2 (+4.6) | 23.3 (−3.0) | 4641.8 (+7.2) |
| AMMABB_LOWNOX | 325.7 (+2.5) | 23.6 (−1.9) | 4555.2 (+5.2) |
The introduction of African BB (as defined in the GFEDv2, GFEDv3 and AMMABB simulations) increases BO
3 between 1.7 and 4.6%, resulting in a mean BO
3 of 326.1 ± 5.3 Tg O
3 (with an associated uncertainty of ~1.6%). The corresponding change in τO
3 is from −1.9% to −3.0%, resulting in a mean τO
3 of 23.5 ± 0.2 days (with an associated uncertainty of ~0.9%). The corresponding differences in BCO, τCO, and τCH
4 are given in
Table 5, along with the total mass of CH
4 that is oxidized in each simulation. For CH
4 the total mass that is oxidized is equal to the amount oxidized by OH, with a fixed soil sink of 30 Tg yr
−1 and a stratospheric sink of 40 Tg yr
−1, as outlined in Stevenson
et al. [
16]. Comparing values reveals that African BB decreases the global lifetimes of both trace gases across the range of CO/N ratios contained in the different BB inventories (~43–69),
i.e., regardless of the seasonal CO emission flux. This gives mean atmospheric lifetimes across all three BB inventories of τCO = 53.4 ± 0.4 days and τCH
4 = 8.70 ± 0.02 years, which translates to an uncertainty of ~0.7% and ~0.2%, respectively.
Table 5.
The global tropospheric burdens and lifetimes of CO, and CH4 for the different BB emission inventories adopted for Africa during 2006.
Table 5.
The global tropospheric burdens and lifetimes of CO, and CH4 for the different BB emission inventories adopted for Africa during 2006.
| | BCO (Tg CO) | CO Lifetime (days) | Mass CH4 Oxidised | CH4 Lifetime (years) |
|---|
| NONE | 302.6 | 54.4 | 499.4 | 8.75 |
| GFEDv2 | 320.8 (+6.0) | 53.6 (+1.5) | 501.8 (+0.5) | 8.72 (−0.4) |
| GFEDv3 | 321.2 (+6.1) | 53.6 (+1.5) | 502.1 (+0.5) | 8.71 (−0.5) |
| AMMABB | 332.2 (+9.9) | 52.9 (+2.8) | 504.3 (+1.0) | 8.68 (−0.8) |
| AMMABB_LOWNOX | 339.1 (+12.0) | 54.3 (+0.2) | 495.1 (-0.9) | 8.82 (+0.8) |
Increasing NOx emissions cause an associated increase in pO3, although a moderating constraint on the variability in both pO3 and BO3 is the enhanced loss of reactive nitrogen via wet deposition as nitric acid (~1 Tg N yr−1extra when adopting the AMMA BB inventory). Thus the oxidative capacity of the global troposphere is enhanced as a result of African BB.
The uncertainty associated with the mean τCH
4 is significant due to the relatively modest increases which occur in the yearly growth rate of fractions of a percent [
20], where the variability in the mass of CH
4 oxidized across all simulations translates to a variability of °0.25 ppbv yr
−1 in the growth rate. The reduction in τCH4 is largest for AMMABB which may be counterintuitive because it also exhibits the highest CO emissions (
cf.
Table 2). From the CO consumption argument [
21] one would expect an increase in τCH4 due to the increased scavenging of OH by CO in the tropics, where the majority of CH
4 is oxidized. The AMMABB_LOWNOX simulation shows that increases in τCH4 are only observed if the CO/N ratio is relatively high (~90), which reduces the chemical production of ozone as shown in
Table 4. Such high CO/N ratios are typically associated with peat burning, charcoal production or animal waste burning [
6] and thus should be considered an upper limit in terms of realistic CO/N ratios. To show the sensitivity of global tropospheric composition to the CO/N ratio the annual zonal mean differences between the AMMABB and AMMABB_LOWNOX simulations are shown in
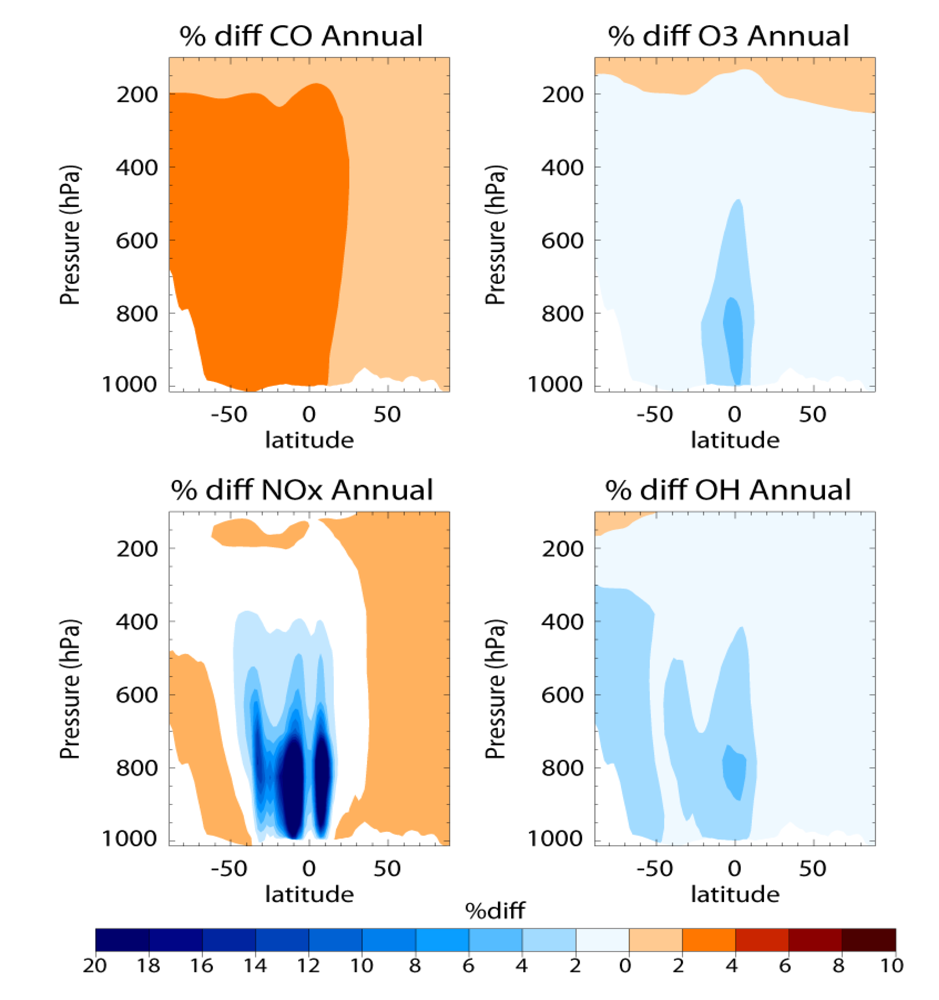
Figure 12. That there is a simultaneous increase in CO and decrease in OH under low NO
x conditions indicates that there is less oxidation capacity in the troposphere for the oxidation of CH
4 resulting in a subsequent increase in its atmospheric lifetime.
Therefore, some mitigation of the effects of high CO emissions from BB occurs due to the associated increase in NOx (and thus O3 and OH). It cannot be expected that the high CO/N ratios defined in the AMMABB_LOWNOX simulation would occur on a continental scale therefore it is likely that increases in τCH4 occur regardless of the inter-annual variability in BB emissions that originate from Africa. It should be noted that the feedback on photolysis rates due to smoke and aerosol particles is not accounted for in these simulations, but are expected to decrease the photo-chemical production of tropospheric O3 in the lower troposphere, thus allowing more CO to be transported out of the region.
Figure 12.
The relative percentage differences in the zonal annual mean concentrations for (a) CO; (b) O3; (c) NOx; and (d) OH between the AMMABB and AMMABB_LOWNOX simulations. The differences are calculated as (AMMABB_LOWNOX-AMMABB)/AMMABB ×100.
Figure 12.
The relative percentage differences in the zonal annual mean concentrations for (a) CO; (b) O3; (c) NOx; and (d) OH between the AMMABB and AMMABB_LOWNOX simulations. The differences are calculated as (AMMABB_LOWNOX-AMMABB)/AMMABB ×100.
For Africa, most burning is associated with either savannah and grasslands or woodlands, whose estimated CO/N ratios are 28.8 and 37.0, respectively [
6]. The ratios given in
Table 3 can be considered aggregates across a wide variety of different vegetation types for the African region, as shown by the regional variability in CO/N ratios shown in
Figure 3 and
Figure 4. Recently van Leeuwen and van der Werf [
61] have shown that when accounting for environmental variables such as precipitation and temperature, CO emissions could be higher for savanna and grasslands per Kg biomass burned significantly impacting emission ratios, although measurements during peak burning periods are necessary to constrain values. In this study we have shown that the global effect of such enhancements would be to increase BCO and BO
3 by a few percent, whilst decreasing the atmospheric lifetimes of long-lived trace species.
6. Conclusions
In this study we have investigated the sensitivity of atmospheric composition in the tropics and Southern Hemisphere towards the biomass burning emission inventory adopted for African biomass burning (namely the GFEDv2, GFEDv3 and AMMABB inventories). Comparing the monthly emission totals for the African continent between the different biomass burning inventories for 2006 shows that the intensity, location and associated CO/N ratios of large scale biomass burning events vary significantly, although the seasonality between inventories is rather similar. For the most active burning season of June-July-August-September this leads to differences of >100% in the monthly emission fluxes from Africa. When comparing the GFEDv2 monthly and 8-day emission inventories the integrated monthly emission estimates vary by ±2 Gg and ±0.2 Tg for nitrogen oxides and carbon monoxide (CO), respectively. These differences are introduced as a result of increasing the temporal resolution of the burning events, although the annual emission flux remains approximately equal. The spatial distribution of burning events within Africa is also highly variable when comparing different inventories, especially for the north of the Equator during the boreal winter.
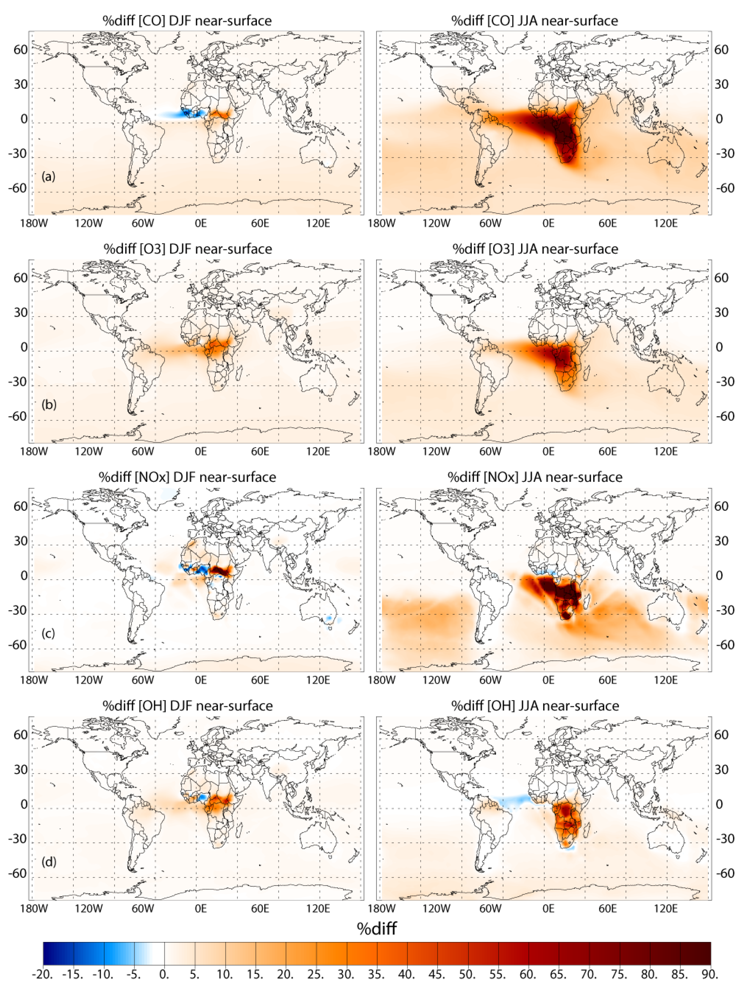
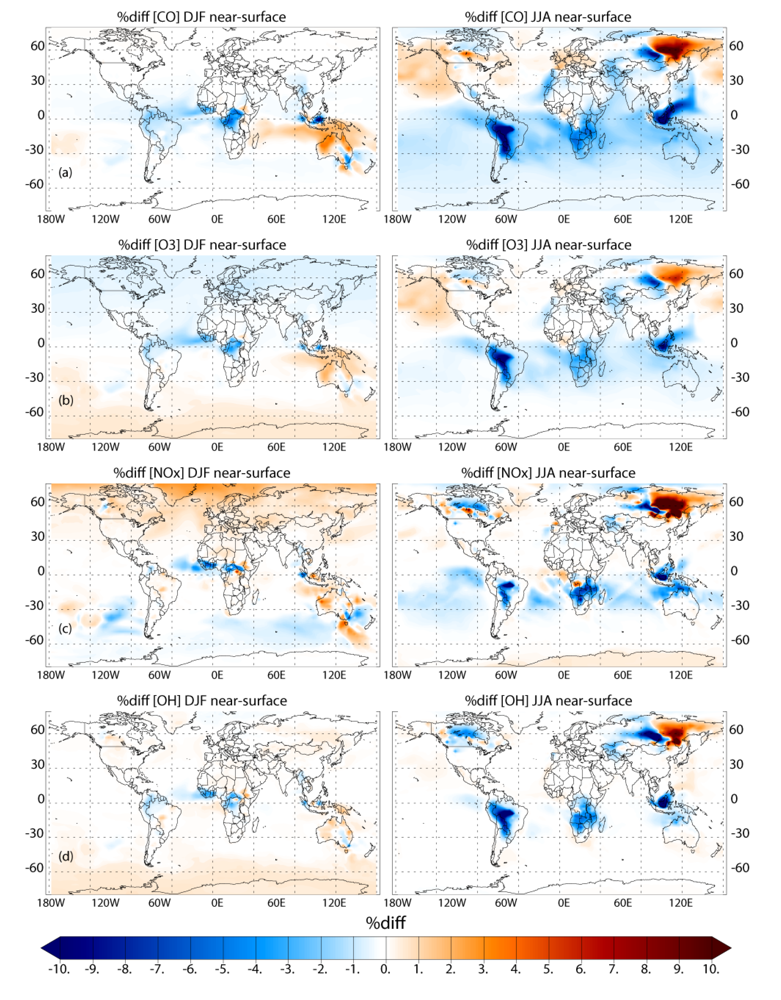
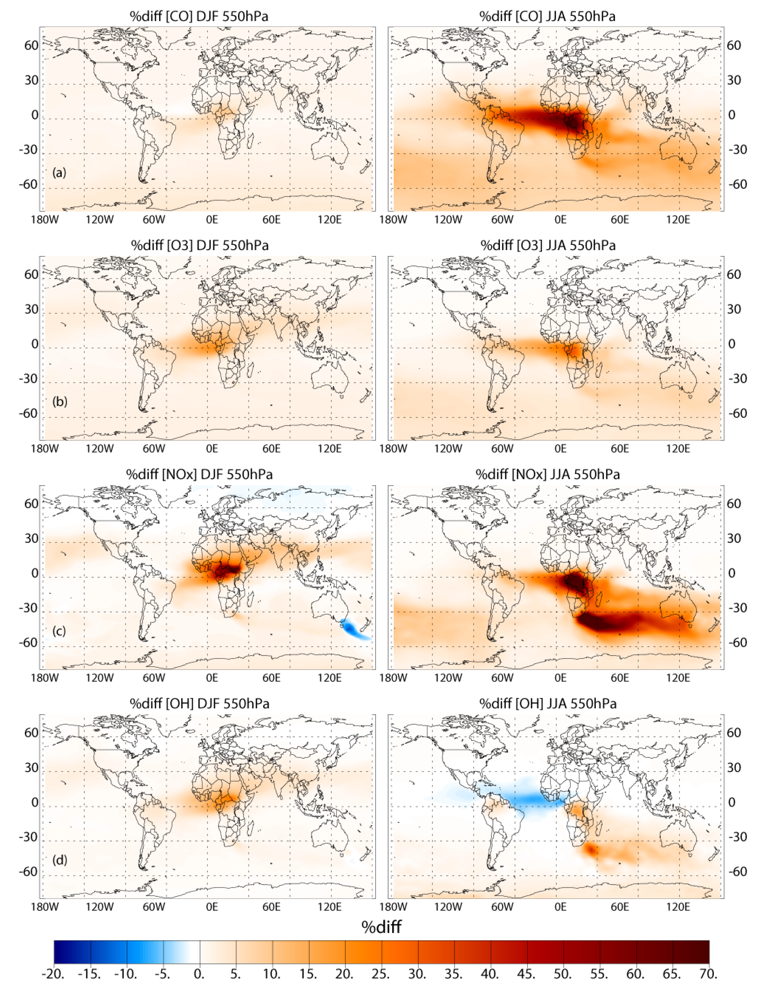
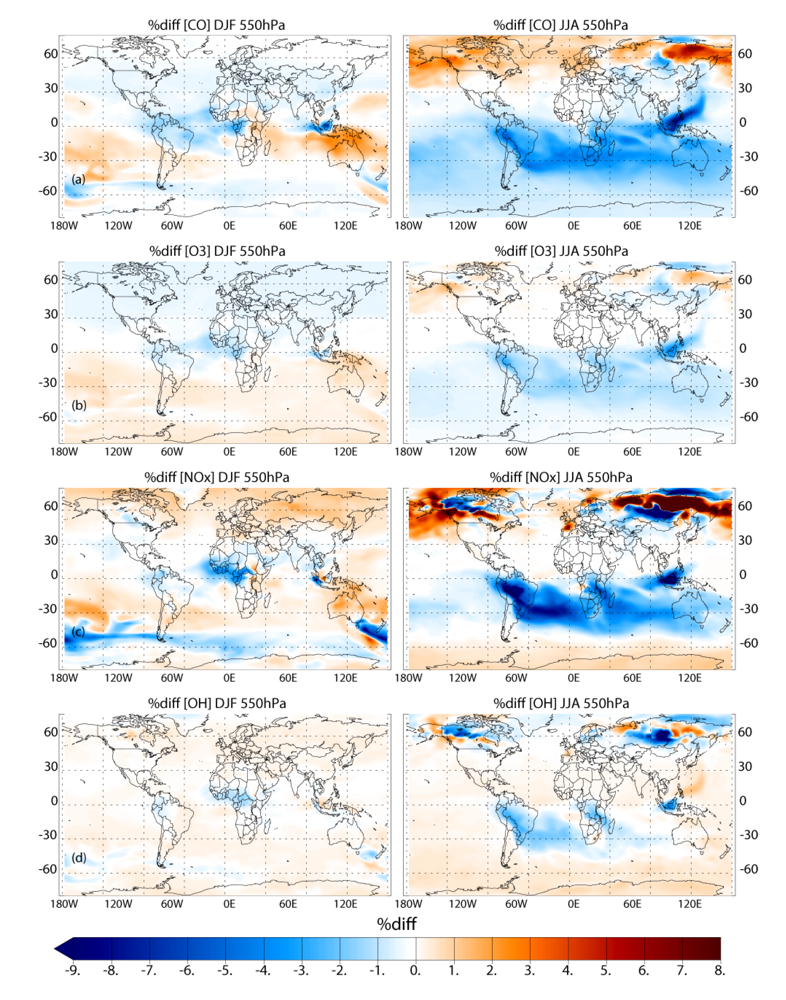
Applying the different biomass burning emission inventories in a 3D global chemistry-transport model leads to significant differences in the near-surface ozone (O3) and CO, especially during June-July-August in the Southern Hemisphere (SH). When comparing the simulations which adopt the GFEDv3 and AMMABB emission inventories, differences in near-surface O3 and CO of ~50–90% occur during June-July-August near the source regions. In the remote SH differences are of the order of ~5–15%. Increasing the update frequency of the temporal distribution of the burning events from monthly to every 8 days shows decreases of between ~0 and 10% for near-surface O3 and CO near the source regions during both seasons. More modest changes of a few percent occur for the outflow regions. This occurs due to both the changes in the geographic variability of the most intense fires and small differences in the total monthly emission fluxes as defined between inventories. Increasing the injection heights at which trace gas emission from fires are introduced in the tropics (30°S–30°N) results in differences of a few percent limited to regions which exhibit the most intense fires, as found in the literature.
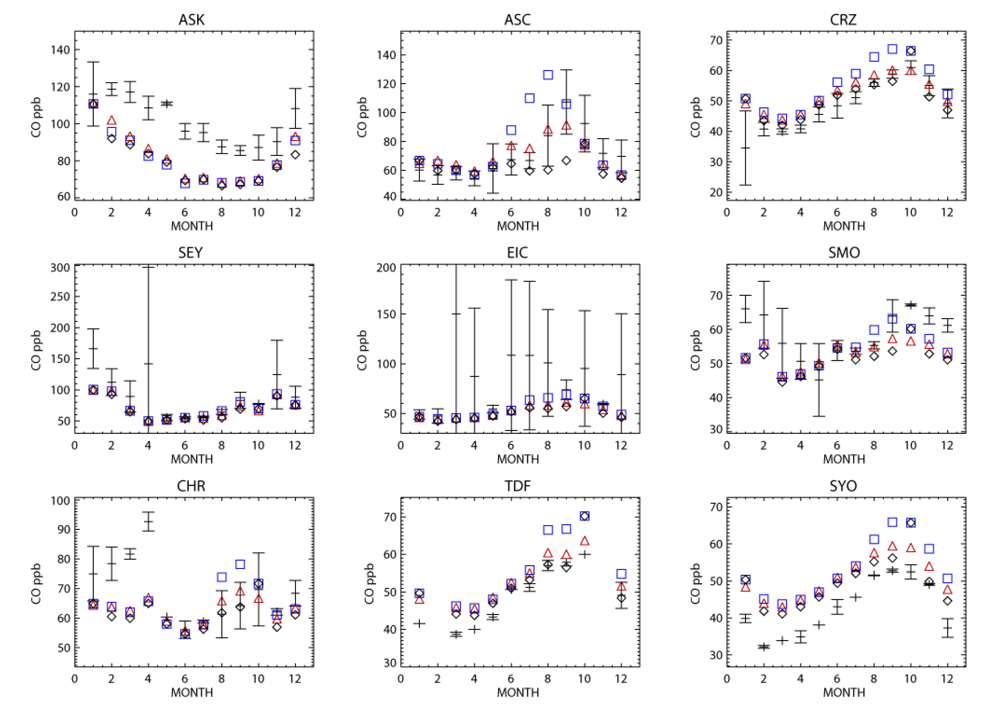
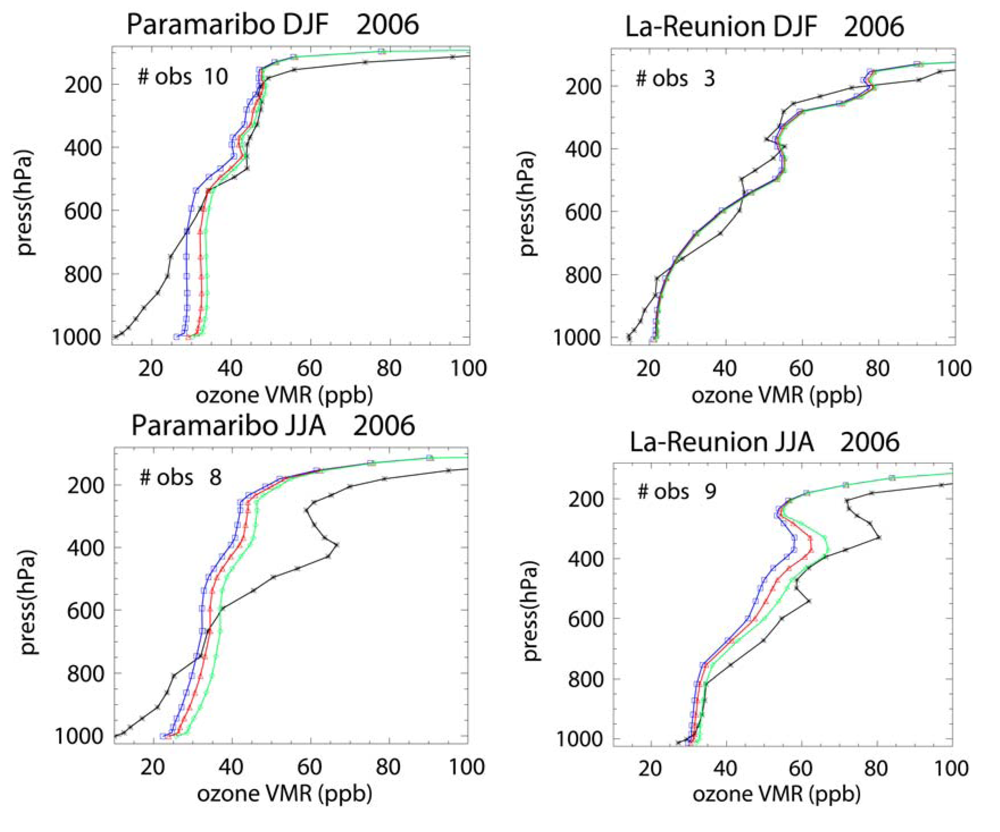
For the long range transport of gases out of Africa substantial differences of ~50–70% in the resident mixing ratios of CO and NOx are simulated in the outflow regions at ~550 hPa, which has a significant impact of the atmospheric burdens of these trace species in the SH. A comparison of the monthly mean mixing ratios for CO from the simulations against ground-based flask measurements shows that the AMMABB inventory tends to overestimate background concentrations during June-July-August-September, with the GFEDv3 emission inventory showing the best correlation. For tropospheric O3 comparisons with composites of tropical ozone soundings taken at locations to the east of Africa show that the AMMABB emission inventory shows a better agreement for the middle and upper troposphere, although this is also dependant on both the chemical mechanism which is employed, the efficiency of the convective and advective mixing and the accuracy of the meteorological data used to drive the chemistry-transport model.
The influence of Africa biomass burning on the atmospheric burdens of CO and O3 results in increases of between 6.0–12.0% and 1.9–3.0% across inventories, respectively. This introduces an associated uncertainty into the total tropospheric burdens of ~1.6% and ~0.9%, respectively. For the atmospheric lifetimes of CO, O3 and CH4 the values are 53.4 ± 0.4 days, 23.5 ± 0.2 days and 8.70 ± 0.2 years are calculated, respectively, with an associated uncertainty of ~0.9%, ~0.7% and ~0.2%. Decreases in τCH4 occur across all BB inventories regardless of the differences in the total CO emitted from African BB. This is due to the associated differences in the CO/N ratio being rather small between the inventories on the horizontal resolution employed for the study. Thus the increase in the CO emission is partially mitigated by the simultaneous increase in the oxidative capacity around the source regions and, subsequently, in the outflow. Only when the CO/N ratio exceeds ~90 does the increase in the tropospheric CO burden result in decreases in the lifetime of CH4. This has important implications when explaining the relationship between the inter-annual variability in the global CH4 growth rate and the inter-annual variability in BB emissions from Africa.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










