The Influence of Absolute Mass Loading of Secondary Organic Aerosols on Their Phase State

Abstract
:1. Introduction
2. Experiments
2.1. Reagents and Equipment
2.2. Chamber Experiments (Generation of SOA)
2.3. Bounce Analysis
3. Results and Discussion
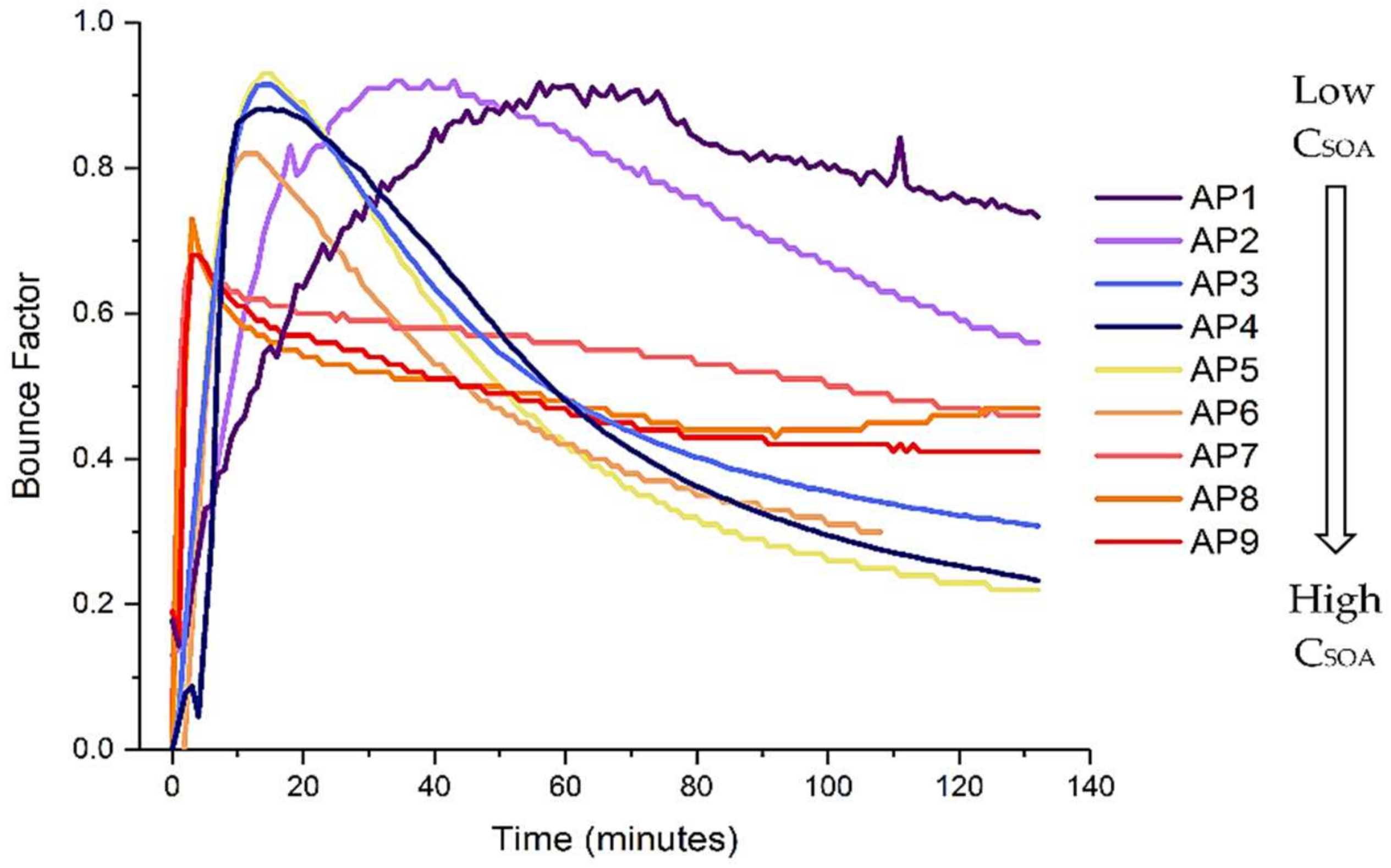
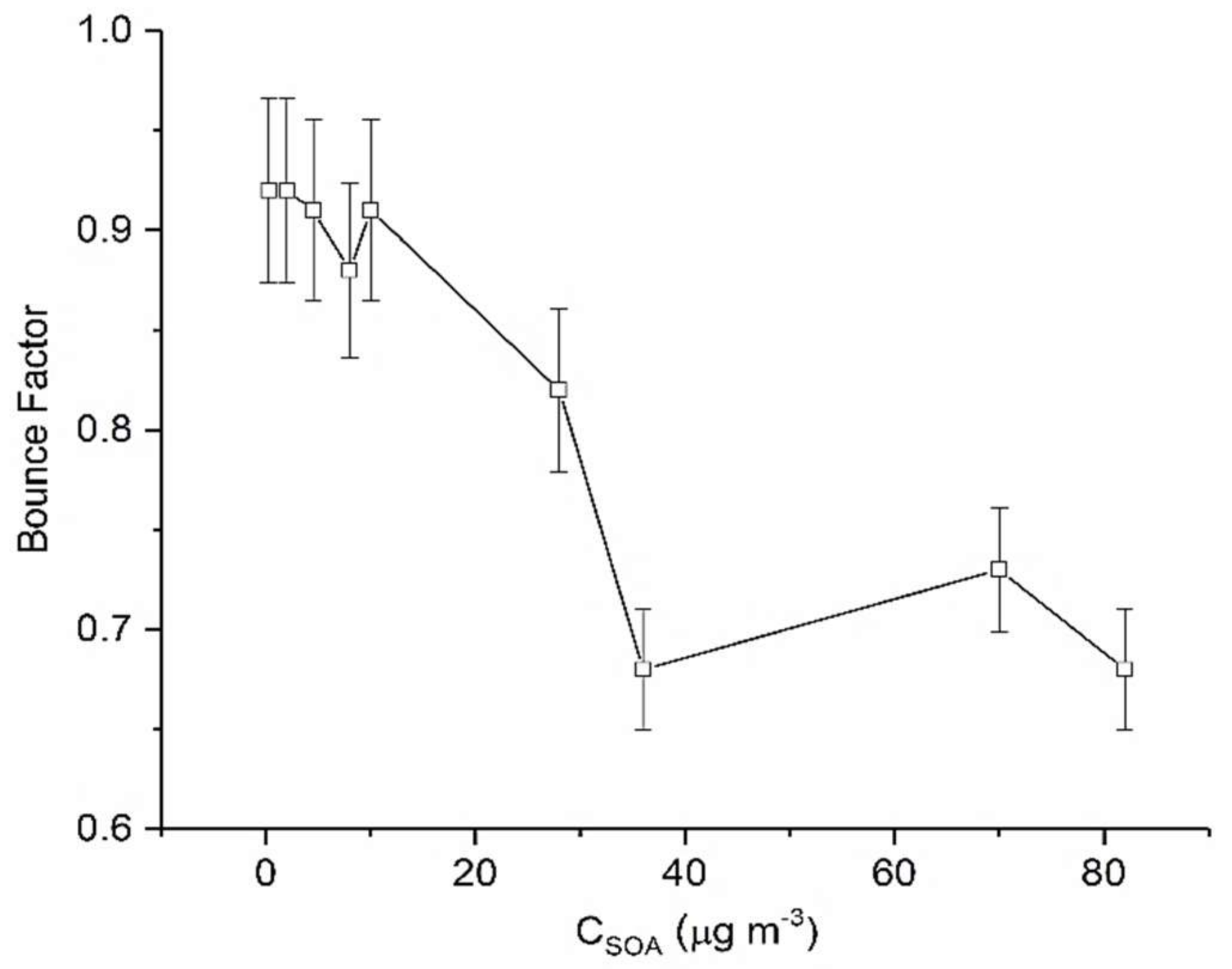
3.1. α-Pinene
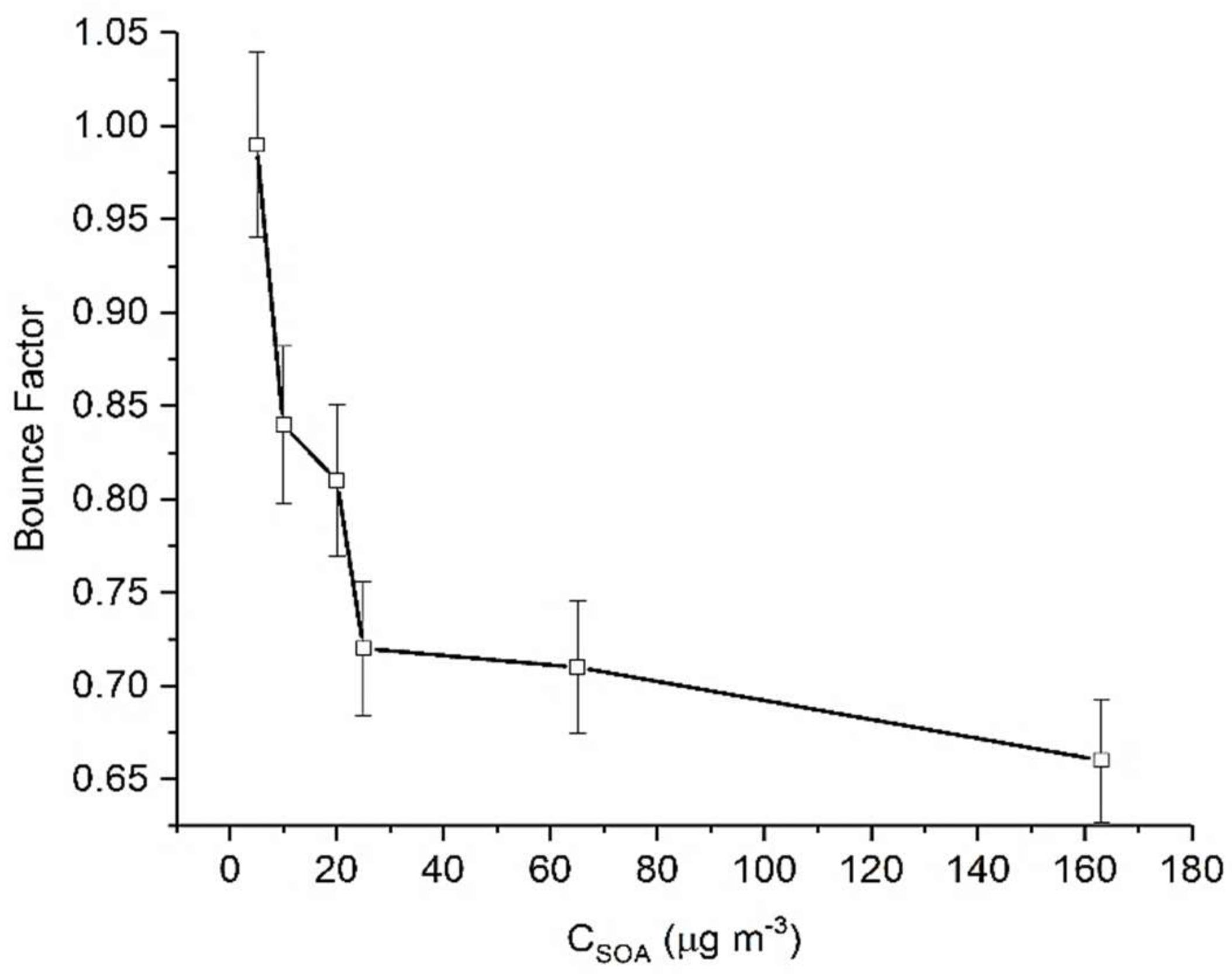
3.2. Limonene
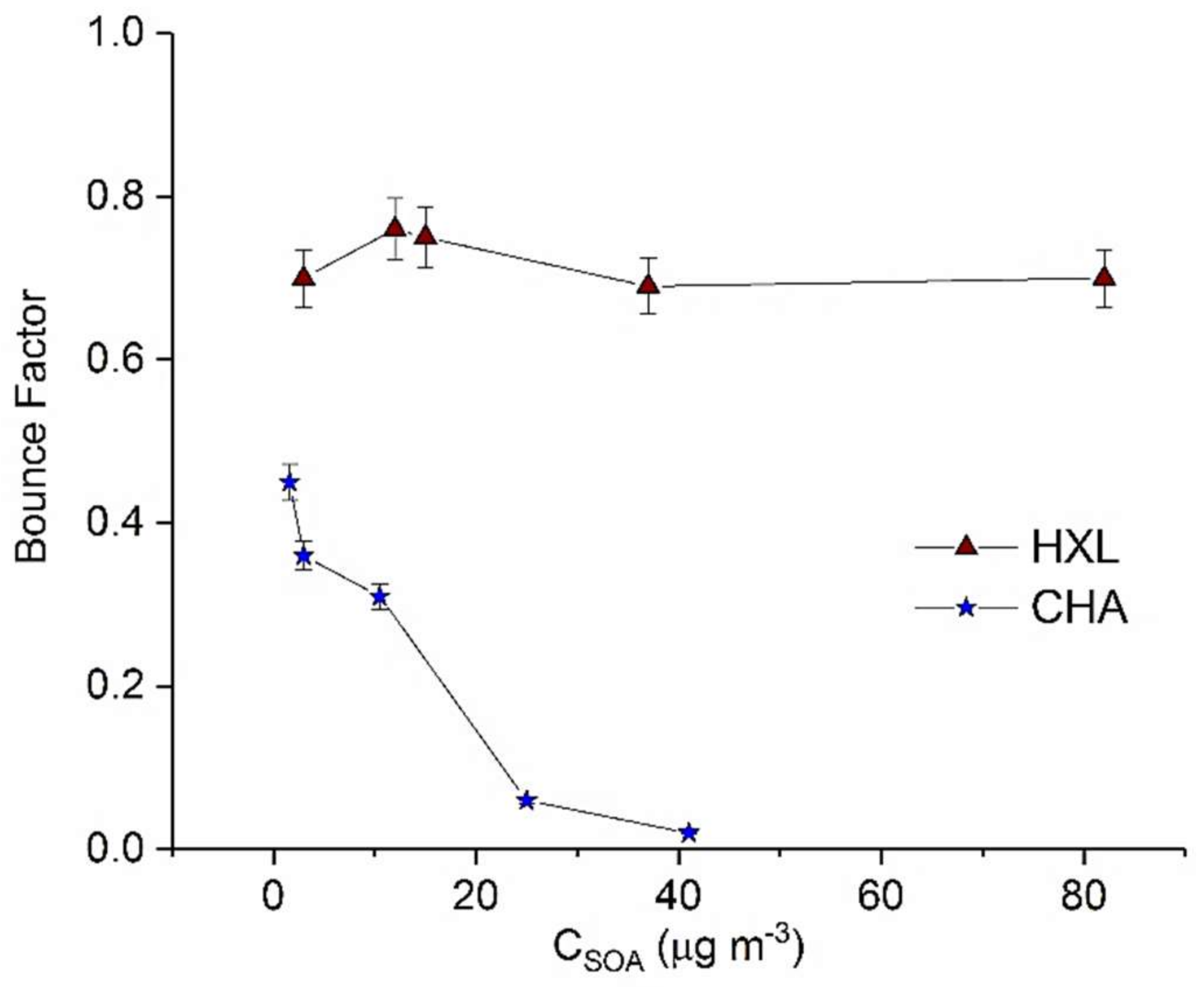
3.3. Cis-3-Hexenyl Acetate (CHA) and Cis-3-Hexen-1-ol (HXL)
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jimenez, J.L.; Canagaratna, M.R.; Donahue, N.M.; Prevot, A.S.H.; Zhang, Q.; Kroll, J.H.; DeCarlo, P.F.; Allan, J.D.; Coe, H.; Ng, N.L.; et al. Evolution of organic aerosols in the atmosphere. Science 2009, 326, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Hallquist, M.; Wenger, J.C.; Baltensperger, U.; Rudich, Y.; Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N.M.; George, C.; Goldstein, A.H.; et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155–5236. [Google Scholar] [CrossRef] [Green Version]
- Ehn, M.; Thornton, J.A.; Kleist, E.; Sipilä, M.; Junninen, H.; Pullinen, I.; Springer, M.; Rubach, F.; Tillmann, R.; Lee, B.; et al. A large source of low-volatility secondary organic aerosol. Nature 2014, 506, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.A.; Dockery, D.W. Health effects of fine particulate air pollution: Lines that connect. J. Air Waste Manag. Assoc. 2006, 56, 709–742. [Google Scholar] [CrossRef] [PubMed]
- Poschl, U. Atmospheric aerosols: Composition, transformation, climate and health effects. Angew. Chem. Int. Ed. 2005, 44, 7520–7540. [Google Scholar] [CrossRef] [PubMed]
- Shiraiwa, M.; Li, Y.; Tsimpidi, A.P.; Karydis, V.A.; Berkemeier, T.; Pandis, S.N.; Lelieveld, J.; Koop, T.; Pöschl, U. Global distribution of particle phase state in atmospheric secondary organic aerosols. Nat. Commun. 2017, 8, 15002. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, M.; Easter, R.C.; Liu, X.; Zelenyuk, A.; Singh, B.; Zhang, K.; Ma, P.-L.; Chand, D.; Ghan, S.; Jimenez, J.L.; et al. Global transformation and fate of SOA: Implications of low-volatility SOA and gas-phase fragmentation reactions. J. Geophys. Res. Atmos. 2015, 120, 4169–4195. [Google Scholar] [CrossRef]
- Yu, H.; Kaufman, Y.J.; Chin, M.; Feingold, G.; Remer, L.A.; Anderson, T.L.; Balkanski, Y.; Bellouin, N.; Boucher, O.; Christopher, S.; et al. A review of measurement-based assessments of the aerosol direct radiative effect and forcing. Atmos. Chem. Phys. 2006, 6, 613–666. [Google Scholar] [CrossRef]
- Berkemeier, T.; Shiraiwa, M.; Poschl, U.; Koop, T. Competition between water uptake and ice nucleation by glassy organic aerosol particles. Atmos. Chem. Phys. 2014, 14, 12513–12531. [Google Scholar] [CrossRef]
- Scott, C.E.; Spracklen, D.V.; Pierce, J.R.; Riipinen, I.; D’Andrea, S.D.; Rap, A.; Carslaw, K.S.; Forster, P.M.; Artaxo, P.; Kulmala, M.; et al. Impact of gas-to-particle partitioning approaches on the simulated radiative effects of biogenic secondary organic aerosol. Atmos. Chem. Phys. 2015, 15, 12989–13001. [Google Scholar] [CrossRef]
- Wang, B.; O’Brien, R.E.; Kelly, S.T.; Shilling, J.E.; Moffet, R.C.; Gilles, M.K.; Laskin, A. Reactivity of liquid and semisolid secondary organic carbon with chloride and nitrate in atmospheric aerosols. J. Phys. Chem. A 2014, 119, 4498–4508. [Google Scholar] [CrossRef] [PubMed]
- Perraud, V.; Bruns, E.A.; Ezell, M.J.; Johnson, S.N.; Yu, Y.; Alexander, M.L.; Zelenyuk, A.; Imre, D.; Chang, W.L.; Dabdub, D.; et al. Nonequilibrium atmospheric secondary organic aerosol formation and growth. Proc. Natl. Acad. Sci. USA 2012, 109, 2836–2841. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.E.; Neu, A.; Epstein, S.A.; MacMillan, A.C.; Wang, B.; Kelly, S.T.; Nizkorodov, S.A.; Laskin, A.; Moffet, R.C.; Gilles, M.K. Physical properties of ambient and laboratory-generated secondary organic aerosol. Geophys. Res. Lett. 2014, 41, 4347–4353. [Google Scholar] [CrossRef]
- Power, R.M.; Simpson, S.H.; Reid, J.P.; Hudson, A.J. The transition from liquid to solid-like behaviour in ultrahigh viscosity aerosol particles. Chem. Sci. 2013, 4, 2597–2604. [Google Scholar] [CrossRef]
- Shiraiwa, M.; Zuend, A.; Bertram, A.K.; Seinfeld, J.H. Gas-particle partitioning of atmospheric aerosols: Interplay of physical state, non-ideal mixing and morphology. Phys. Chem. Chem. Phys. 2013, 15, 11441–11453. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, M.; Martin, S.T. Phase of atmospheric secondary organic material affects its reactivity. Proc. Natl. Acad. Sci. USA 2012, 109, 17354–17359. [Google Scholar] [CrossRef] [PubMed]
- Pajunoja, A.; Lambe, A.T.; Hakala, J.; Rastak, N.; Cummings, M.J.; Brogan, J.F.; Hao, L.; Paramonov, M.; Hong, J.; Prisle, N.L.; et al. Adsorptive uptake of water by semisolid secondary organic aerosols. Geophys. Res. Lett. 2015, 42, 3063–3068. [Google Scholar] [CrossRef]
- Virtanen, A.; Kannosto, J.; Kuuluvainen, H.; Arffman, A.; Joutsensaari, J.; Saukko, E.; Hao, L.; Yli-Pirila, P.; Tiitta, P.; Holopainen, J.K.; et al. Bounce behavior of freshly nucleated biogenic secondary organic aerosol particles. Atmos. Chem. Phys. 2011, 11, 8759–8766. [Google Scholar] [CrossRef]
- Slade, J.H.; Knopf, D.A. Multiphase oh oxidation kinetics of organic aerosol: The role of particle phase state and relative humidity. Geophys. Res. Lett. 2014, 41, 5297–5306. [Google Scholar] [CrossRef]
- Slade, J.H.; Shiraiwa, M.; Arangio, A.; Su, H.; Pöschl, U.; Wang, J.; Knopf, D.A. Cloud droplet activation through oxidation of organic aerosol influenced by temperature and particle phase state. Geophys. Res. Lett. 2017, 44, 1583–1591. [Google Scholar] [CrossRef]
- Pankow, J.F. An absorption-model of gas-particle partitioning of organic-compounds in the atmosphere. Atmos. Environ. 1994, 28, 185–188. [Google Scholar] [CrossRef]
- Chan, A.W.H.; Kroll, J.H.; Ng, N.L.; Seinfeld, J.H. Kinetic modeling of secondary organic aerosol formation: Effects of particle- and gas-phase reactions of semivolatile products. Atmos. Chem. Phys. 2007, 7, 4135–4147. [Google Scholar] [CrossRef]
- Vaden, T.D.; Imre, D.; Beranek, J.; Shrivastava, M.; Zelenyuk, A. Evaporation kinetics and phase of laboratory and ambient secondary organic aerosol. Proc. Natl. Acad. Sci. USA 2011, 108, 2190–2195. [Google Scholar] [CrossRef] [PubMed]
- Loza, C.L.; Coggon, M.M.; Nguyen, T.B.; Zuend, A.; Flagan, R.C.; Seinfeld, J.H. On the mixing and evaporation of secondary organic aerosol components. Environ. Sci. Technol. 2013, 47, 6173–6180. [Google Scholar] [CrossRef] [PubMed]
- Abramson, E.; Imre, D.; Beranek, J.; Wilson, J.; Zelenyuk, A. Experimental determination of chemical diffusion within secondary organic aerosol particles. Phys. Chem. Chem. Phys. 2013, 15, 2983–2991. [Google Scholar] [CrossRef] [PubMed]
- Cappa, C.D.; Wilson, K.R. Evolution of organic aerosol mass spectra upon heating: Implications for oa phase and partitioning behavior. Atmos. Chem. Phys. 2011, 11, 1895–1911. [Google Scholar] [CrossRef]
- Sato, K.; Fujitani, Y.; Inomata, S.; Morino, Y.; Tanabe, K.; Ramasamy, S.; Hikida, T.; Shimono, A.; Takami, A.; Fushimi, A.; et al. Lower than expected volatility of secondary organic aerosols formed during α-pinene ozonolysis. Atmos. Chem. Phys. Discuss. 2017, 2017, 1–17. [Google Scholar] [CrossRef]
- Virtanen, A.; Joutsensaari, J.; Koop, T.; Kannosto, J.; Yli-Pirila, P.; Leskinen, J.; Makela, J.M.; Holopainen, J.K.; Poschl, U.; Kulmala, M.; et al. An amorphous solid state of biogenic secondary organic aerosol particles. Nature 2010, 467, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Koop, T.; Bookhold, J.; Shiraiwa, M.; Poschl, U. Glass transition and phase state of organic compounds: Dependency on molecular properties and implications for secondary organic aerosols in the atmosphere. Phys. Chem. Chem. Phys. 2011, 13, 19238–19255. [Google Scholar] [CrossRef] [PubMed]
- Renbaum-Wolff, L.; Grayson, J.W.; Bateman, A.P.; Kuwata, M.; Sellier, M.; Murray, B.J.; Shilling, J.E.; Martin, S.T.; Bertram, A.K. Viscosity of alpha-pinene secondary organic material and implications for particle growth and reactivity. Proc. Natl. Acad. Sci. USA 2013, 110, 8014–8019. [Google Scholar] [CrossRef] [PubMed]
- Shiraiwa, M.; Ammann, M.; Koop, T.; Poschl, U. Gas uptake and chemical aging of semisolid organic aerosol particles. Proc. Natl. Acad. Sci. USA 2011, 108, 11003–11008. [Google Scholar] [CrossRef] [PubMed]
- Lignell, H.; Hinks, M.L.; Nizkorodov, S.A. Exploring matrix effects on photochemistry of organic aerosols. Proc. Natl. Acad. Sci. USA 2014, 111, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Zobrist, B.; Marcolli, C.; Pedernera, D.A.; Koop, T. Do atmospheric aerosols form glasses? Atmos. Chem. Phys. 2008, 8, 5221–5244. [Google Scholar] [CrossRef]
- Li, Y.J.; Liu, P.; Gong, Z.; Wang, Y.; Bateman, A.P.; Bergoend, C.; Bertram, A.K.; Martin, S.T. Chemical reactivity and liquid/nonliquid states of secondary organic material. Environ. Sci. Technol. 2015, 49, 13264–13274. [Google Scholar] [CrossRef] [PubMed]
- Pajunoja, A.; Malila, J.; Hao, L.Q.; Joutsensaari, J.; Lehtinen, K.E.J.; Virtanen, A. Estimating the viscosity range of soa particles based on their coalescence time. Aerosol Sci. Technol. 2014, 48, I–IV. [Google Scholar] [CrossRef]
- Saukko, E.; Lambe, A.T.; Massoli, P.; Koop, T.; Wright, J.P.; Croasdale, D.R.; Pedernera, D.A.; Onasch, T.B.; Laaksonen, A.; Davidovits, P.; et al. Humidity-dependent phase state of soa particles from biogenic and anthropogenic precursors. Atmos. Chem. Phys. 2012, 12, 7517–7529. [Google Scholar] [CrossRef] [Green Version]
- Shilling, J.E.; Chen, Q.; King, S.M.; Rosenoern, T.; Kroll, J.H.; Worsnop, D.R.; DeCarlo, P.F.; Aiken, A.C.; Sueper, D.; Jimenez, J.L.; et al. Loading-dependent elemental composition of alpha-pinene soa particles. Atmos. Chem. Phys. 2009, 9, 771–782. [Google Scholar] [CrossRef]
- Grieshop, A.P.; Donahue, N.M.; Robinson, A.L. Is the gas-particle partitioning in alpha-pinene secondary organic aerosol reversible? Geophys. Res. Lett. 2007, 34. [Google Scholar] [CrossRef]
- Chan, M.N.; Chan, A.W.H.; Chhabra, P.S.; Surratt, J.D.; Seinfeld, J.H. Modeling of secondary organic aerosol yields from laboratory chamber data. Atmos. Chem. Phys. 2009, 9, 5669–5680. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, S.; Donahue, N.M.; Chuang, W.; Hildebrandt Ruiz, L.; Ng, N.L.; Wang, Y.; Hao, J. Evaluation of one-dimensional and two-dimensional volatility basis sets in simulating the aging of secondary organic aerosol with smog-chamber experiments. Environ. Sci. Technol. 2015, 49, 2245–2254. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.M.; Zahardis, J.; Petrucci, G.A. Establishing the contribution of lawn mowing to atmospheric aerosol levels in american suburbs. Atmos. Chem. Phys. 2014, 14, 797–812. [Google Scholar] [CrossRef]
- Jain, S.; Petrucci, G.A. A new method to measure aerosol particle bounce using a cascade electrical low pressure impactor. Aerosol Sci. Technol. 2015, 49, 390–399. [Google Scholar] [CrossRef]
- Geddes, S.; Nichols, B.; Flemer, S.; Eisenhauer, J.; Zahardis, J.; Petrucci, G.A. Near-infrared laser desorption/ionization aerosol mass spectrometry for investigating primary and secondary organic aerosols under low loading conditions. Anal. Chem. 2010, 82, 7915–7923. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.; Odum, J.R.; Bowman, F.; Collins, D.; Klockow, D.; Flagan, R.C.; Seinfeld, J.H. Formation of organic aerosols from the oxidation of biogenic hydrocarbons. J. Atmos. Chem. 1997, 26, 189–222. [Google Scholar] [CrossRef]
- Odum, J.R.; Jungkamp, T.P.W.; Griffin, R.J.; Flagan, R.C.; Seinfeld, J.H. The atmospheric aerosol-forming potential of whole gasoline vapor. Science 1997, 276, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Turpin, B.J.; Lim, H.-J. Species contributions to pm2.5 mass concentrations: Revisiting common assumptions for estimating organic mass. Aerosol Sci. Technol. 2001, 35, 602–610. [Google Scholar] [CrossRef]
- Pathak, R.K.; Stanier, C.O.; Donahue, N.M.; Pandis, S.N. Ozonolysis of α-pinene at atmospherically relevant concentrations: Temperature dependence of aerosol mass fractions (yields). J. Geophys. Res. Atmos. 2007, 112, D03201. [Google Scholar] [CrossRef]
- Nakao, S.; Tang, P.; Tang, X.; Clark, C.H.; Qi, L.; Seo, E.; Asa-Awuku, A.; Cocker, D. Density and elemental ratios of secondary organic aerosol: Application of a density prediction method. Atmos. Environ. 2013, 68, 273–277. [Google Scholar] [CrossRef]
- Virtanen, A.; Marjamäki, M.; Ristimäki, J.; Keskinen, J. Fine particle losses in electrical low-pressure impactor. J. Aerosol Sci. 2001, 32, 389–401. [Google Scholar] [CrossRef]
- Järvinen, A.; Aitomaa, M.; Rostedt, A.; Keskinen, J.; Yli-Ojanperä, J. Calibration of the new electrical low pressure impactor (ELPI+). J. Aerosol Sci. 2014, 69, 150–159. [Google Scholar] [CrossRef]
- Kolesar, K.R.; Chen, C.; Johnson, D.; Cappa, C.D. The influences of mass loading and rapid dilution of secondary organic aerosol on particle volatility. Atmos. Chem. Phys. 2015, 15, 9327–9343. [Google Scholar] [CrossRef]
- King, S.M.; Rosenoern, T.; Shilling, J.E.; Chen, Q.; Martin, S.T. Increased cloud activation potential of secondary organic aerosol for atmospheric mass loadings. Atmos. Chem. Phys. 2009, 9, 2959–2971. [Google Scholar] [CrossRef]
- Gao, Y.Q.; Hall, W.A.; Johnston, M.V. Molecular composition of monoterpene secondary organic aerosol at low mass loading. Environ. Sci. Technol. 2010, 44, 7897–7902. [Google Scholar] [CrossRef] [PubMed]
- Pfaffenberger, L.; Barmet, P.; Slowik, J.G.; Praplan, A.P.; Dommen, J.; Prevot, A.S.H.; Baltensperger, U. The link between organic aerosol mass loading and degree of oxygenation: An alpha-pinene photooxidation study. Atmos. Chem. Phys. 2013, 13, 6493–6506. [Google Scholar] [CrossRef]
- Hosny, N.A.; Fitzgerald, C.; Vysniauskas, A.; Athanasiadis, A.; Berkemeier, T.; Uygur, N.; Poschl, U.; Shiraiwa, M.; Kalberer, M.; Pope, F.D.; et al. Direct imaging of changes in aerosol particle viscosity upon hydration and chemical aging. Chem. Sci. 2016, 7, 1357–1367. [Google Scholar] [CrossRef]
- Booth, A.M.; Murphy, B.; Riipinen, I.; Percival, C.J.; Topping, D.O. Connecting bulk viscosity measurements to kinetic limitations on attaining equilibrium for a model aerosol composition. Environ. Sci. Technol. 2014, 48, 9298–9305. [Google Scholar] [CrossRef] [PubMed]
- Finlayson-Pitts, B.J. Reactions at surfaces in the atmosphere: Integration of experiments and theory as necessary (but not necessarily sufficient) for predicting the physical chemistry of aerosols. Phys. Chem. Chem. Phys. 2009, 11, 7760–7779. [Google Scholar] [CrossRef] [PubMed]
- Shiraiwa, M.; Yee, L.D.; Schilling, K.A.; Loza, C.L.; Craven, J.S.; Zuend, A.; Ziemann, P.J.; Seinfeld, J.H. Size distribution dynamics reveal particle-phase chemistry in organic aerosol formation. Proc. Natl. Acad. Sci. USA 2013, 110, 11746–11750. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Fisseha, R.; Putman, A.L.; Rahn, T.A.; Mazzoleni, L.R. High molecular weight soa formation during limonene ozonolysis: Insights from ultrahigh-resolution ft-icr mass spectrometry characterization. Atmos. Chem. Phys. 2012, 12, 5523–5536. [Google Scholar] [CrossRef]
- Bateman, A.P.; Nizkorodov, S.A.; Laskin, J.; Laskin, A. Time-resolved molecular characterization of limonene/ozone aerosol using high-resolution electrospray ionization mass spectrometry. Phys. Chem. Chem. Phys. 2009, 11, 7931–7942. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.; Imre, D.; Beranek, J.; Shrivastava, M.; Zelenyuk, A. Evaporation kinetics of laboratory-generated secondary organic aerosols at elevated relative humidity. Environ. Sci. Technol. 2015, 49, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Maksymiuk, C.S.; Gayahtri, C.; Gil, R.R.; Donahue, N.M. Secondary organic aerosol formation from multiphase oxidation of limonene by ozone: Mechanistic constraints via two-dimensional heteronuclear nmr spectroscopy. Phys. Chem. Chem. Phys. 2009, 11, 7810–7818. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Paulson, S.E. Real refractive indices and volatility of secondary organic aerosol generated from photooxidation and ozonolysis of limonene, alpha-pinene and toluene. Atmos. Chem. Phys. 2013, 13, 7711–7723. [Google Scholar] [CrossRef]
- Hamilton, J.F.; Lewis, A.C.; Carey, T.J.; Wenger, J.C.; Garcia, E.B.I.; Munoz, A. Reactive oxidation products promote secondary organic aerosol formation from green leaf volatiles. Atmos. Chem. Phys. 2009, 9, 3815–3823. [Google Scholar] [CrossRef]
- Kleist, E.; Mentel, T.F.; Andres, S.; Bohne, A.; Folkers, A.; Kiendler-Scharr, A.; Rudich, Y.; Springer, M.; Tillmann, R.; Wildt, J. Irreversible impacts of heat on the emissions of monoterpenes, sesquiterpenes, phenolic bvoc and green leaf volatiles from several tree species. Biogeosciences 2012, 9, 5111–5123. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Zahardis, J.; Petrucci, G.A. Soft ionization chemical analysis of secondary organic aerosol from green leaf volatiles emitted by turf grass. Environ. Sci. Technol. 2014, 48, 4835–4843. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.F.; Lewis, A.C.; Carey, T.J.; Wenger, J.C. Characterization of polar compounds and oligomers in secondary organic aerosol using liquid chromatography coupled to mass spectrometry. Anal. Chem. 2008, 80, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Criegee, R. Mechanism of ozonolysis. Angew. Chem. Int. Ed. 1975, 14, 745–752. [Google Scholar] [CrossRef]
- Grosjean, E.; Grosjean, D. The gas-phase reaction of alkenes with ozone: Formation yields of carbonyls from biradicals in ozone-alkene-cyclohexane experiments. Atmos. Environ. 1998, 32, 3393–3402. [Google Scholar] [CrossRef]
- Horie, O.; Moortgat, G.K. Gas-phase ozonolysis of alkenes. Recent advances in mechanistic investigations. Acc. Chem. Res. 1998, 31, 387–396. [Google Scholar] [CrossRef]
- Johnson, D.; Marston, G. The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere. Chem. Soc. Rev. 2008, 37, 699–716. [Google Scholar] [CrossRef] [PubMed]
- Vereecken, L.; Francisco, J.S. Theoretical studies of atmospheric reaction mechanisms in the troposphere. Chem. Soc. Rev. 2012, 41, 6259–6293. [Google Scholar] [CrossRef] [PubMed]
- Bones, D.L.; Reid, J.P.; Lienhard, D.M.; Krieger, U.K. Comparing the mechanism of water condensation and evaporation in glassy aerosol. Proc. Natl. Acad. Sci. USA 2012, 109, 11613–11618. [Google Scholar] [CrossRef] [PubMed]
- Hodas, N.; Zuend, A.; Mui, W.; Flagan, R.C.; Seinfeld, J.H. Influence of particle-phase state on the hygroscopic behavior of mixed organic-inorganic aerosols. Atmos. Chem. Phys. 2015, 15, 5027–5045. [Google Scholar] [CrossRef]
- Shiraiwa, M.; Seinfeld, J.H. Equilibration timescale of atmospheric secondary organic aerosol partitioning. Geophys. Res. Lett. 2012, 39, L24801. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parent VOC | Code | VOC (µL) | VOC (ppbv) | Ozone (ppbv) | RH (%) | CSOA Max (µg m−3) |
|---|---|---|---|---|---|---|
 | AP1 | 1 | 20 | 25 | 23 | 0.25 |
| AP2 | 1 | 20 | 58 | 22 | 2 | |
| AP3 | 3 | 60 | 58 | 21 | 4.5 | |
| AP4 | 5 | 100 | 89 | 21 | 8 | |
| AP5 | 3 | 60 | 57 | 22 | 10 | |
| AP6 | 5 | 100 | 58 | 23 | 28 | |
| AP7 | 10 | 200 | 77 | 21 | 36 | |
| AP8 | 10 | 200 | 200 | 21 | 70 | |
| AP9 | 10 | 200 | 550 | 23 | 82 | |
 | L1 | 1 | 20 | 12 | 23 | 5 |
| L2 | 2.5 | 50 | 13 | 23 | 10 | |
| L3 | 5 | 100 | 13 | 23 | 20 | |
| L4 | 2.5 | 50 | 47 | 25 | 25 | |
| L5 | 2.5 | 50 | 160 | 24 | 65 | |
| L6 | 5 | 100 | 235 | 23 | 163 | |
 | CHA1 | 40 | 750 | 280 | 20 | 1.6 |
| CHA2 | 50 | 1000 | 280 | 22 | 3 | |
| CHA3 | 40 | 750 | 420 | 21 | 10.5 | |
| CHA4 | 50 | 1000 | 990 | 22 | 25 | |
| CHA5 | 40 | 750 | 620 | 21 | 41 | |
 | HXL1 | 13 | 350 | 195 | 22 | 3 |
| HXL2 | 25 | 670 | 200 | 20 | 12 | |
| HXL3 | 13 | 350 | 350 | 21 | 15 | |
| HXL4 | 25 | 670 | 350 | 22 | 37 | |
| HXL5 | 25 | 670 | 600 | 19 | 82 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, S.; Fischer, K.B.; Petrucci, G.A. The Influence of Absolute Mass Loading of Secondary Organic Aerosols on Their Phase State. Atmosphere 2018, 9, 131. https://doi.org/10.3390/atmos9040131
Jain S, Fischer KB, Petrucci GA. The Influence of Absolute Mass Loading of Secondary Organic Aerosols on Their Phase State. Atmosphere. 2018; 9(4):131. https://doi.org/10.3390/atmos9040131
Chicago/Turabian StyleJain, Shashank, Kevin B. Fischer, and Giuseppe A. Petrucci. 2018. "The Influence of Absolute Mass Loading of Secondary Organic Aerosols on Their Phase State" Atmosphere 9, no. 4: 131. https://doi.org/10.3390/atmos9040131
APA StyleJain, S., Fischer, K. B., & Petrucci, G. A. (2018). The Influence of Absolute Mass Loading of Secondary Organic Aerosols on Their Phase State. Atmosphere, 9(4), 131. https://doi.org/10.3390/atmos9040131