4.1. Hydrochemical Pattern Resulting from Groundwater Flow Systems
Based on the available chemical data, the hydrochemical characteristics of HTCH lake water and groundwater were discussed.
The chemical data for lake water show that TDS, a comprehensive indicator of hydrochemistry, is relatively high and ranges from 4464.5 mg/L to 5165.7 mg/L with the mean value of 4719.2 mg/L. The pH values lie between 9.97 and 10.03 with an average of 10.00, indicating an alkaline nature. The water type is Na–Cl(HCO
3), as shown in Piper plot (
Figure 3). Na
+ and Cl
− are the dominant elements: Na
+ accounts for, on average, 88.6% of total cations, and Cl
− accounts for, on average, 51.3% of total anions. This is in accordance with hydrochemical characteristics of inland salt lake in other regions [
16].
The TDS of groundwater range between 171.8 and 724.0 mg/L with mean and median values of 300.7 mg/L and 234.9 mg/L, respectively. Groundwater is slightly alkaline and has pH values varying from 7.69 and 9.39. The measured Eh values of groundwater range from −137.50 to −55.60 mV with the mean value of −90.34 mV. Ca2+, and the Na+ concentrations in groundwater vary in a wide range from 2.6 to 64.3 mg/L and 14.9 to 225.3 mg/L, respectively. The Cl− and NO3− concentrations also exhibit a wide range from 3.5 to 60.9 mg/L and from ND (not detected) to 67.8 mg/L, respectively.
The Piper plot is widely used to graphically display the bulk major ion compositions of groundwater, and it is, therefore, used to classify groundwater [
31].
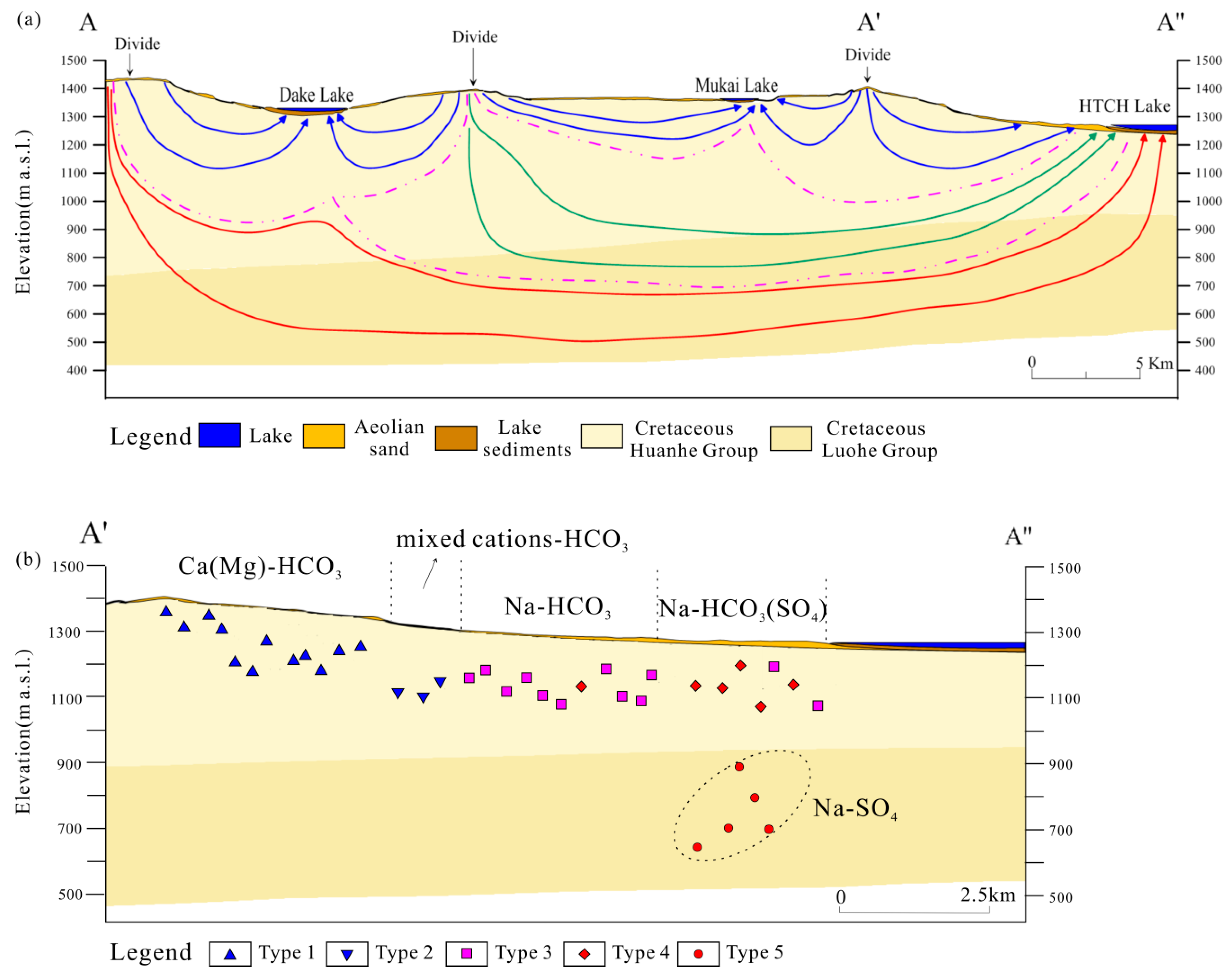
Figure 3 shows that they are found to be chemically classified into five distinct types: Ca(Mg)–HCO
3 (type 1), mixed cations-HCO
3 (type 2), Na–HCO
3 (type 3), Na–HCO
3(SO
4) (type 4) and Na–SO
4 (type 5). In this classification scheme, type 1 through type 5 represent a natural chemical evolutionary sequence (
Figure 3). The TDS and major ion concentrations for the 5 water types are also show illustrated in the Box plots of
Figure 4. The calculated average values of the geochemical data for the 5 types of groundwater, including TDS, temperature, pH, Eh, major ions and stable isotopes (δD, δ
18O and
87Sr/
86Sr) are summarized in
Table S3. The hydrochemical characteristics of each type of groundwater are discussed in detail below.
Type 1 waters have a Ca(Mg)–HCO
3 composition and average TDS of 234 mg/L (
Table S3). These waters contain more than 70% (in meq/L) Ca
2+ + Mg
2+ of the total cations content. HCO
3− is the major anion component, which accounts for more than 60% of total anions contents. This type predominantly occurs in the topographic highs of the basin, as shown in
Figure 2b. Type 2 waters are mixed cations-HCO
3, and no dominant cation exists, belonging to the transitional type. The anion relationship is rHCO
3− > rSO
42− > rCl
−. Type 3 is Na–HCO
3 water. Na
+ is the major cation, which constitutes more than 54% of the total cations; HCO
3−, the major anion, accounts for more than 67% of the total anions. These waters are low in SO
42− content that makes up less than 23% of the total anions. Type 4 is Na–HCO
3(SO
4) water with the mean TDS of 305.2 mg/L. Na
+ is the predominant cation, and it accounts for greater than 77% of the total cation contents; HCO
3− is the predominant anion, and it occurs in amounts greater than 48% of the total anion contents. The SO
42− constitutes more than 23% of the total anions, which is considerably higher relative to type 3 waters. These three types, including type 2, 3 and 4, are predominantly located in the topographic lows, as shown in
Figure 2b. Type 5 is Na–SO
4 water, with the mean TDS value of 692.8 mg/L. Na
+ contents range from 213.8 to 258.0 mg/L with the average value of 225.03 mg/L and constitute, on average, about 91% of total cations; SO
42− concentrations vary between 322.2 and 608.7 mg/L with the mean of 387.11 mg/L, which accounts for, on average, 68% of total anions. The concentrations of TDS, Na
+ and SO
42− in type 5 are significantly higher than types 1, 2, 3 and 4, as illustrated in
Figure 4. In summary, type 1 is characterized by and dominated by Ca
2+, while types 3, 4 and 5 are characterized by Na
+ among cations; types 1, 2, 3 and 4 are characterized by a predominance of HCO
3−, whereas type 5 is characterized by SO
42− among anions (
Figure 3). In addition, it is should be noted that the mean Cl
− concentrations are much higher in type 1 (16.4 mg/L) than type 2 (9.0 mg/L) and type 3 (8.2 mg/L), as shown in
Table S3 and
Figure 4e, and is not consistent with the general pattern that Cl
− contents increase along the flow line. This is attributed to the fact that some samples in type 1 waters have been strongly affected by anthropogenic contamination, such as agricultural fertilizer, as indicated by higher mean NO
3 contents in type 1 (range: 13.6–67.8 mg/L; mean: 36.1 mg/L) than type 2 (range: 3.1–31.1 mg/L; mean: 18.5 mg/L) and type 3 (range: 13.6–180.1 mg/L; mean: 2.4 mg/L) (
Table S3 and
Figure 5). A similar contamination phenomenon caused by human activities is also observed in the regions close to the study area in the Ordos Plateau [
20,
32].
Broad relationships between the hydrochemical pattern and groundwater flow systems have been demonstrated by various workers. According to theory and observations, the groundwater flow from the recharge to discharge zones may be attended generally by an increase in the TDS and a systematic variation in the dominant cations and anions [
33,
34,
35]. On the other hand, in a complex basin with the development of three types of groundwater flow systems (i.e., local, intermediate and regional), as described by Toth [
36], groundwater from local flow systems may exhibit significantly distinct hydrochemical characteristics from those from intermediate/regional flow systems. This is because of the significant difference in the length of the flow paths and, hence, the contact time between groundwater and rock, especially in the lower reaches of a basin where a regional flow system lies below a local system [
37,
38]. Generally, systems may be characterized as local by low-TDS bicarbonate waters, as intermediate by intermediate-TDS-sulfate waters and as regional by high-TDS chloride-rich waters.
When we correlate the water types with the geographic locations, types 1, 2, 3 and 4 characterized by a predominance of HCO
3− were found to be located in the shallow aquifer (<200 m), whereas all samples of type 5 dominated by SO
42− occurred in the deep parts (around 350–600 m), as shown in
Figure 2b and
Figure 5. Such a vertical pattern of groundwater chemistry is in accordance with the propositions of Freeze [
37] and Toth [
38]. In addition, groundwater is of a meteoric origin, and its chemical composition is predominantly controlled by a water–rock interaction within the aquifer in the study area that is relatively simple and macroscopically homogeneous sandstone (see
Section 4.3). Therefore, the significant geochemical differences between shallow and deep groundwater seem to support the hypothesis of nested flow systems with different flow distances and flow times (
Figure 2). That is, the groundwater chemistry suggests that shallow groundwater (types 1, 2, 3 and 4) belongs to a local flow system with a relatively short travel time and distance, while deep groundwater (type 5) belongs to a regional flow system with a longer travel time and distance. The TDS and Cl
− content, traditional indicators of groundwater residence time, are significantly higher in deep groundwater compared to shallow groundwater, as shown in
Figure 5a,g, which also support this interpretation.
4.2. Groundwater Origin
The environmental isotopes of deuterium (
2H) and oxygen-18 (
18O) are excellent tracers for determining the groundwater origin [
39]. A local meteoric water line (LMWL: δ
2H = 6.45δ
18O − 6.51) for the Ordos Plateau basin defined by Yin et al. [
23] provides the basis for the interpretation of δ
2H and δ
18O values for groundwater in this study. The lower slope of LMWL relative to the global meteoric water line (GMWL) (δ
2H = 8δ
18O − 10) is attributed to the secondary evaporation process during rainfall as a result of an arid environment.
The δ
2H and δ
18O values in shallow groundwater exhibit a relatively wide range from −86.5‰ to −57.5‰ and from −11.3‰ to −7.4‰, respectively, which is within the general range previously observed for shallow groundwater in the Ordos Plateau [
23,
40]. The plot of δ
2H versus δ
18O (
Figure 6) shows that all groundwater samples lie close to the LMWL and GMWL, indicating that they are of local meteoric origin. However, they can be divided into two groups. One group (group 1) is the type 1 groundwater located in the recharge area of local flow system; the average stable isotopic composition (−63.3‰ for δ
2H and −8.3‰ for δ
18O) in these groundwater corresponds to that of modern rainfall in the Ordos Plateau with a weighted mean of −58.5‰ for δ
2H and −8.1‰ for δ
18O (
Figure 7) [
23]. This suggests that they are recharged primarily by modern precipitation or precipitation in the late Holocene period. The other group (group 2) includes the other four types of groundwater (types 2, 3, 4 and 5). They are depleted in heavy isotopes (mean −80.9‰ for δ
2H and −10.4‰ for δ
18O) in comparison with type 1 groundwater and present meteoric water (
Figure 7), suggesting that they are primarily recharged during the late Pleistocene and early Holocene periods when climate was relatively colder and wetter than the present, after having considered that the altitude effect was not significant in the whole Ordos Plateau [
23,
40]. In addition, in group 2, type 5 waters are found to have isotopic compositions very similar to type 4 and eight samples in type 3 because they are both old waters, while type 2 waters and four samples in type 3 display the isotopic compositions within those of type 1 and those 8 samples in type 3 (type 4, 5), which may be caused by a mixing between type 1 and type 3 based on the chemical characteristics and hydrogeological setting.
4.3. Mechanisms Controlling Groundwater Chemical Evolution
This section aims to identify the dominant mechanisms responsible for the chemical compositions of groundwater of various types. Understanding geochemical processes in groundwater is a prerequisite for the correction of 14C ages and for recording groundwater recharge and flow. Processes controlling the hydrochemical evolution were determined using hydrochemistry, stable isotope and inverse geochemical modeling.
4.3.1. Constraints from Water Chemistry
Generally, the chemical compositions of groundwater are primarily controlled by various factors, including the chemical composition of recharge water, water–rock interaction, dissolution/precipitation of mineral phases, evaporation, mixing processes and human activities [
1,
2]. The Gibbs diagram [
41], which exhibits the ratios of Na
2+/(Na
2+ + Ca
2+) and Cl
−/(Cl
− + HCO
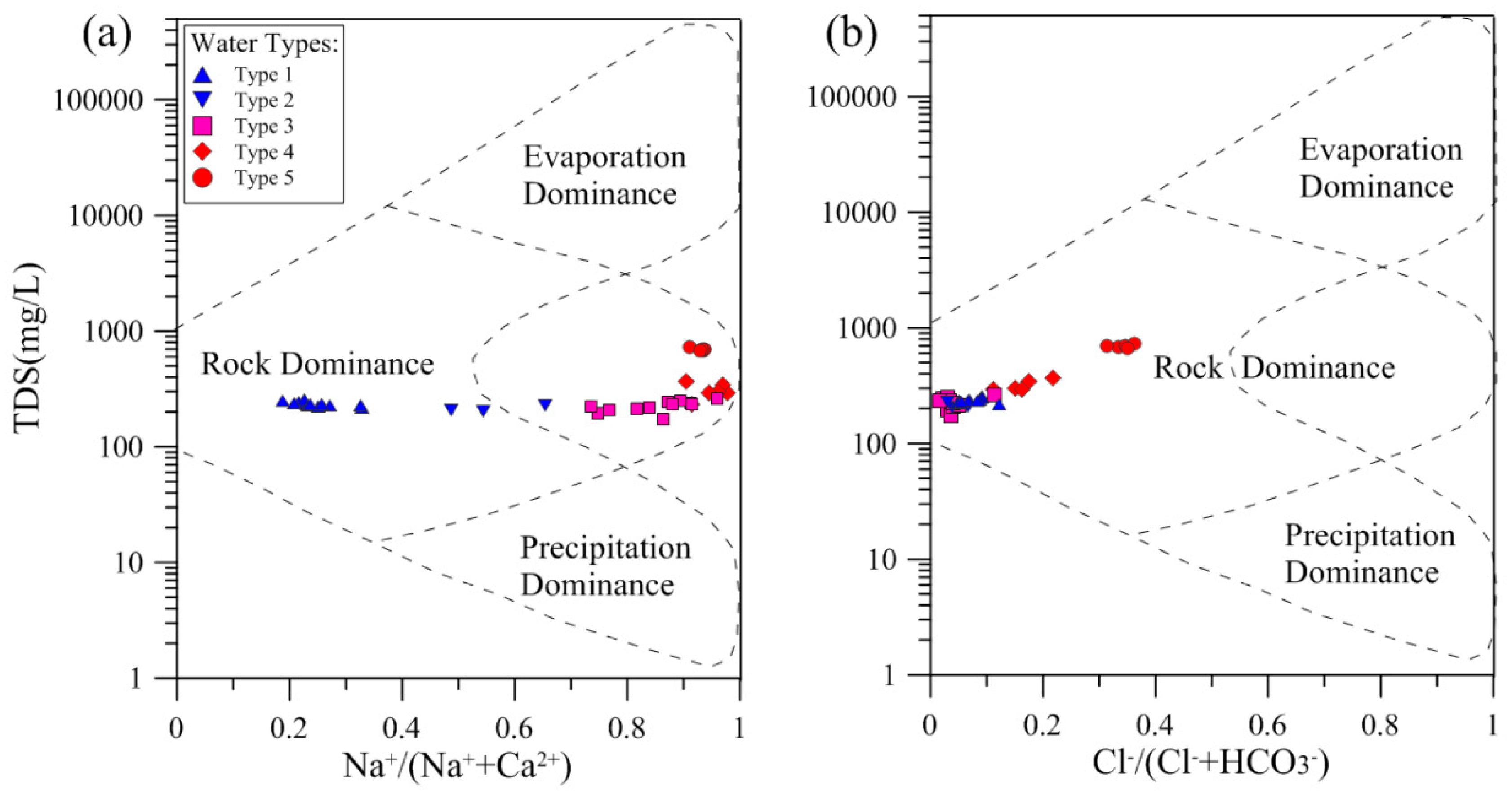
3−) as a function of TDS, is an effective tool to assess the natural mechanisms controlling water chemistry, including the atmospheric precipitation, the water–rock interaction and the evaporation-crystallization process.
Figure 8 shows that all groundwater samples fall into the zone of rock dominance, suggesting that water–rock interactions are the major mechanism controlling groundwater chemistry. The Cretaceous sandstones consist mainly of quartz; K-feldspar; plagioclase; calcite; and minor amounts of dolomite, ankerite, siderite, pyrite, hematite, analcite, Glauber’s salt and clay minerals, as presented in the
Table S4. Therefore, potential water–rock interactions include the dissolution/weathering of these minerals forming aquifer and cation exchange occurring on the clay minerals. In addition, the dissolution of halite and gypsum can also take place in the aquifer, although they are not found in the study area. This is due to the fact that they have been observed in the Cretaceous aquifer in the Ordos Plateau [
15].
In order to further reveal the origin of solutes in groundwater, the relationship between the dissolved chemical constituents is employed [
42]. The relationship between Na
+ and Cl
− is commonly used as a first order indicator of water–rock interactions [
43]. The plot of Na
+ versus Cl
− (
Figure 9a) shows that only three samples in type 1 groundwater samples (about 25% of type 1 waters) lie close to the 1:1 relation line expected from the dissolution of halite, indicating the occurrence of halite dissolution in these waters, while other samples cluster above the 1:1 line and molar Na/Cl ratios vary between 1.5 and 30.2 with a mean value of 8.7. This indicates the existence of additional sources contributing to the dissolved Na
+, which potentially include the weathering of Na-bearing silicates, a dissolution of Na-bearing evaporites and cation exchange between Ca
2+/Mg
2+ in groundwater and Na
+ on clay minerals after taking into consideration the mineralogical compositions of aquifer studied [
44,
45,
46].
The plot of (Ca
2+ + Mg
2+) versus (HCO
3– + SO
42–) (
Figure 9b) indicates that type 1 groundwater is distributed around the 1:1 relation line, suggesting that Ca, Mg and HCO
3 in these waters are derived mainly from simple dissolution of calcite, dolomite and gypsum. The mean SI values for calcite and dolomite are 0.71 and 0.93, respectively, indicative of a slight oversaturation of groundwater with carbonates minerals and the importance of carbonates dissolution in these waters. These reactions commonly occur in the recharge areas in many sedimentary aquifer environments, and the reaction equations are as follows [
2]:
In contrast, type 2, 3, 4 and 5 waters exhibit a significant deviation from the 1:1 relation line and plot above it, indicating the simple dissolution of carbonate and gypsum alone could not interpret their chemical compositions. This may be caused by the cation exchange, silicates weathering or evaporites dissolution [
47].
Figure 9c shows that majority of these waters plot above or below the theoretical line of weathering of plagioclase (y = x), suggesting silicate weathering is not the major source of Na
+. As shown in
Figure 9d, type 2, 3, 4 and 5 waters have a much higher Na/Ca ratio relative to type 1 water and TDS slightly increases as groundwater evolves from type 1 to types 2, 3 and 4. This seems to imply that a cation exchange has taken place as groundwater flows in the aquifer. The plot of (Na
+ − Cl
−) against (Ca
2+ + Mg
2+) could be employed to evaluate the cation exchange [
46,
48]. If the relationship between these two parameters is linear with a slope of −1.0, suggesting a cation exchange is significant in controlling geochemical compositions. As shown in
Figure 9e, types 1, 2, 3 and 4 are located near the 1:1 relation line; in contrast, type 5 groundwater lie away from the 1:1 line. This mirror image of mono- (Na
+) and bivalent cations (Ca
2+, Mg
2+), along with the almost constant HCO
3– contents along the groundwater flow path (
Figure 5e), indicate that a cation exchange controls the hydrochemical evolution of shallow groundwater (types 1, 2, 3 and 4) along the flow path; the reaction equations are as follows:
The use of two Chloro-Alkaline indices (CAI-1 and CAI-2) proposed by Schoeller is helpful for indicating the cation exchange process [
49]. CAI-1 and CAI-2 are defined as the ratio of (Cl − (Na + K)) to Cl and (HCO
3 + SO
4 + CO
3 + NO
3), respectively. The positive indices suggest that the ion exchange occurs between Na
+ in the groundwater and Ca
2+/Mg
2+ in the aquifer sediments, while the negative values suggest that the reverse ion exchange takes place. As shown in
Figure 9f, both of the indices (CAI-1 and CAI-2) are found to be below zero in the type 2, 3 and 4 groundwater samples, implying that a high concentration of Na
+ and a low concentration of Ca
2+ are attributed to the ion-exchange process in these waters.
Figure 9g shows that deep type 5 water displays a significant deviation from the 1:1 line expected from gypsum dissolution and plots above the 1:1 line, indicating that gypsum is not the major process in this type of water and that SO
42− has additional sources. The SI values for gypsum in these waters range from −1.85 to −1.67 with an average of −1.79, furthering supporting this interpretation. However, these waters have a relatively higher average SI value for gypsum than type 1 (−2.33), type 2 (−2.46), type 3 (−2.84) and type 4 (−2.84), as shown in
Table S3, suggesting a stronger degree of gypsum dissolution in this type than in the other four types.
Figure 9h shows that type 5 groundwater plots relatively close to the 1:1 relation line, suggesting Na
+, SO
42− and dominant ions in type 5 primarily derive from the dissolution of evaporite including halite and Glauber’s salt (Na
2SO
4). The reaction equation of Glauber’s salt dissolution is as follows:
Another significant characteristic of the groundwater is the extremely low K
+ content between 0.6 and 4.4 mg/L (mean 2.3) and the very low K/Na molar ratios of near 0. The low levels of potassium may be attributed to its tendency to participate in the formation of secondary minerals and the lower reactivity of K-feldspars compared to plagioclase [
8,
43].
The dissolved silica concentrations (range: 1.66–11.90 mg/L; mean: 7.14 mg/L) are generally lower than the typical case where silicate weathering is a major process in groundwater (approx. 20 to >100 mg/L) [
9], suggesting that the silicate weathering is a minor source of Na
+ in groundwater.
4.3.2. Constraints from Strontium Isotope
The Sr isotopes are extensively applied as tracers to effectively delineated mineral weathering reactions in subsurface hydrology [
50,
51,
52]. This is attributed to the following properties. The strontium can be removed from the water through mineral precipitation or cation exchange reaction, but the Sr isotopic composition of water remains largely unaffected because its mass-dependent fraction is usually negligible in nature. Moreover, Sr
2+ is geochemically similar to Ca
2+ and usually resides in carbonates, gypsums and K-bearing or Ca-bearing silicates [
51,
53]. These common rock-forming minerals generally exhibit a wide and predictable range of
87Sr/
86Sr ratios that are controlled by initial values, Rb/Sr ratios and the age of minerals. Silicate minerals, particularly potassic silicates (e.g., biotite and K-feldspar), that exceed a few million years old have more radiogenic
87Sr/
86Sr ratios due to the addition of radiogenic
87Sr through the decay of
87Rb (t
1/2 = 4.8 × 10
10 years). In contrast, Ca-rich minerals such as calcite or gypsum display relatively less radiogenic
87Sr/
86Sr ratios and remain relatively constant over time.
Groundwater primarily originates from the local precipitation in the study area, as indicated by δD and δ
18O. Therefore, the strontium geochemistry of groundwater is initially controlled by the precipitation. The Sr concentrations in precipitation samples collected from the Otak meteorological station (
Figure 1b), nearest to the study area, range from 10 to 250 μg/L with the volume-weighted mean (VWM) value of 50 μg/L [
54]. The Sr concentrations in groundwater vary in a wide range from 377 to 1000 μg/L, with an average value of around 630 μg/L. The much higher Sr
2+ concentration in groundwater compared to those observed for local precipitation demonstrates that Sr
2+ in groundwater have other potential sources besides precipitation, which may include the dissolution of carbonate and silicate minerals that are major Sr-bearing minerals in the aquifer matrix.
The Sr sources might control the
87Sr/
86Sr ratios and can be well constrained by the relationship between the Sr isotopic compositions and concentration ratios for groundwater [
55].
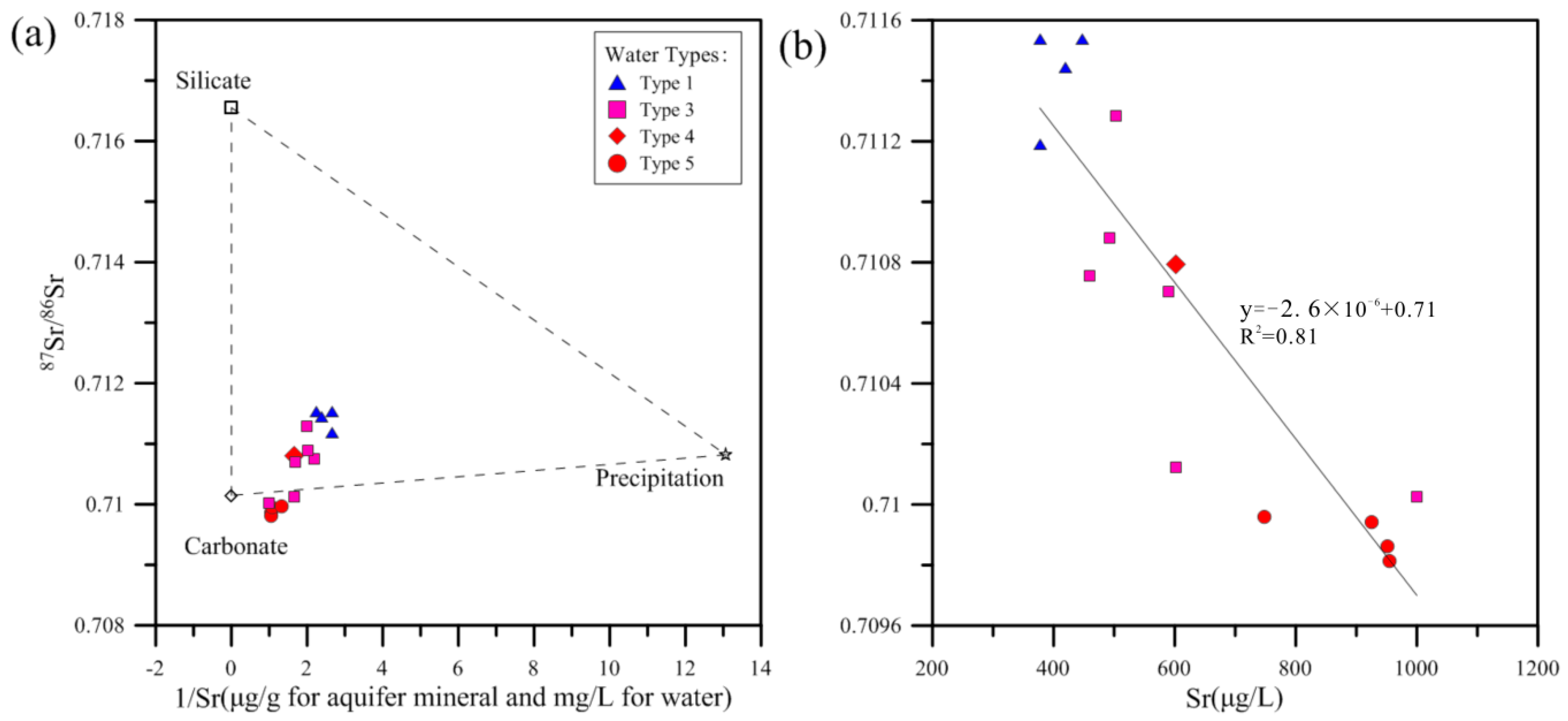
Figure 10a shows their relationship in the plot of
87Sr/
86Sr ratios versus 1/Sr. The geochemical compositions of three potential end-members are also shown: (a) carbonate fraction of aquifer rocks, represented by an
87Sr/
86Sr ratio and Sr concentrations of 0.710148 and 309 μg/g, respectively [
56]; (b) silicate fractions of aquifer rocks, characterized by a significantly more radiogenic
87Sr/
86Sr ratio (0.716552) and a relatively lower Sr content (284 μg/g) relative to carbonate minerals [
57]; and (c) local precipitation, characterized by an
87Sr/
86Sr ratio and Sr concentration of 0.710760 and 309 μg/g, respectively [
54].
Figure 10a shows almost all the groundwater samples points are located within the triangle consisting of local precipitation, carbonate and silicate fractions, and the plot is obviously closer to the carbonate fraction end-member than the other two end-members, which indicates the carbonate dissolution exerts the significantly more important control on the Sr
2+ in groundwater than the other two sources.
Figure 10b shows that
87Sr/
86Sr ratios tend to gradually decrease with increasing Sr
2+ concentrations in groundwater, furthering demonstrating that the major source of the dissolved Sr
2+ is not the weathering of silicates with more radiogenic
87Sr/
86Sr ratios. This is due to the fact that
87Sr/
86Sr ratios will increase with increasing Sr
2+ concentrations if silicate weathering is an important process controlling the groundwater hydrochemical evolution [
9].
Figure 10b displays that types 3, 4 and 5 have relatively lower
87Sr/
86Sr ratios compared to type 1, which may result from the gypsum dissolution as groundwater moves from recharge areas within the aquifer. This is supported by the fact that the mean SO
42− concentrations are higher for type 3 (28.6 mg/L), type 4 groundwater (67.1 mg/L) and type 5 (342.8 mg/L) than type 1 groundwater (18.9 mg/L) as shown in
Table S3.
As the silicate weathering is not significant within the aquifer, excess Na+ relative to the Cl− concentration in type 3 and 4 groundwater is believed to be the primary result of the cation exchange process and the dissolution of Glauber’s salt. Types 3 and 4 are characterized by a predominance of Na+ and HCO3−; therefore, cation exchange is the major source of excess Na in these waters. That is, Sr geochemical signatures further confirm the conclusion that a cation exchange significantly contributes to dissolved Na+ in type 3 and type 4 groundwater, deduced from the groundwater chemistry discussed above.
4.3.3. Constraints from Sulfur Isotope
Groundwater sulfate can be derived from various sources, which include the accession of inorganic SO
4 via the atmospheric deposition of marine SO
4 aerosols, the dissolution of SO
4 minerals such as gypsum, the oxidation of reduced sulfide minerals such as pyrite or the mineralization of organic S compounds within the soil zone [
58,
59]. The sulfur isotopes values for SO
4 (δ
34S
SO4) of these sources are markedly different. The δ
34S values of sedimentary reduced sulfides (e.g., pyrite) typically range from approximately −50 to +10‰, while the evaporite minerals tend to be relatively enriched in δ
34S values between around 10 and 20‰ [
13]. Therefore, the sources of SO
42− in groundwater can be better determined by sulfur isotope values of SO
42− [
60,
61].
In this study, an isotopic (δ
34S
SO4) analysis was carried out for type 5 groundwater (Na–SO
4) with relatively higher SO
42− concentrations in order to identify the SO
42− source in groundwater. Groundwater primarily originates from the local precipitation in the study area. Hence, the sulfur geochemistry of groundwater is initially controlled by the precipitation. Precipitation has SO
42− concentrations between 9.56 and 28.67 mg/L, and the mean δ
34S value for SO
4 in precipitation is around 3.7‰ in the Ordos Plateau [
62]. The SO
42− concentrations range from 322.2 to 367.4 mg/L in type 5 groundwater, which is significantly higher compared to local precipitation. This indicates the presence of additional SO
42− sources in groundwater except precipitation. The relatively higher δ
34S values of these waters (12.4‰ to 14.1‰, mean 13.0‰) than those of precipitation also support this conclusion. These waters have δ
34S values within the typical range of evaporite minerals, indicating that SO
42− in type 5 is primarily from the dissolution of evaporite such as gypsum and Glauber’s salt in the aquifer. This interpretation is consistent with the hydrochemical data discussed above.
4.3.4. Constraints from Inverse Geochemical Modeling
An inverse geochemical modeling was performed in order to further constrain and quantify the main natural geochemical processes identified above: the dissolution of calcite, dolomite, anhydrite, halite and Glauber’s salt; the precipitation of calcite and dolomite; and a cation exchange with clays. The weathering of silicate minerals is thought to be unimportant for the chemical evolution and, hence, is not considered in the models.
During the inverse modeling, at least two chemical analyses of groundwater at different points of the flow path and a set of plausible phases (mineral and/or gases) that potentially react along this path are required. In the models, the average chemical parameter values from the statistical analyses of each type of groundwater, as presented in
Table S3, were used to represent the “initial” and “final” waters along the groundwater flow paths. A total of five models (models 1, 2, 3, 4 and 5) have been run for an interpretation of the chemical compositions of the five types of groundwater present in the study area. The five simulated paths are represented by the transition between the following end-members: (1) pure water to type 1 water; (2) type 1 water to type 2 water; (3) type 2 to type 3 water; (4) type 3 to type 4 water; and (5) type 4 to type 5 water. The phases used were calcite, dolomite, gypsum and halite in all the models based on the lithological and mineralogical information. CO
2 gas was only included in Model 1 from the precipitation to type 1 water to simulate carbonate dissolution under the action of CO
2 because only this system is supposed to be open for CO
2 in the recharge area. CaX, MgX and NaX phases were only included in Models 2, 3 and 4 to simulate the cation exchange; Glauber’s salt was only considered in Model 5 to simulate Glauber’s salt dissolution. The constraints used included Ca
2+, Mg
2+, Na
+, HCO
3−, SO
42− and Cl
− in all models.
Table S5 presents the selected results of the inverse modeling from all possible models based on the statistical parameters (the sum of residuals and maximum fractional error) calculated by PHREEQC, representing different possible combinations of reactants and products that can account for the changes in the groundwater chemistry. This result demonstrates that the amount of a given mineral phase that must dissolve or the precipitation to produce the observed variations in groundwater chemistry between two end points along the flow path.
Model 1 is the geochemical path from the local precipitation to a typical recharge water of type 1 (Ca·Mg–HCO3); the pure water is assumed to be representative of precipitation in this model. It is summarized as follows:
Precipitation (pure water) + CO2 gas + Calcite + Dolomite + Gypsum + Halite→Ca·Mg–HCO3 type water
Model 2 is the geochemical path from type 1 to type 2 (mixed cations-HCO3) and is summarized as follows:
Ca·Mg–HCO3 type water + Na from cation exchange + Gypsum→mixed cations-HCO3 type water + calcite precipitation (or Ca loss for ion exchange) + dolomite precipitation (or Mg loss for ion exchange)
Model 3 reproduces the geochemical path from type 2 groundwater to type 3 (Na–HCO3) and is summarized as follows:
Mixed cations-HCO3 type water + Na from cation exchange + Gypsum→Na–HCO3 type water + Ca loss for ion exchange + Mg loss for ion exchange
Model 4 reproduces the geochemical path from type 3 water to type 4 (Na–HCO3·SO4) and is summarized as follows:
Na–HCO3 type water + Na from cation exchange + Gypsum + Halite→Na–HCO3·SO4 type water + calcite precipitation (Ca loss for ion exchange) + dolomite precipitation (Mg loss for ion exchange)
Model 5 represents the geochemical path from type 4 water to type 5 (Na–SO4), and it is summarized as follows:
Na–HCO3·SO4 type water + Glauber’s salt + Gypsum + (Halite)→Na–SO4 type water + calcite dolomite precipitation
Summarizing, the geochemical modeling results support the hypothetical geochemical processes derived from the qualitative analyses for the groundwater chemical and isotopic compositions. From the modeling results, it can be confirmed that the dissolution of carbonate and evaporite minerals, as well as a cation exchange within the aquifer primarily control the chemical evolution of groundwater in the study area.
4.4. Groundwater Age
Groundwater age can be used to further constrain groundwater origin and flow systems. Radiocarbon (
14C) is the most widely applied dating method by far because of the almost ubiquitous presence of dissolved inorganic carbon (DIC) in groundwater [
13].
In spite of the widespread use of
14C, the interpretation of
14C model age still is limited by many uncertainties in determining the
14C content of dissolved carbon in the recharge areas and in accounting for various chemical and physical processes affecting the
14C content along groundwater flow paths in aquifers [
63,
64,
65,
66]. Using the measurements of
14C of the DIC in groundwater,
14C
DIC, the basic equation for groundwater dating by using
14C is as follows [
63]:
where t is the groundwater age; 5730 is the modern half-life of
14C;
14C
DIC is the measured
14C value of the DIC in groundwater sample; and
14C
0 is the initial
14C content of DIC after correction for geochemical reactions without radioactivity decay.
The estimation of
14C
0 is essential for groundwater
14C dating. Numerous single-sample-based traditional geochemical adjustment models have been proposed to calculate
14C
0 so far, such as Vogel, Tamer, Pearson, Fontes and Garnier models and so on [
67,
68,
69,
70]. These different models may give significantly distinct values of
14C
0. The incorrect use of the models for chemical and isotopic measurements in groundwater will lead to a wide range of estimations of
14C
0 and greatly limits the usefulness of radiocarbon as a dating tool for groundwater [
63]. Therefore, choosing the most appropriate models is of critical importance for improving the estimation of
14C age in a particular groundwater system, depending on which models most completely account for the geochemical processes affecting the chemical and isotopic compositions of the waters [
65].
The Han and Plummer diagram of δ
13C versus
14C [
63] can aid in determining which adjustment model is the most appropriate and should be applied for the specific groundwater system.
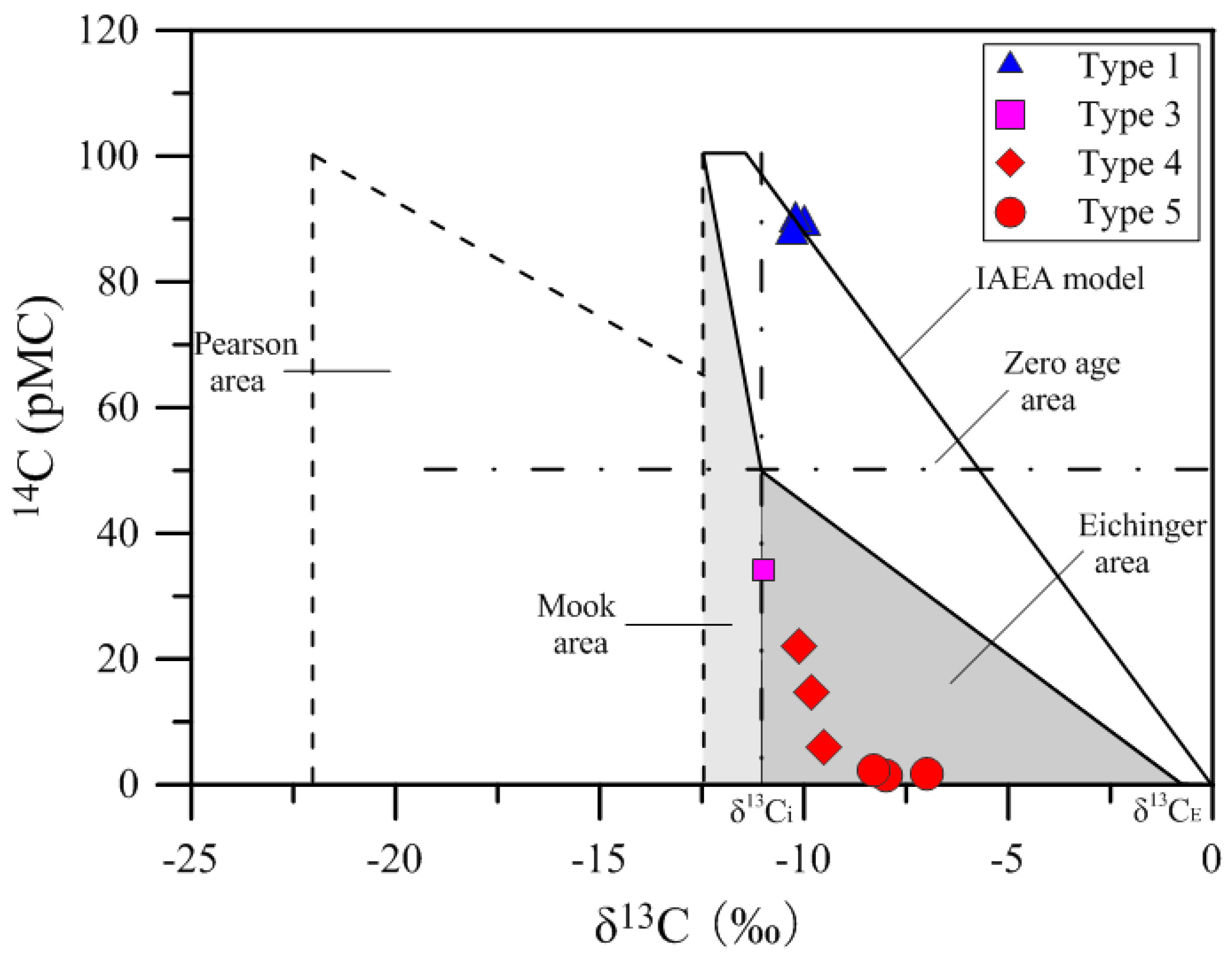
Figure 11 shows a plot of
14C versus δ
13C (Han and Plummer diagram) for the groundwater samples studied, indicating that all samples can be divided into two groups. One group is composed of type 1 waters; they plot close to the IAEA (International Atomic Energy Agency) line, suggesting they are young waters with
14C age close to zero. Another group includes type 4 and 5 waters, which have δ
13C
DIC ranging between δ
13C
i (approx. −11.05‰) and δ
13C
E (approx. −0.15‰) and are plotted in the Eichinger area. This indicates that the carbon isotopic compositions of DIC in these waters have been affected by a carbon exchange between DIC in groundwater and solid carbonate under closed system, and the Eichinger model [
71], accounting for a carbon exchange between solid carbonate and DIC under a closed system, should be applied to estimate
14C
0 for type 4 and 5 waters. In fact, the models of Wigley and Evans also take into consideration the carbon exchange between DIC and carbonate mineral, but they are less precise compared to the Eichinger model because they neglect the contribution of CO
2 (aq) to DIC (the models assume that DIC = HCO
3−). Therefore, Eichinger’s model considering the contribution of CO
2 (aq) to DIC is selected to obtain a relatively accurate age in this study.
According to Han and Plummer, the Han and Plummer’s model and IAEA model are also applicable for groundwater samples studied [
63]. Han and Plummer’s model is a revised version of the Fontes and Garnier model [
72], which considers not only carbon mixing but also carbon exchange occurring both in the saturated and unsaturated zone. This model is a combination of three models: Tamers’, Mook’s and Eichinger’s models. If the carbon exchange between DIC and solid carbonate occurs but without an exchange with soil CO
2, the models reduces to Eichinger’s model. The carbon exchange between DIC and solid carbonate predominantly under closed system conditions occurs in the groundwater system studied; therefore, the Han and Plummer model will give a similar result to Eichinger’s model. Unlike the Han and Plummer’s model, assuming either open or closed systems, the IAEA model assumes mixed open and closed system evolutions [
73]. That is, this model assumes that the isotopic composition of DIC evolved initially under completely open system condition and then evolved under closed system condition.
The measured and assumed parameters for estimating
14C
0 using various adjustment models are summarized in
Table S6. The measured parameters include temperature, pH, DIC, contents of dissolved CO
2 (aq), HCO
3− and CO
3−. The assumed parameters include
14C and δ
13C values of soil gas CO
2 and solid carbonate mineral. The
14C of soil CO
2 (
14C
g) and carbonate mineral (
14C
s) are assumed to be 100 pMC and 0 pMC, respectively. The δ
13C value for carbonate mineral (δ
13C
s) is assumed to be 0‰. The vegetation in the Ordos Plateau is predominantly C3 species, and the soil profiles in Yulin, approximately 125 km away from study area, show that δ
13C values range from −22‰ to −20‰ with an average of −21‰ [
74,
75]. Therefore, the δ
13C value for soli gas CO
2 (δ
13C
g) is assumed to be −21‰ in the study area. In addition, the calculated fraction factor for determination of
14C
0 is also presented in
Table S6. The ε
a/g and ε
b/g are
13C fractionate factors of dissolved CO
2 and HCO
3− with respect to gaseous CO
2, respectively. The ε
a/s and ε
b/s are the
13C fractionate factors of dissolved CO
2 and HCO
3− with respect to carbonate mineral.
The measured
14C
DIC values ranged from 1.4 to 89.6 pMC, with an average of 34.9 pMC (
Table S7). Based on above these parameters, the calculated
14C
0 and
14C age including the apparent and corrected ages are presented in
Table S7. The models of Eichinger and of Han and Plummer yield similar ages, which are relatively lower than those determined by IAEA model. The results derived from these three models are overall comparable, suggesting that
14C method provides the relatively reliable and reasonable ages to represent the mean groundwater residence time. The average values of three models were considered as the final ages. For shallow groundwater in the recharge areas (type 1), the corrected
14C age is approximately 0 year BP (Before Present); for those in discharge areas (type 3 and 4), the corrected
14C age ranges from 3929 to 16,996 years BP with the mean value of 9450 years, which is consistent with the
3H content that is below the detection limit of 1.3 TU (
Table S2). For deep groundwater (type 5), the corrected
14C age varies from 23,145 to 27,436 years BP with the average of 25,075 years, which is also in agreement with
3H content below the detection limit (
Table S2). Summarizing, the age data further demonstrates that shallow groundwater in the recharge areas were recharged during the Holocene period, while those in the discharge areas and deep groundwater were recharged during the late Pleistocene period. That is, groundwater age agrees well with the δD and δ
18O data discussed above. In addition, it is found that deep groundwater is about 2.7 times higher than shallow groundwater in the discharge areas. It is well-known that the groundwater age should increase along the groundwater flow direction. Therefore, such a vertical distribution of groundwater age further corroborates the hypothesis on the development of hierarchically nested groundwater flow systems. That is, groundwater age indicates that shallow and deep groundwater belong to local and regional flow system, respectively.
It is well-known that
14C ages are not identical to calendar age because atmospheric
14C concentration changes temporarily as a result of variation in the production rate, resulting from geomagnetic, solar modulation of the cosmic-ray flux and the carbon cycle [
63,
76]. In order to convert
14C ages into calendar ages relative to 1950 AD, many radiocarbon age calibration curves have been constructed using a variety of methods. In this study, the calibration curve IntCal13, representing the midlatitude Northern Hemisphere atmospheric reservoirs, was applied to estimate calendar age. Before it is applied, a conversion of conventional
14C ages calculated by using the modern
14C half-life (5730 years) to the Libby half-life (5568 years) is required because the calibration curves are obtained by first using the Libby half-life. The calibration curve IntCal13 and groundwater samples are shown in
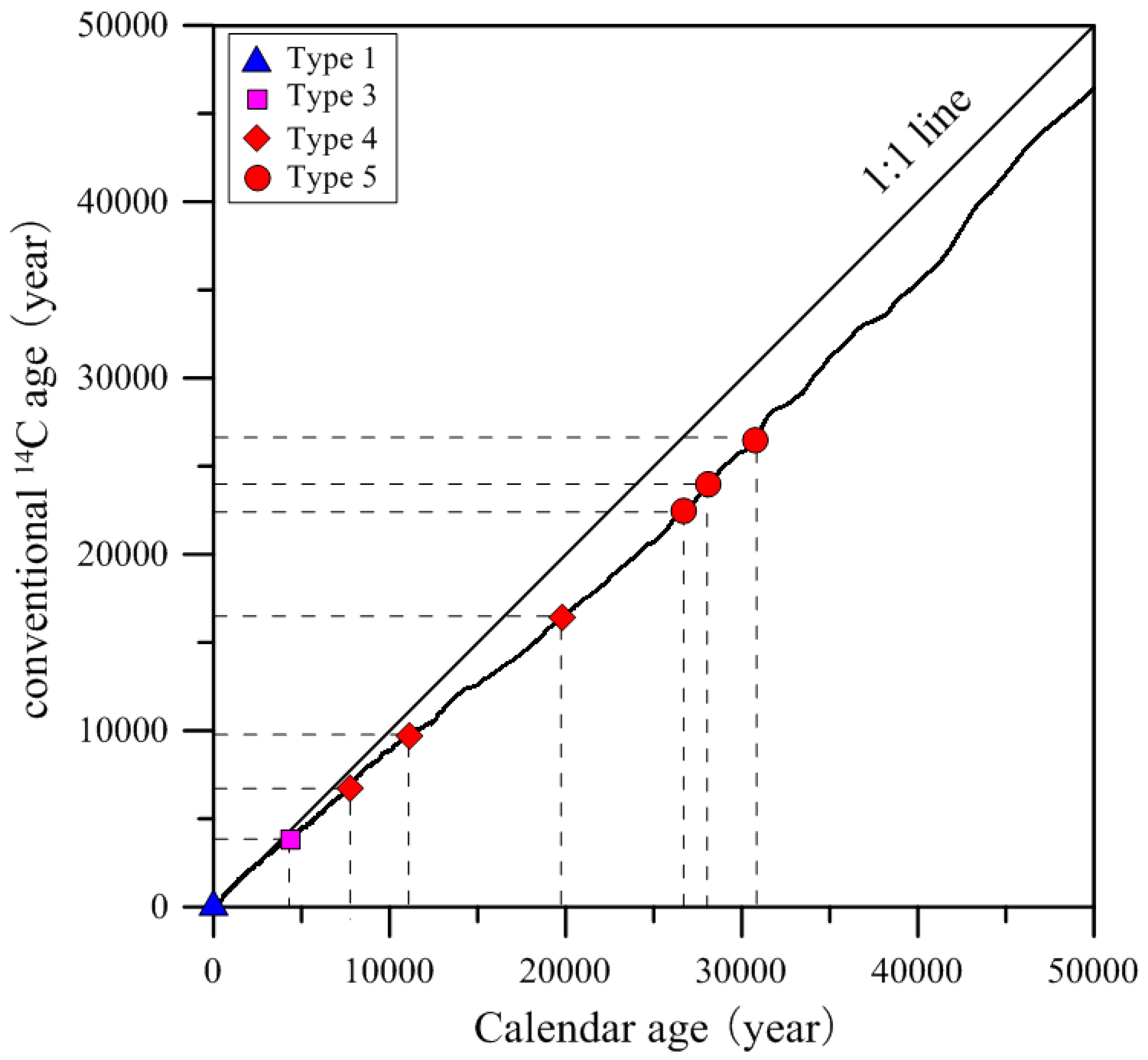
Figure 12, and the average calendar years of groundwater are also summarized in
Table S7. The results of the calendar ages for groundwater show that shallow groundwater in the recharge areas is approximately 0 year BP, while those in discharge area ranges from 4.340 to 19,965 years BP with the mean value of 10,829 years; deep groundwater varies from 26,736 to 28,125 years BP with the average of 28,588 years.



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











