
Trace Element Mobility during Corg-Enhanced Denitrification in Two Different Aquifers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Area
2.1.1. Water Production in the Lower Rhine Embayment
2.1.2. Water Production in the Haltern Fm
2.2. Sampling
2.3. Sediment Analysis
2.4. Isotopic and Water Analysis
2.5. Circulation Column Experiments
3. Results
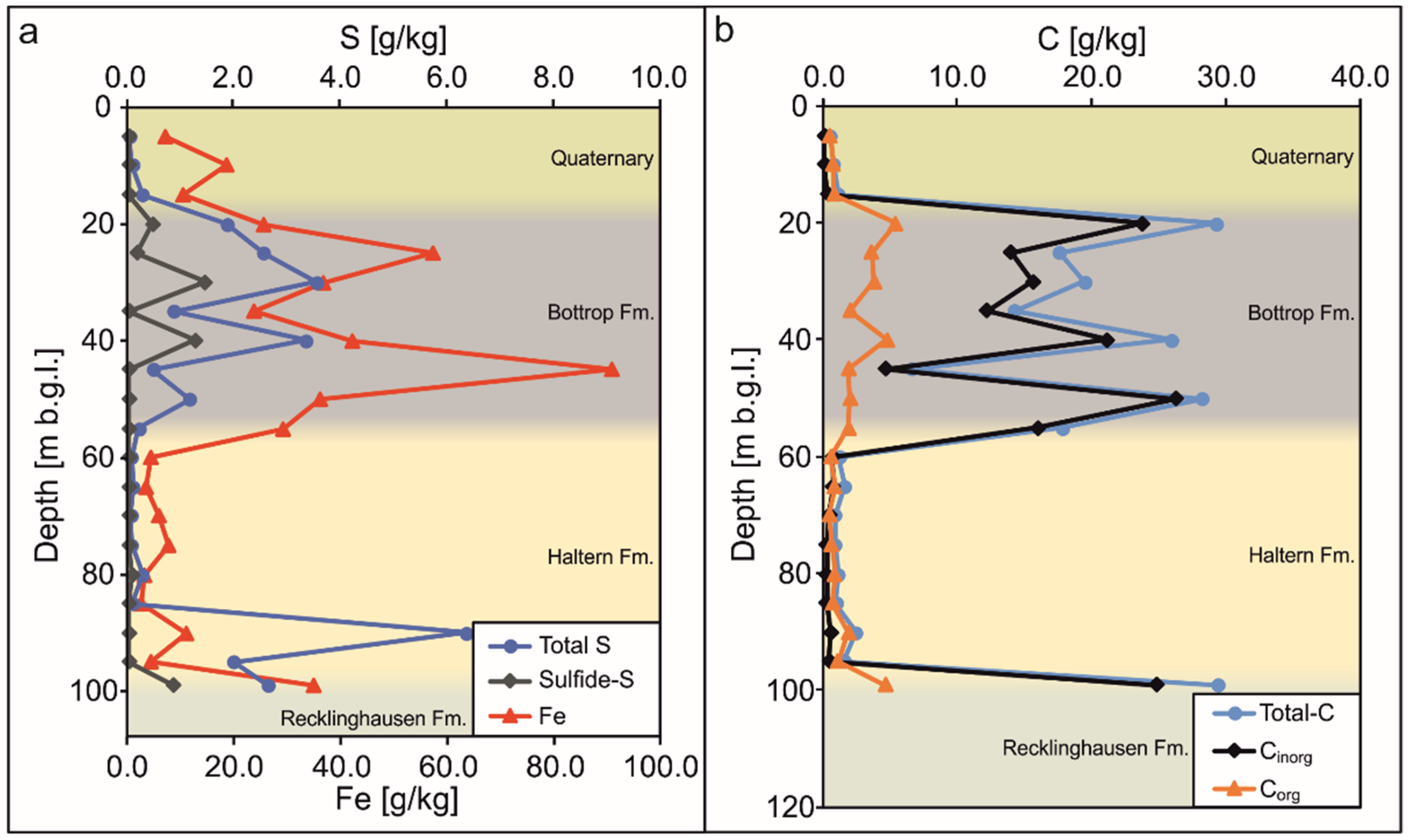
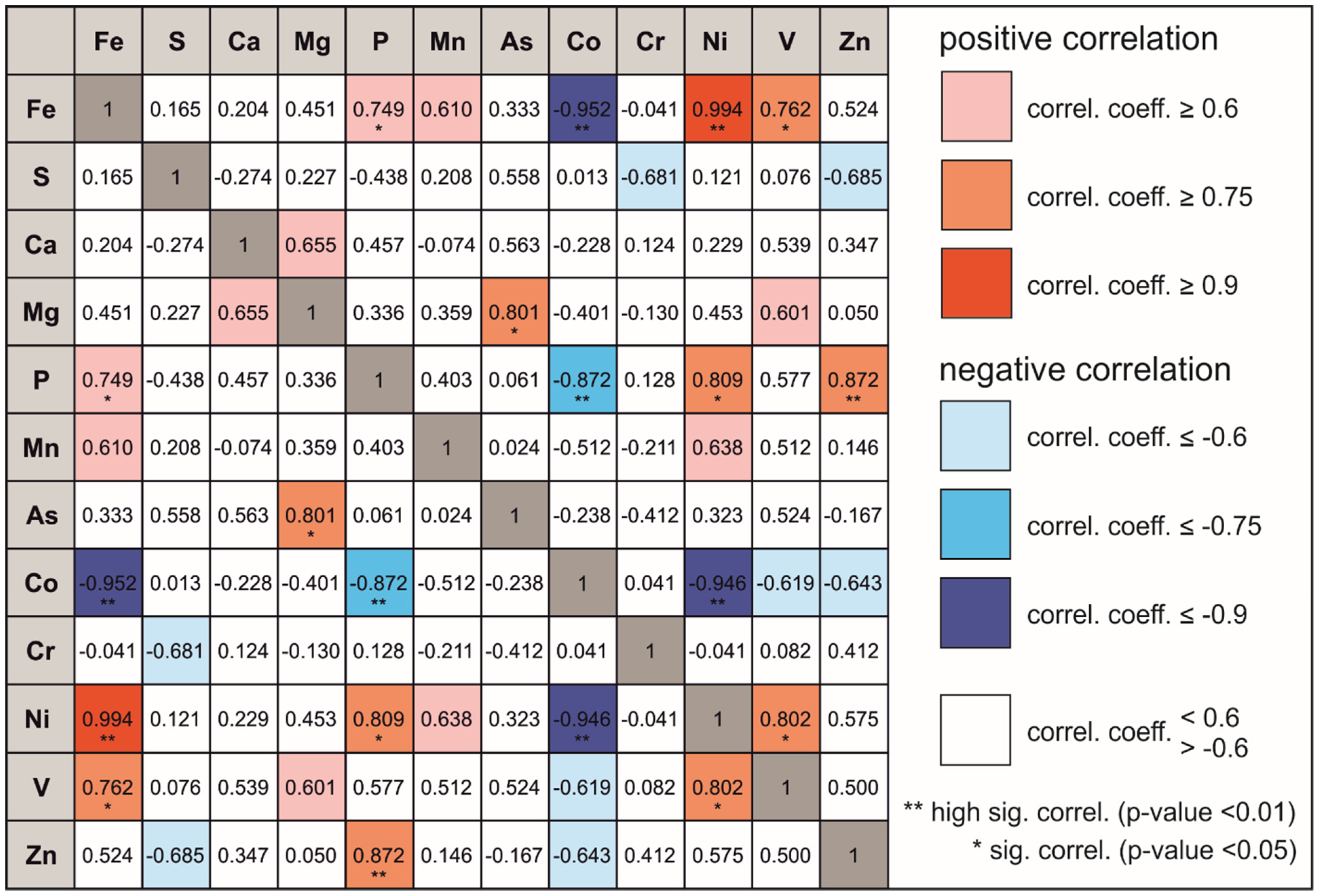
3.1. Sediment Analysis
3.1.1. Quaternary–Tertiary Aquifer
3.1.2. Haltern Fm. Aquifer
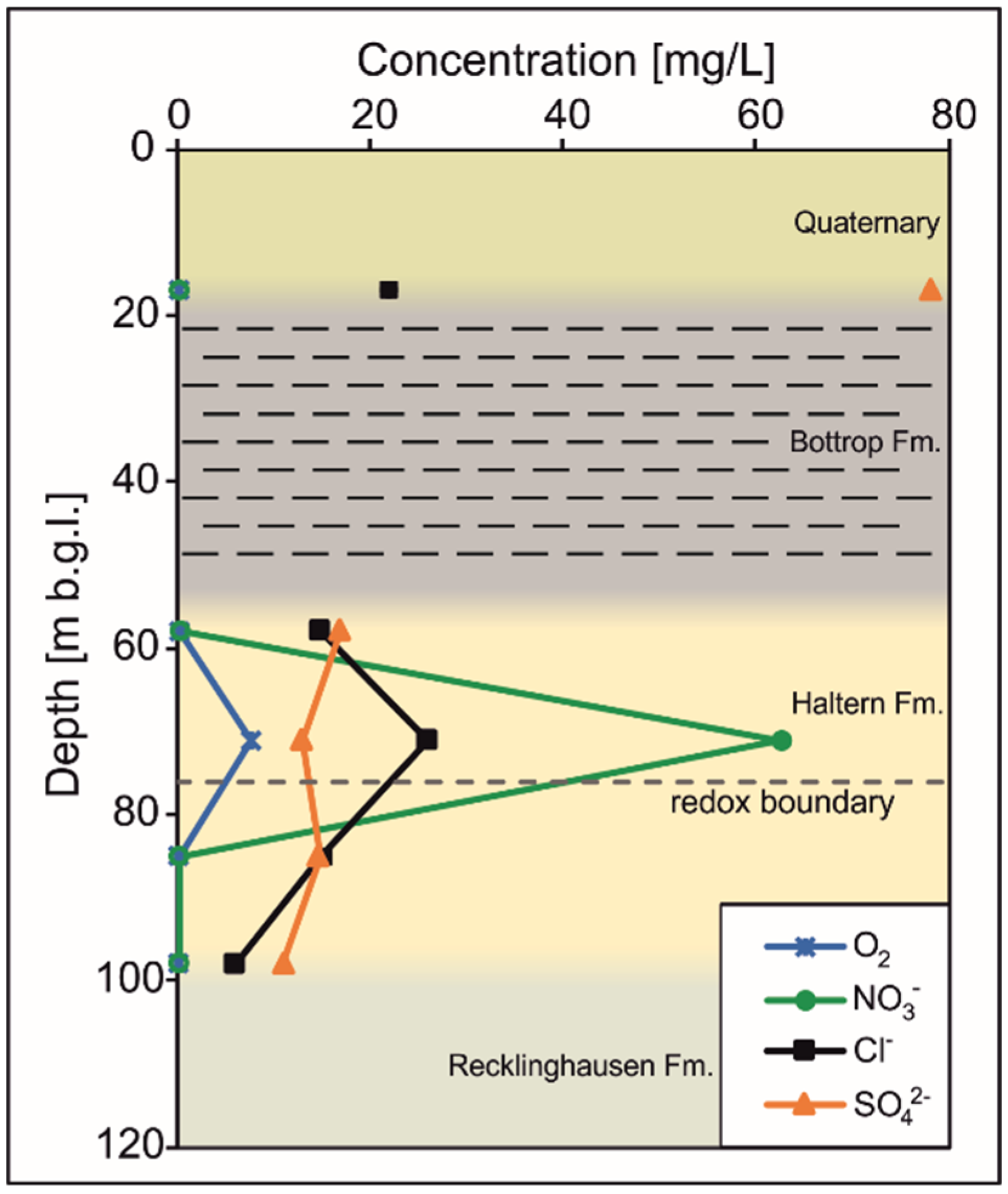
3.2. Isotopic and Water Analysis
3.2.1. Quaternary–Tertiary Aquifer
3.2.2. Haltern Fm. Aquifer
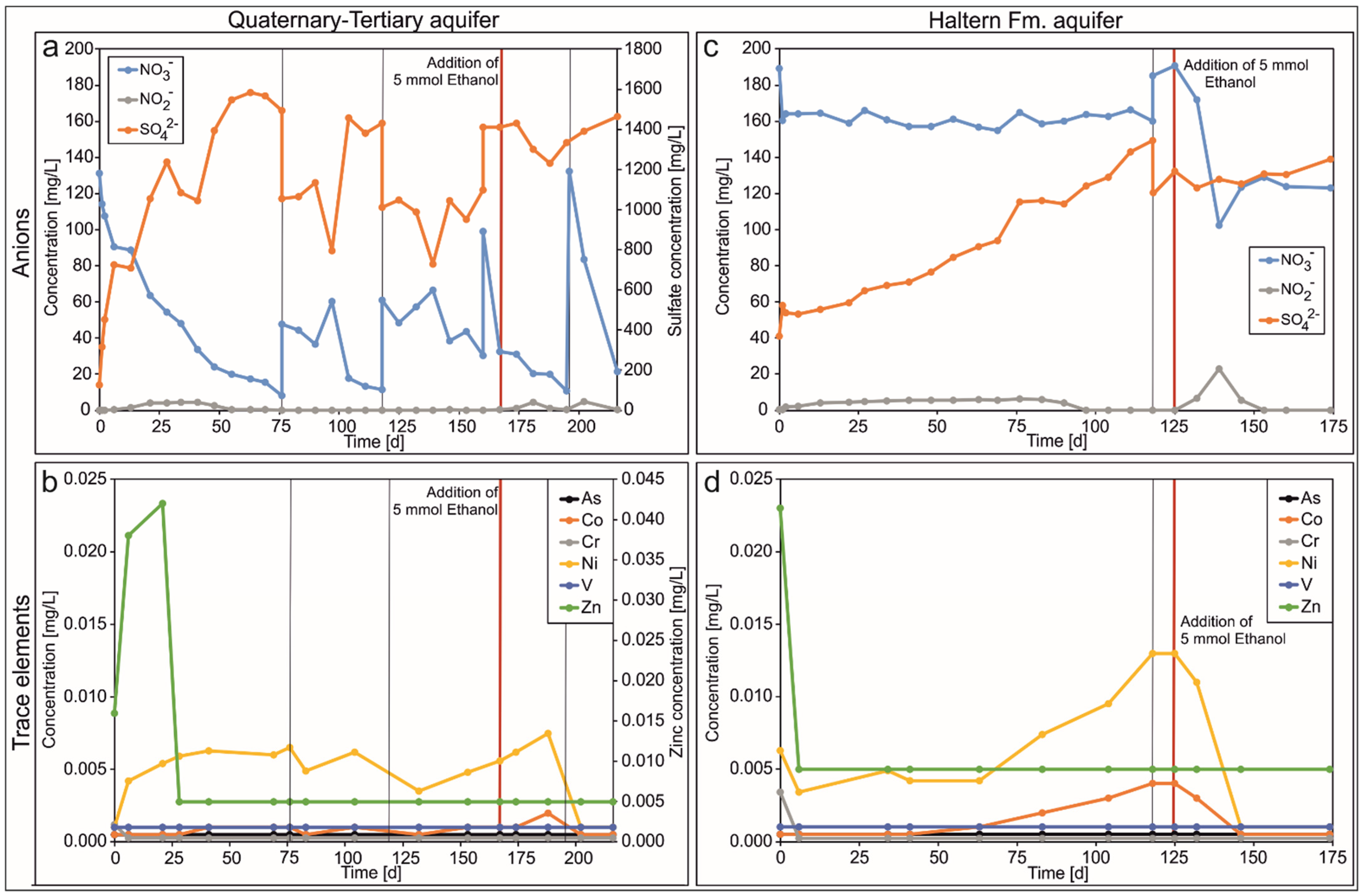
3.3. Circulation Column Experiments
4. Discussion
4.1. Sediment Analysis
4.2. Isotopic and Water Analysis
4.3. Column Experiments
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hosono, T.; Tokunaga, T.; Kagabu, M.; Nakata, H.; Orishikida, T.; Lin, I.-T.; Shimadab, J. The use of δ15N and δ18O tracers with an understanding of groundwater flow dynamics for evaluating the origins and attenuation mechanisms of nitrate pollution. Water Res. 2013, 47, 2661–2675. [Google Scholar] [CrossRef] [PubMed]
- Almasri, M.N. Nitrate contamination of groundwater: A conceptual management framework. Environ. Impact Assess. Rev. 2007, 27, 220–242. [Google Scholar] [CrossRef]
- Carrey, R.; Otero, N.; Vidal-Gavilan, G.; Ayora, C.; Soler, A.; Gómez-Alday, J.J. Induced nitrate attenuation by glucose in groundwater: Flow-through experiment. Chem. Geol. 2014, 370, 19–28. [Google Scholar] [CrossRef]
- Rivett, M.O.; Buss, S.R.; Morgan, P.; Smith, J.W.N.; Bemment, C.D. Nitrate attenuation in groundwater: A review of biogeochemical controlling processes. Water Res. 2008, 42, 4215–4232. [Google Scholar] [CrossRef] [PubMed]
- Vitousek, P.M.; Aber, J.D.; Howarth, R.W.; Likens, G.E.; Matson, P.A.; Schindler, D.W.; Schlesinger, W.H.; Tilman, D.G. Human alteration of the global nitrogen cycle: Sources and consequences. Ecol. Appl. 1997, 7, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Ortmeyer, F.; Mas-Pla, J.; Wohnlich, S.; Banning, A. Forecasting nitrate evolution in an alluvial aquifer under distinct environmental and climate change scenarios (Lower Rhine Embayment, Germany). Sci. Total Environ. 2021, 768, 144463. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.A.; Spalding, R.F. Enhanced in situ denitrification for a municipal well. Water Res. 2004, 38, 3382–3388. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Gavilan, G.; Carrey, R.; Solanas, A.; Soler, A. Feeding strategies for groundwater enhanced biodenitrification in an alluvial aquifer: Chemical, microbial and isotope assessment of a 1D flow-through experiment. Sci. Total Environ. 2014, 494–495, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Korom, S.F. Natural denitrification in the saturated zone: A review. Water Resour. Res. 1992, 28, 1657–1668. [Google Scholar] [CrossRef]
- Ge, S.; Peng, Y.; Wang, S.; Lu, C.; Cao, X.; Zhu, Y. Nitrite accumulation under constant temperature in anoxic denitrification process: The effects of carbon sources and COD/ NO3–N. Bioresour. Technol. 2012, 114, 137–143. [Google Scholar] [CrossRef]
- Schroeder, A.; Souza, D.H.; Fernandes, M.; Rodrigues, E.B.; Trevisan, V.; Skoronski, E. Application of glycerol as carbon source for continuous drinking water denitrification using microorganism from natural biomass. J. Environ. Manag. 2020, 256, 109964. [Google Scholar] [CrossRef]
- Costa, D.D.; Albino, A.; Fernandes, M.; Lopes, R.; De Lourdes, M.; Magalh, B. Using natural biomass microorganisms for drinking water denitrification. J. Environ. Manag. 2018, 217, 520–530. [Google Scholar] [CrossRef]
- Schipper, L.A.; Vojvodic, M. Nitrate removal from groundwater and denitrification rates in a porous treatment wall amended with sawdust. Ecol. Eng. 2000, 14, 269–278. [Google Scholar] [CrossRef]
- Trois, C.; Pisano, G.; Oxarango, L. Alternative solutions for the bio-denitrification of landfill leachates using pine bark and compost. J. Hazard. Mater. 2010, 178, 1100–1105. [Google Scholar] [CrossRef]
- Carrey, R.; Rodríguez-Escales, P.; Soler, A.; Otero, N. Tracing the role of endogenous carbon in denitrification using wine industry by-product as an external electron donor: Coupling isotopic tools with mathematical modeling. J. Environ. Manag. 2018, 207, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Margalef-Marti, R.; Carrey, R.; Soler, A.; Neus Otero, N. Evaluating the potential use of a dairy industry residue to induce denitrification in polluted water bodies: A flow-through experiment. J. Environ. Manag. 2019, 245, 86–94. [Google Scholar] [CrossRef]
- Ortmeyer, F.; Begerow, D.; Guerreiro, M.A.; Wohnlich, S.; Banning, A. Comparison of denitrification induced by various organic substances—Reaction rates, microbiology and temperature effect. Water Resour. Res. in revision.
- Böttcher, J.; Strebel, O.; Voerkelius, S.; Schmidt, H.-L. Using isotope fractionation of nitrate-nitrogen and nitrate-oxygen for evaluation of microbial denitrification in a sandy aquifer. J. Hydrol. 1990, 114, 413–424. [Google Scholar] [CrossRef]
- Nikolenko, O.; Jurado, A.; Borges, A.V.; Knöller, K.; Brouyère, S. Isotopic composition of nitrogen species in groundwater under agriculture areas: A review. Sci. Total Environ. 2018, 621, 1415–1432. [Google Scholar] [CrossRef] [PubMed]
- Houben, G.J.; Sitnikova, M.A.; Post, V.E. Terrestrial sedimentary pyrites as a potential source of trace metal release to groundwater—A case study from Emsland, Germany. Appl. Geochem. 2017, 76, 99–111. [Google Scholar] [CrossRef]
- Abraitis, P.K.; Pattrick, R.A.D.; Vaughan, D.J. Variations in the compositional, textural and electrical properties of natural pyrite: A review. Int. J. Miner. Process. 2004, 74, 41–59. [Google Scholar] [CrossRef]
- Diehl, S.; Goldhaber, M.B.; Koenig, A.E.; Lowers, H.A.; Ruppert, L.F. Distribution of arsenic, selenium, and other trace elements in high pyrite Appalachian coals: Evidence for multiple episodes of pyrite formation. Int. J. Coal Geol. 2012, 94, 238–249. [Google Scholar] [CrossRef]
- Karikari-Yeboah, O.; Skinner, W.; Addai-Mensah, J. The impact of preload on the mobilisation of multivalent trace metals in pyrite-rich sediment. Environ. Monit. Assess. 2018, 190, 398. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.; Postma, D. Nickel Mobilization in a groundwater well field: Release by pyrite oxidation and desorption from manganese oxides. Environ. Sci. Technol. 1997, 31, 2589–2595. [Google Scholar] [CrossRef]
- Banning, A.; Rüde, T.R. Enrichment processes of arsenic in oxidic sedimentary rocks—From geochemical and genetic characterization to potential mobility. Water Res. 2010, 44, 5512–5531. [Google Scholar] [CrossRef]
- Banning, A.; Rüde, T.R.; Dölling, B. Crossing redox boundaries—Aquifer redox history and effects on iron mineralogy and arsenic availability. J. Hazard. Mater. 2013, 262, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Rinklebe, J.; Shahenn, S.M. Redox chemistry of nickel in soils and sediments: A review. Chemosphere 2017, 179, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Smedley, P.L.; Kinniburgh, D.G. A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem. 2002, 17, 517–568. [Google Scholar] [CrossRef] [Green Version]
- Banning, A.; Pawletko, N.; Röder, J.; Kübeck, C.; Wisotzky, F. Ex situ groundwater treatment triggering the mobilization of geogenic uranium from aquifer sediments. Sci. Total Environ. 2017, 587–588, 371–380. [Google Scholar] [CrossRef]
- Ortmeyer, F.; Volkova, K.; Wisotzky, F.; Wohnlich, S.; Banning, A. Monitoring nitrate reduction: Hydrogeochemistry and clogging potential in raw water wells. Environ. Monit. Assess. 2021, 193, 112. [Google Scholar] [CrossRef]
- Banning, A.; Coldewey, W.G.; Göbel, P. A procedure to identify natural arsenic sources, applied in an affected area in North Rhine-Westphalia, Germany. Environ. Geol. 2009, 57, 775–787. [Google Scholar] [CrossRef]
- Zhu, J.; Yu, L.; Bakken, L.R.; Mørkved, P.T.; Mulder, J.; Dörsch, P. Controlled induction of denitrification in Pseudomonas aureofaciens: A simplified denitrifier method for dual isotope analysis in NO3−. Sci. Total Environ. 2018, 633, 1370–1378. [Google Scholar] [CrossRef]
- Stock, P.; Order, S.; Burghardt, D. Further optimization of the denitrifier method for the rapid 15N and 18O analysis of nitrate in natural water samples. Rapid Comm. Mass Spectrom. 2020. [Google Scholar] [CrossRef]
- DWA—Deutsche Vereinigung für Wasserwirtschaft, Abwasser und Abfall (Ed.) DWA-Themen: Stickstoffumsatz im Grundwasser; Deutsche Vereinigung für Wasserwirtschaft, Abwasser und Abfall: Hennef, Germany, 2015; p. 87. [Google Scholar]
- Ma, Y.; Hooda, P.S. Chromium, nickel and cobalt. In Trace Elements in Soils, 1st ed.; Hooda, P.S., Ed.; John Wiley & Sons Ltd.: Oxford, UK, 2010; pp. 461–480. [Google Scholar]
- Cox, R.M.; Hutchinson, T.C. Environmental factors influencing the rate of spread of the grass Deschampsia caespitosa invading areas around the Sudbury nickel-copper smelter. Water Air Soil Pollut. 1981, 16, 83–106. [Google Scholar] [CrossRef]
- Otero, N.; Torrentó, C.; Soler, A.; Menció, A.; Mas-Pla, J. Monitoring groundwater nitrate attenuation in a regional system coupling hydrogeology with multi-isotopic methods: The case of Plana de Vic (Osona, Spain). Agric. Ecosyst. Environ. 2009, 133, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Schulte, U.; Strauß, H.; Bergmann, A.; Obermann, P. Isotopenverhältnisse der Schwefel-und Kohlenstoffspezies aus Sedimenten und tiefen Grundwässern der Niederrheinischen Bucht. Grundwasser 1997, 2, 103–110. [Google Scholar] [CrossRef]
- Aravena, R.; Robertson, W.D. Use of multiple isotope tracers to evaluate denitrification in ground water: Study of nitrate from a large-flux septic system plume. Groundwater 1998, 36, 975–982. [Google Scholar] [CrossRef]
- Jorgensen, C.; Jacobsen, O.; Elberling, B.; Aamand, J. Microbial oxidation of pyrite coupled to nitrate reduction in anoxic groundwater sediment. Environ. Sci. Technol. 2009, 43, 4851–4857. [Google Scholar] [CrossRef]
- van Beek, C.G.E.M.; Hettinga, F.A.M.; Straatman, R. The effect of manure spreading and acid deposition upon groundwater quality at Vierlingsbeek, The Netherlands. IAHS Publ. 1989, 185, 155–162. [Google Scholar]
- Trinkwasserverordnung in der Fassung der Bekanntmachung vom 10. März 2016 (BGBl. I S. 459), die Zuletzt durch Artikel 1 der Verordnung vom 20. Dezember 2019 (BGBl. I S. 2934) Geändert Worden Ist. 2001. Available online: http://www.gesetze-im-internet.de/trinkwv_2001/ (accessed on 28 April 2021).
- Corsini, A.; Cavalca, L.; Crippa, L.; Zaccheo, P.; Andreoni, V. Impact of glucose on microbial community of a soil containing pyrite cinders: Role of bacteria in arsenic mobilization under submerged condition. Soil Biol. Biochem. 2010, 42, 699–707. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortmeyer, F.; Wohnlich, S.; Banning, A. Trace Element Mobility during Corg-Enhanced Denitrification in Two Different Aquifers. Water 2021, 13, 1589. https://doi.org/10.3390/w13111589
Ortmeyer F, Wohnlich S, Banning A. Trace Element Mobility during Corg-Enhanced Denitrification in Two Different Aquifers. Water. 2021; 13(11):1589. https://doi.org/10.3390/w13111589
Chicago/Turabian StyleOrtmeyer, Felix, Stefan Wohnlich, and Andre Banning. 2021. "Trace Element Mobility during Corg-Enhanced Denitrification in Two Different Aquifers" Water 13, no. 11: 1589. https://doi.org/10.3390/w13111589
APA StyleOrtmeyer, F., Wohnlich, S., & Banning, A. (2021). Trace Element Mobility during Corg-Enhanced Denitrification in Two Different Aquifers. Water, 13(11), 1589. https://doi.org/10.3390/w13111589