
Ambient-Visible-Light-Mediated Enhanced Degradation of UV Stabilizer Bis(4-hydroxyphenyl)methanone by Nanosheet-Assembled Cobalt Titanium Oxide: A Comparative and DFT-Assisted Investigation



 ,
,  ,
, _Lin.png)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
3. Results and Discussions
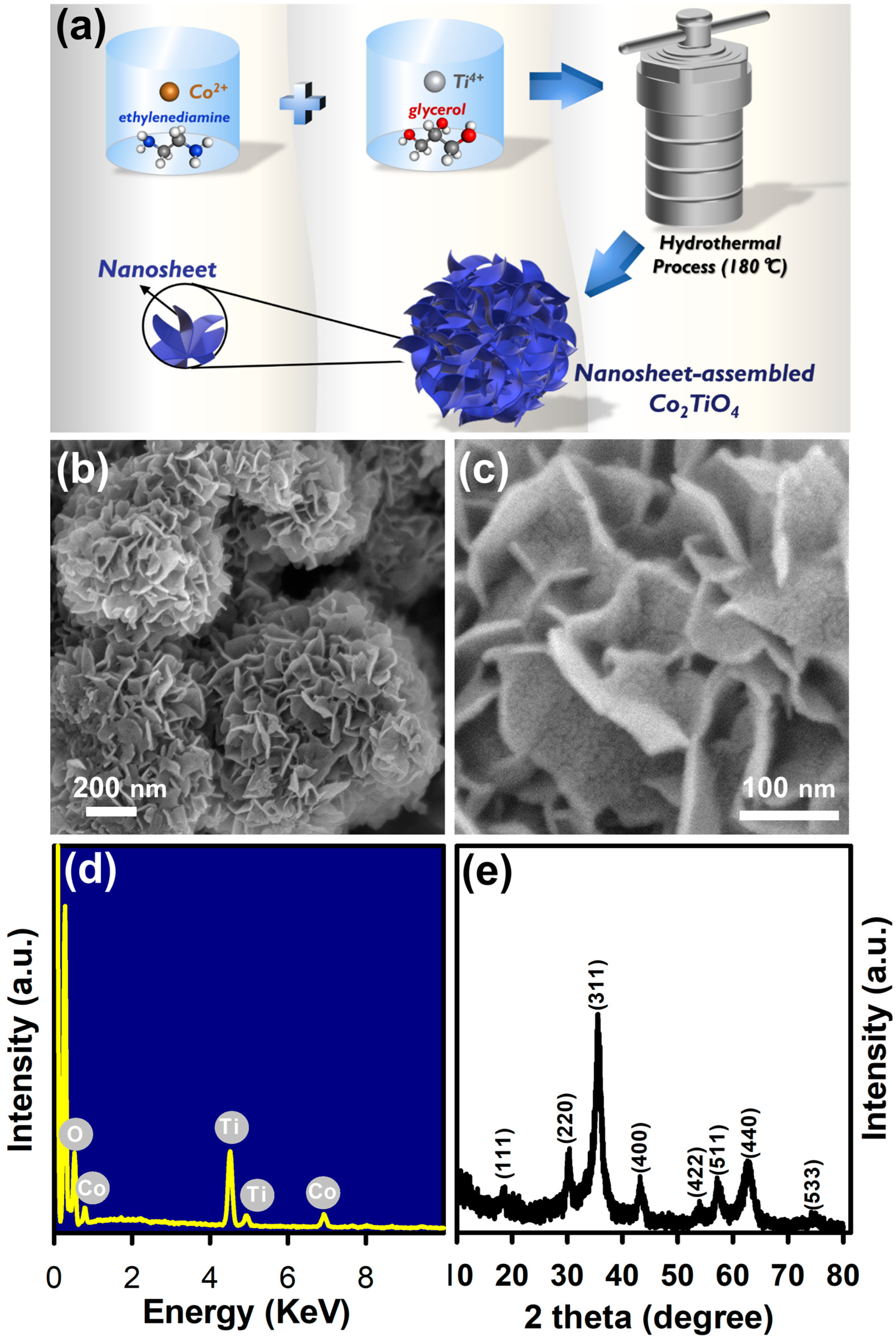
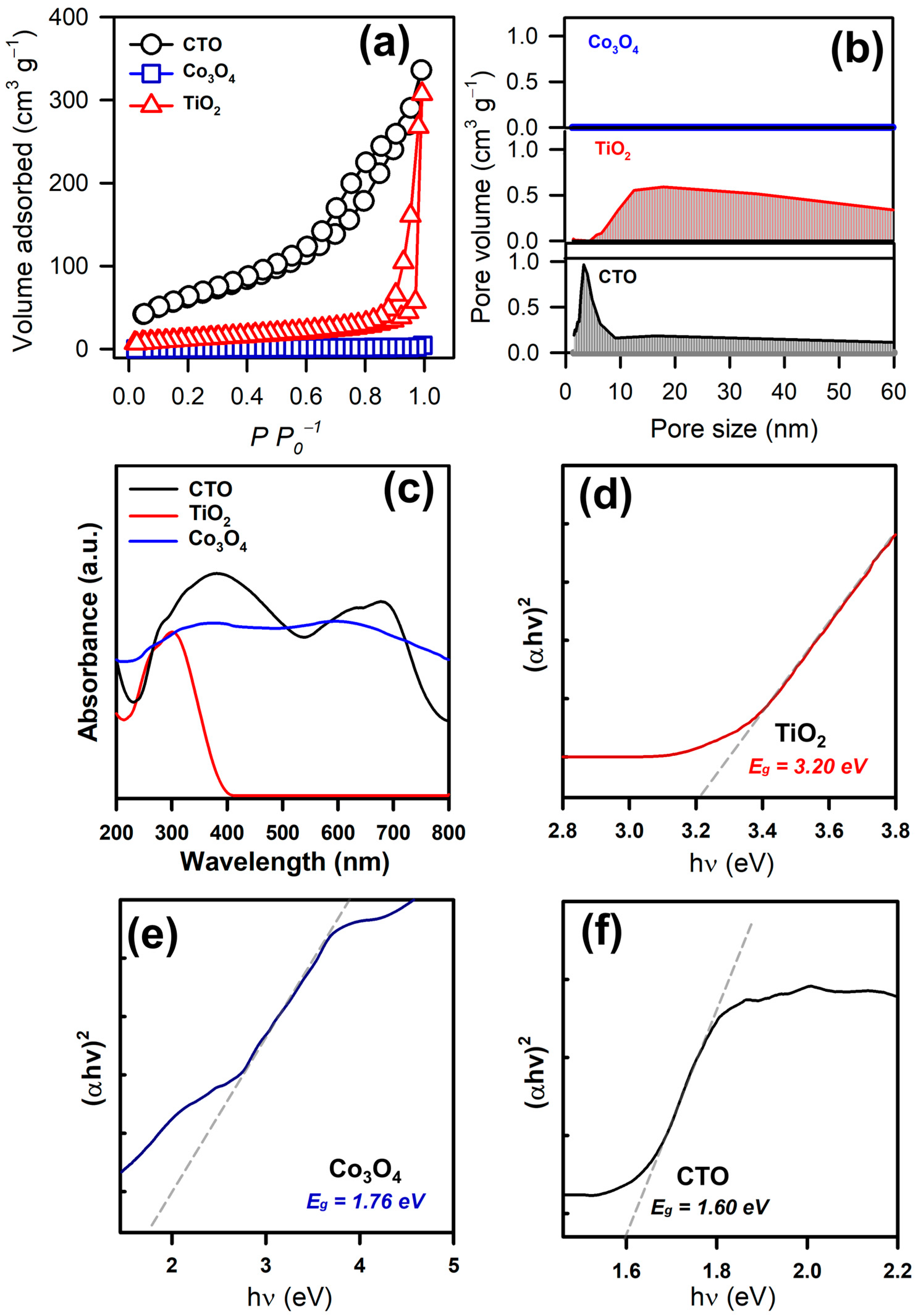
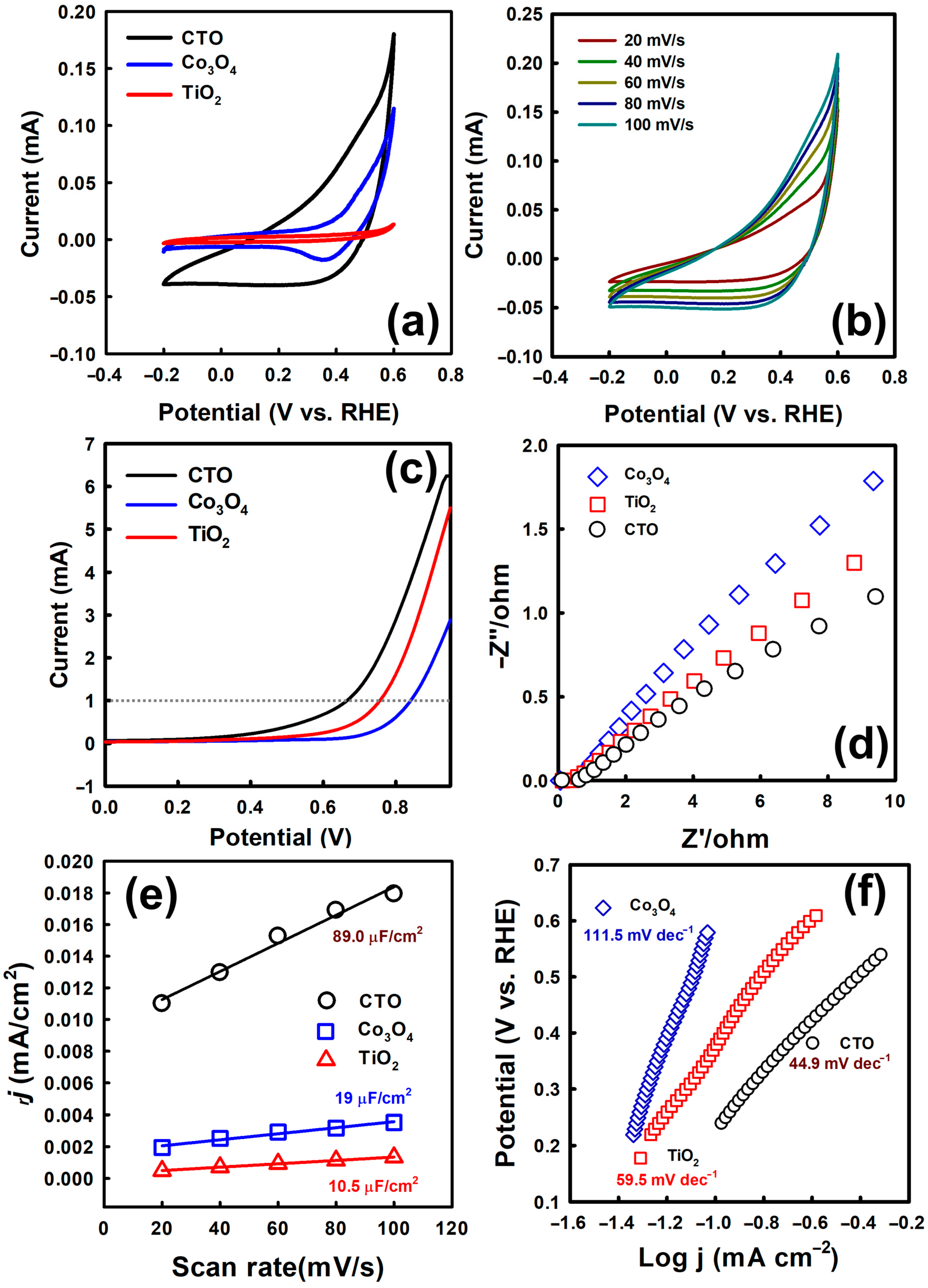
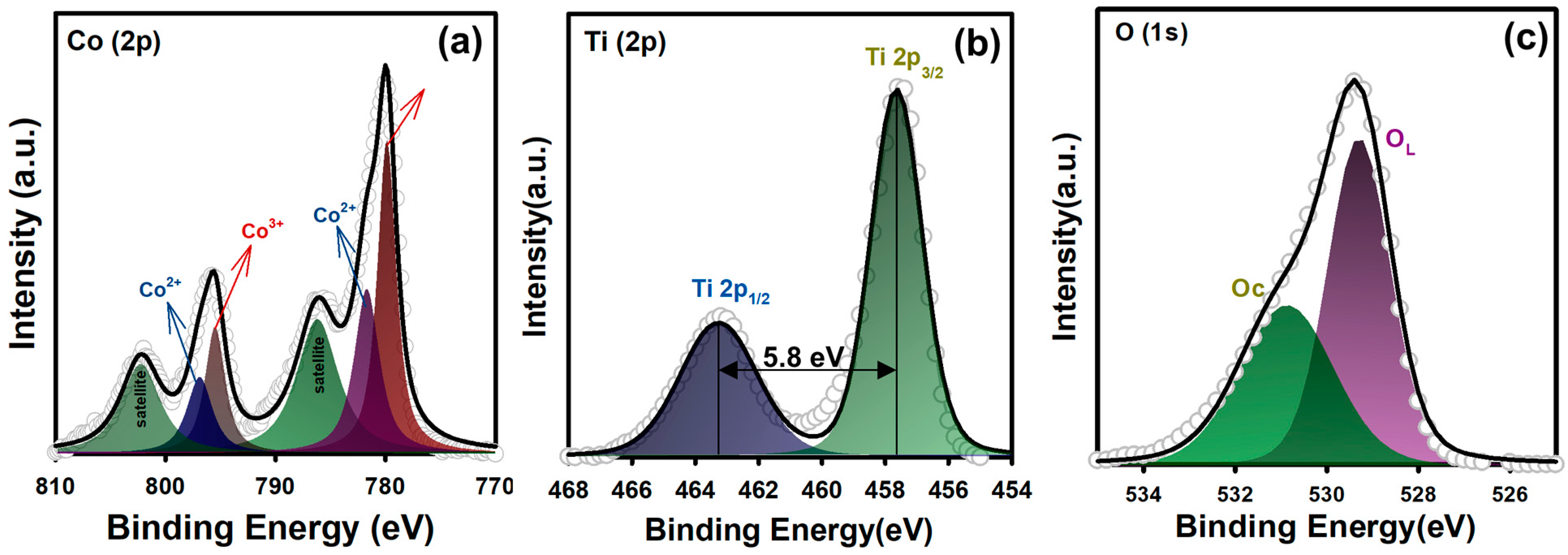
3.1. Physical and Chemical Properties of CTO
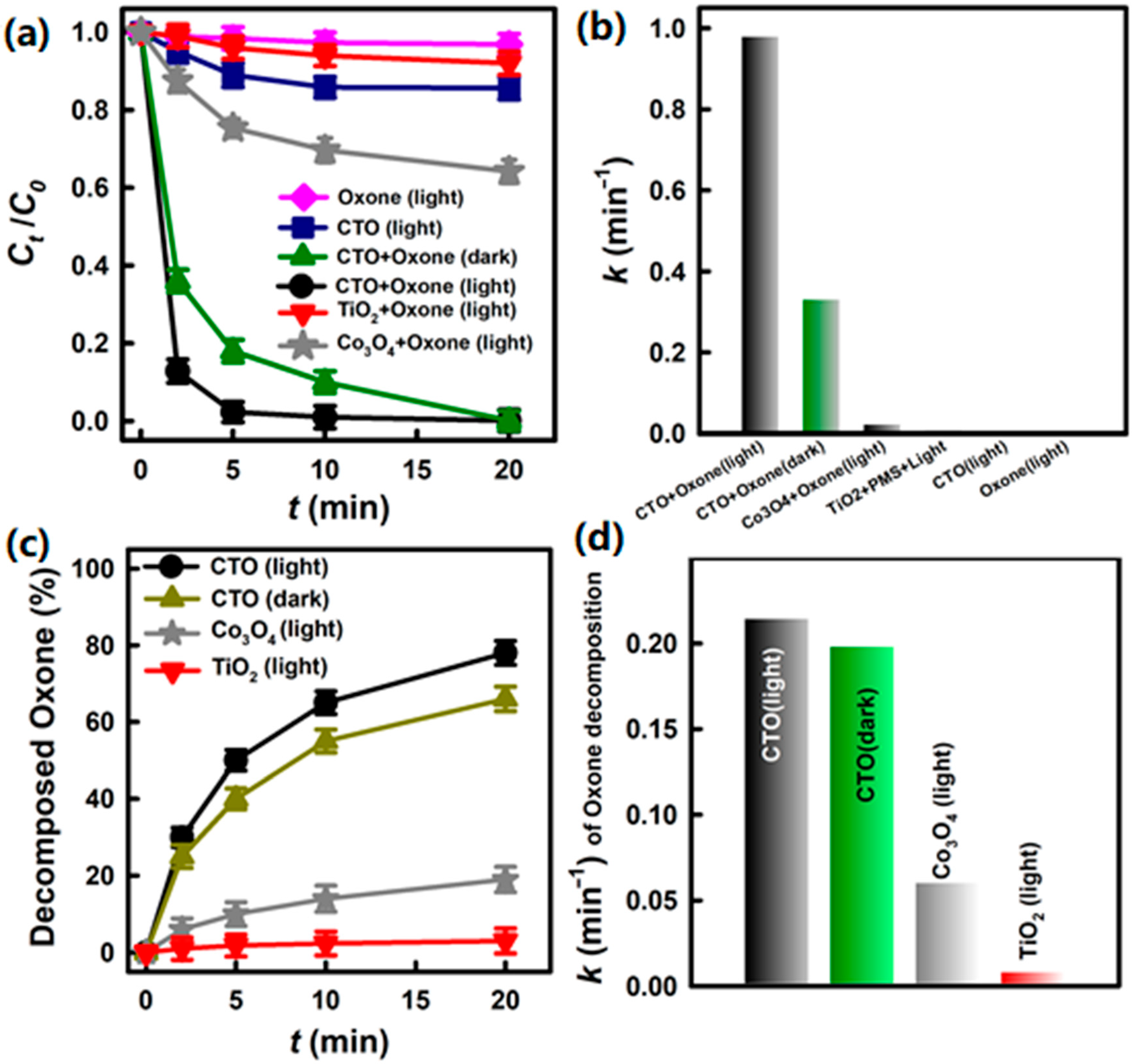
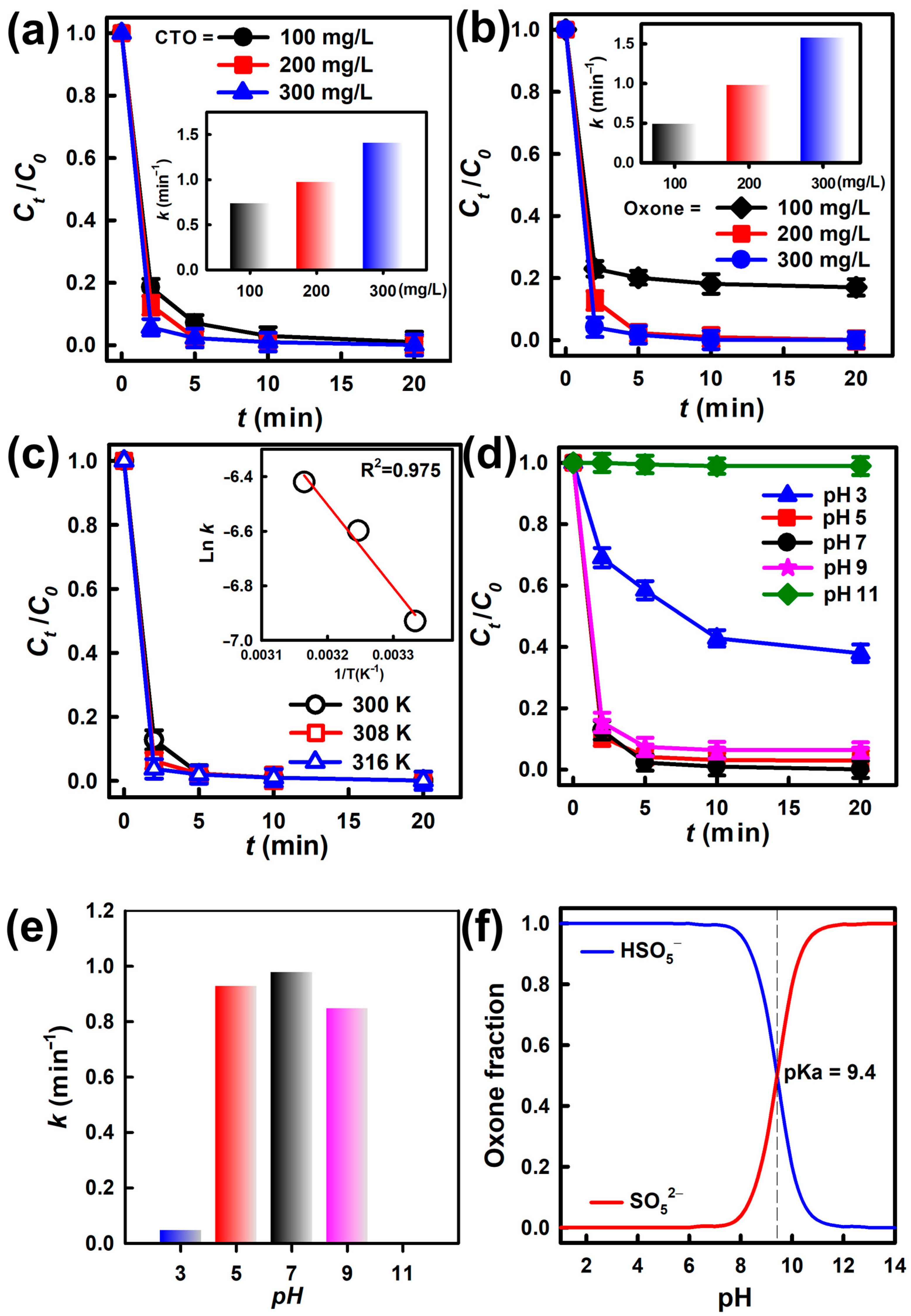
3.2. Degradation of BHPM and Decomposition of Oxone by CTO
3.3. Influence of Catalysts/Oxone Dosages on BHPM Elimination
3.4. Influences of Temperature and pH to BHPM Elimination
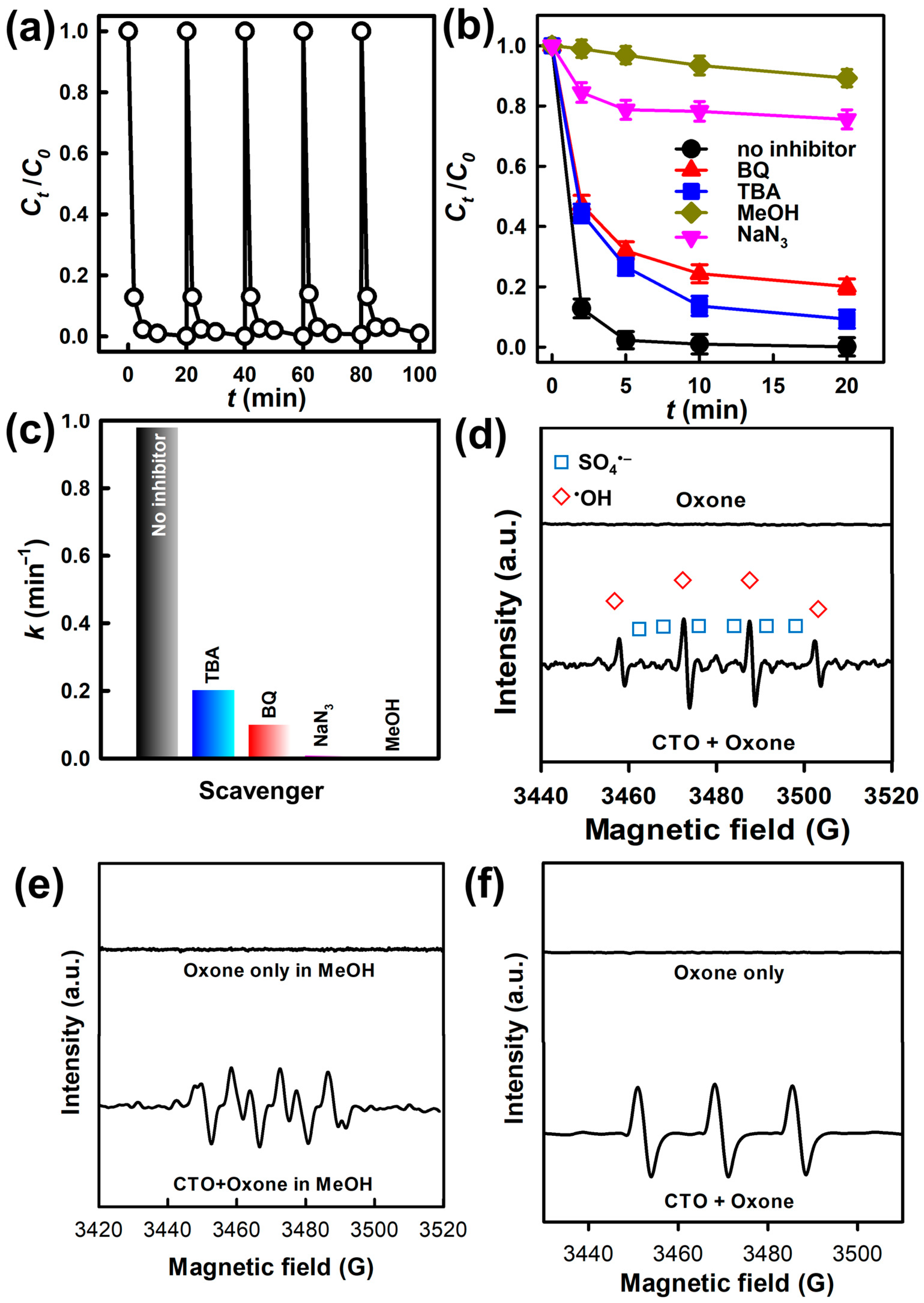
3.5. Reusability of CTO for Degradation of BHPM
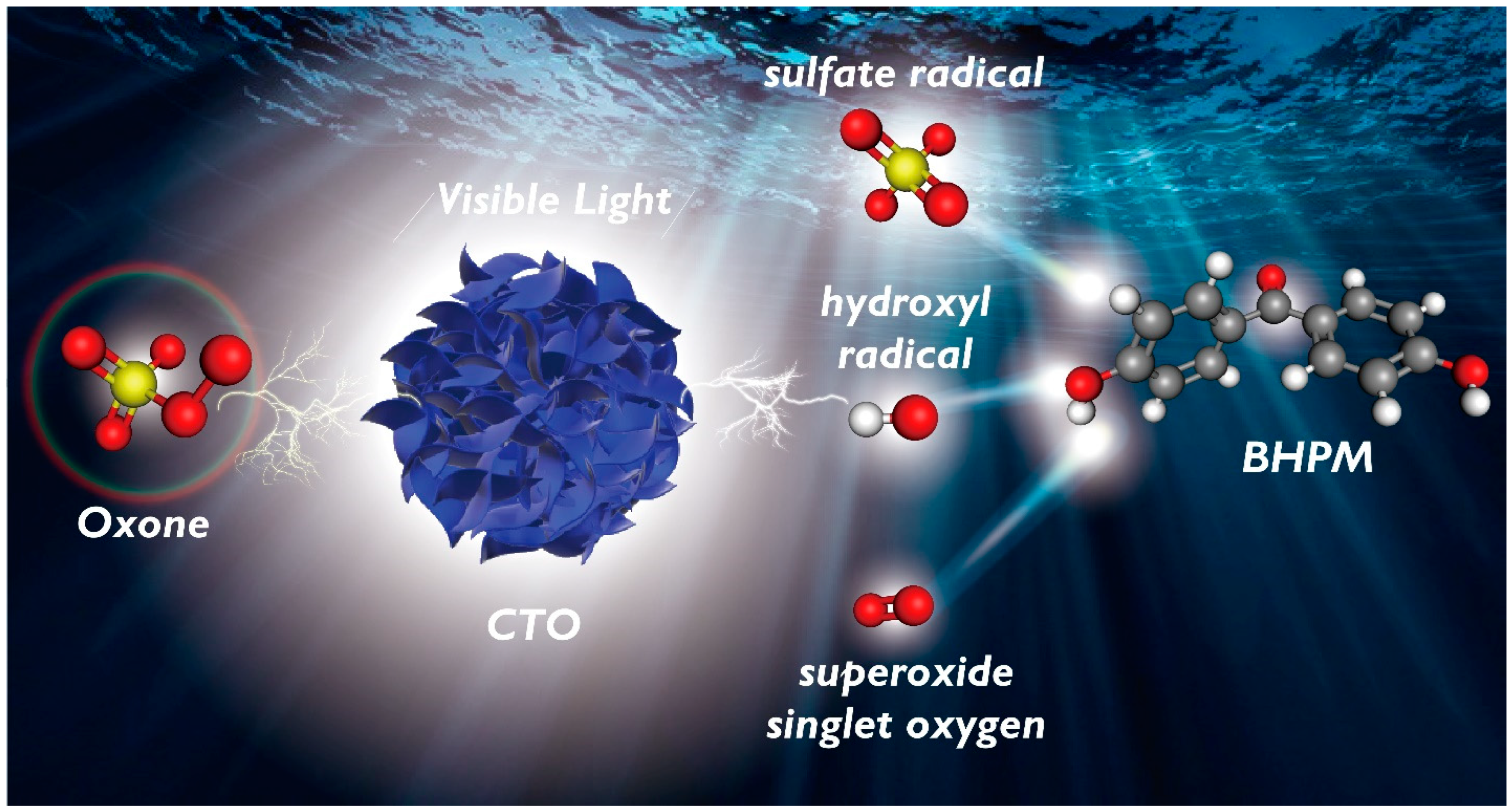
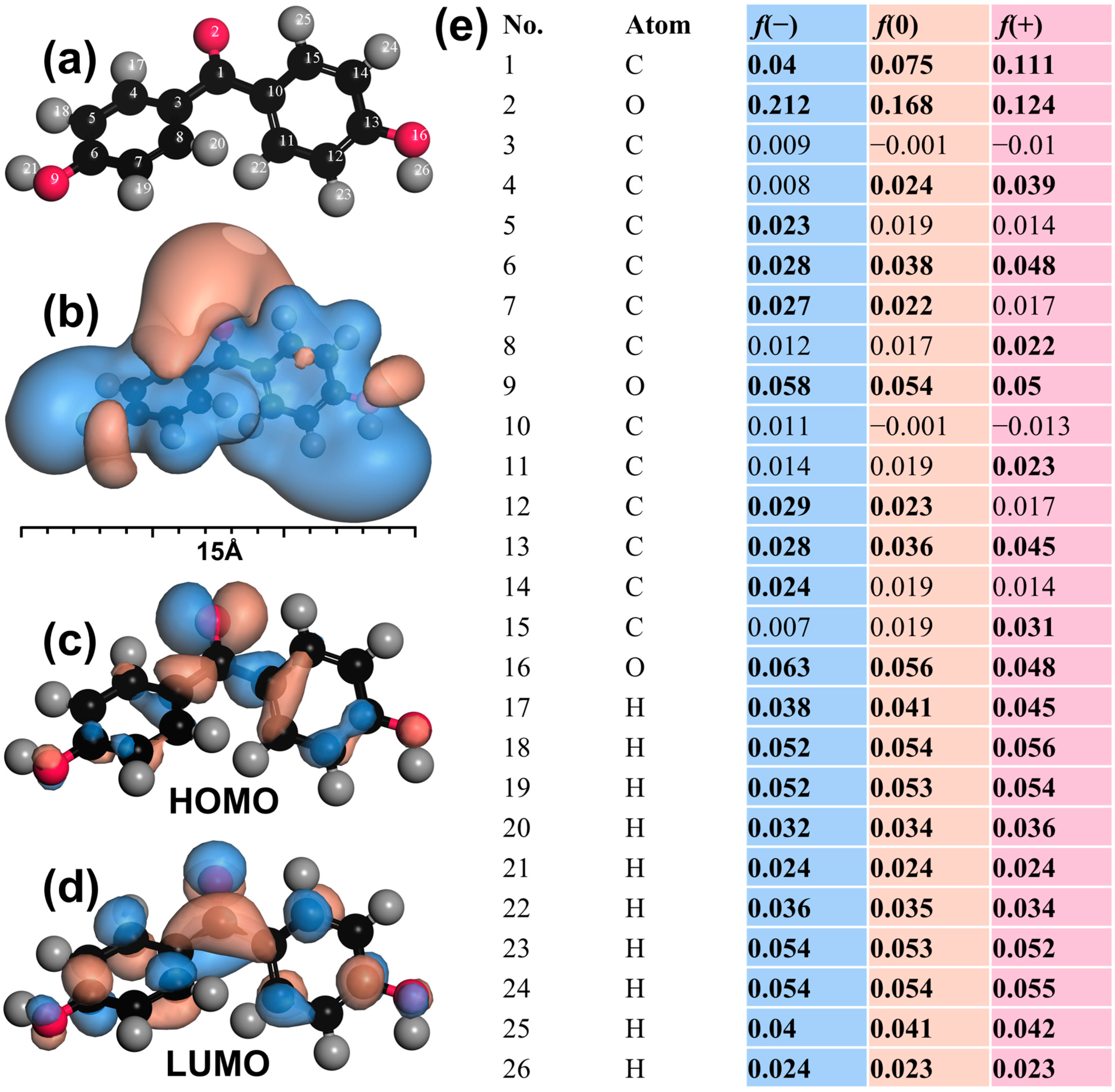
3.6. Mechanistic Insights into BHPM Elimination by CTO-Activated Oxone
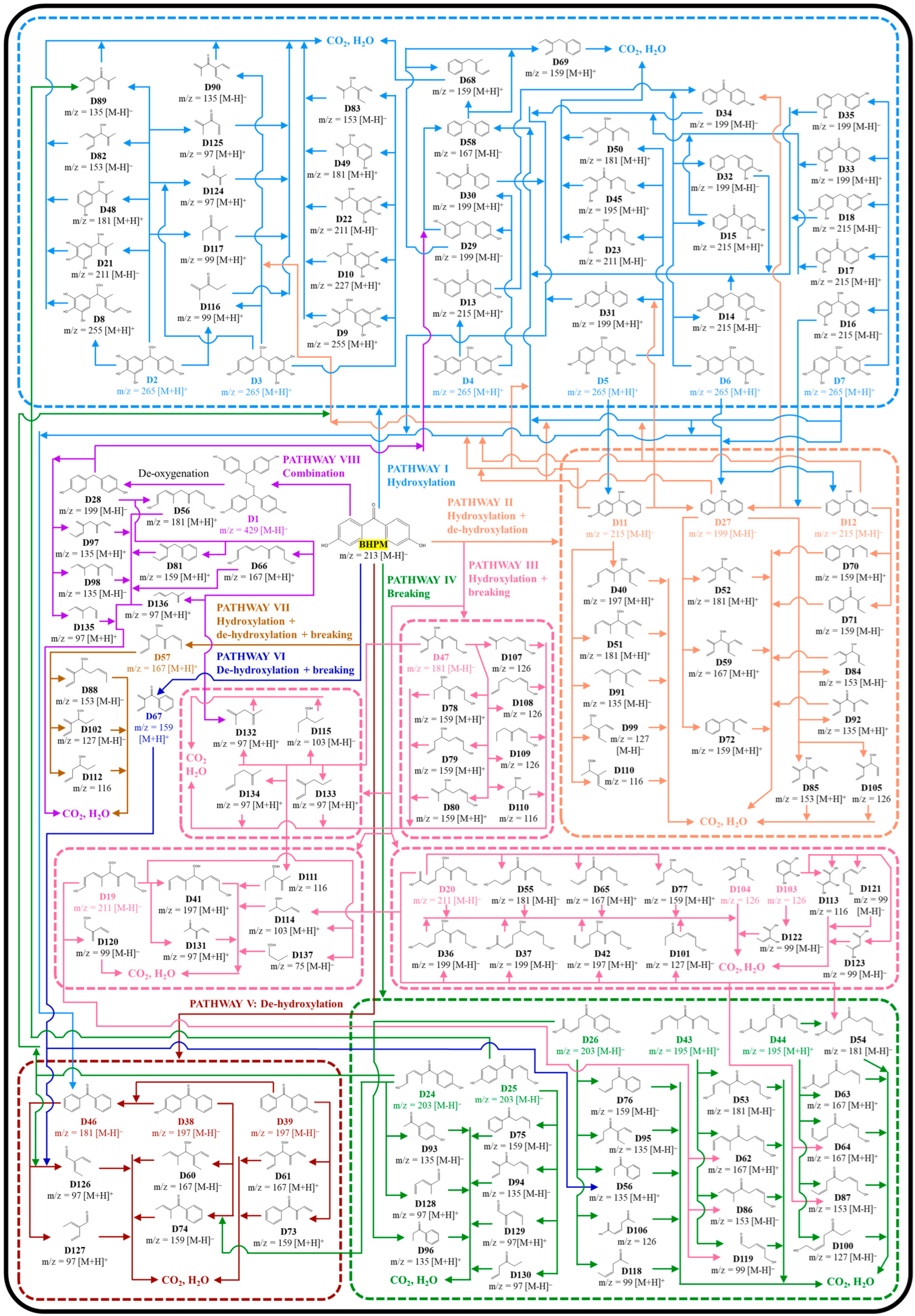
3.7. Possible BHPM Degradation Pathways and Computational Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pintado-Herrera, M.G.; Lara-Martín, P.A.; González-Mazo, E.; Allan, I.J. Determination of silicone rubber and low-density polyethylene diffusion and polymer/water partition coefficients for emerging contaminants. Environ. Toxicol. Chem. 2016, 35, 2162–2172. [Google Scholar] [CrossRef] [PubMed]
- Tsui, M.M.P.; Leung, H.W.; Wai, T.-C.; Yamashita, N.; Taniyasu, S.; Liu, W.; Lam, P.K.S.; Murphy, M.B. Occurrence, distribution and ecological risk assessment of multiple classes of UV filters in surface waters from different countries. Water Res. 2014, 67, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.; Bussink, J.; van de Grampel, H.T.; Wheatley, G.W.; Dorf, E.-U.; Ostlinning, E.; Reinking, K. Polymers, High-Temperature. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Hoboken, NJ, USA, 1914. [Google Scholar]
- Eddine, A.N.; von Kries, J.P.; Podust, M.V.; Warrier, T.; Kaufmann, S.H.E.; Podust, L.M. X-ray Structure of 4,4′-Dihydroxybenzophenone Mimicking Sterol Substrate in the Active Site of Sterol 14α-Demethylase (CYP51)*♦. J. Biol. Chem. 2008, 283, 15152–15159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fast, S.A.; Gude, V.G.; Truax, D.D.; Martin, J.; Magbanua, B.S. A Critical Evaluation of Advanced Oxidation Processes for Emerging Contaminants Removal. Environ. Process. 2017, 4, 283–302. [Google Scholar] [CrossRef]
- Andrew Lin, K.-Y.; Hsu, F.-K.; Lee, W.-D. Magnetic cobalt-graphene nanocomposite derived from self-assembly of MOFs with graphene oxide as an activator for peroxymonosulfate. J. Mater. Chem. A 2015, 3, 9480–9490. [Google Scholar] [CrossRef]
- Li, M.-C.; Ghanbari, F.; Chang, F.-C.; Hu, C.; Lin, K.-Y.A.; Du, Y. Enhanced degradation of 5-sulfosalicylic acid using peroxymonosulfate activated by ordered porous silica-confined Co3O4 prepared via a solvent-free confined space strategy. Sep. Purif. Technol. 2020, 249, 116972. [Google Scholar] [CrossRef]
- Trang, N.H.; Kwon, E.; Lisak, G.; Hu, C.; Andrew Lin, K.-Y. Cobalt ferrite nanoparticle-loaded nitrogen-doped carbon sponge as a magnetic 3D heterogeneous catalyst for monopersulfate-based oxidation of salicylic acid. Chemosphere 2021, 267, 128906. [Google Scholar] [CrossRef]
- Yang, Q.; Choi, H.; Al-Abed, S.R.; Dionysiou, D.D. Iron–cobalt mixed oxide nanocatalysts: Heterogeneous peroxymonosulfate activation, cobalt leaching, and ferromagnetic properties for environmental applications. Appl. Catal. B Environ. 2009, 88, 462–469. [Google Scholar] [CrossRef]
- Cai, C.; Zhang, H.; Zhong, X.; Hou, L. Ultrasound enhanced heterogeneous activation of peroxymonosulfate by a bimetallic Fe–Co/SBA-15 catalyst for the degradation of Orange II in water. J. Hazard. Mater. 2015, 283, 70–79. [Google Scholar] [CrossRef]
- Anipsitakis, G.P.; Stathatos, E.; Dionysiou, D.D. Heterogeneous Activation of Oxone Using Co3O4. J. Phys. Chem. B 2005, 109, 13052–13055. [Google Scholar] [CrossRef]
- Tuan, D.D.; Hu, C.; Kwon, E.; Du, Y.; Lin, K.-Y.A. Coordination polymer-derived porous Co3O4 nanosheet as an effective catalyst for activating peroxymonosulfate to degrade sulfosalicylic acid. Appl. Surf. Sci. 2020, 532, 147382. [Google Scholar] [CrossRef]
- Chen, X.; van Gog, H.; van Huis, M.A. Transformation of Co3O4 nanoparticles to CoO monitored by in situ TEM and predicted ferromagnetism at the Co3O4/CoO interface from first principles. J. Mater. Chem. C 2021, 9, 5662–5675. [Google Scholar] [CrossRef]
- Chen, X.; Chen, J.; Qiao, X.; Wang, D.; Cai, X. Performance of nano-Co3O4/peroxymonosulfate system: Kinetics and mechanism study using Acid Orange 7 as a model compound. Appl. Catal. B 2008, 80, 116–121. [Google Scholar] [CrossRef]
- Guan, Z.-Y.; Kwon, E.; Lee, J.; Lin, Y.-F.; Lin, K.-Y.A. Electrospun cobalt ferrite nanofiber as a magnetic and effective heterogeneous catalyst for activating peroxymonosulfate to degrade sulfosalicylic acid. Sep. Purif. Technol. 2021, 259, 118163. [Google Scholar] [CrossRef]
- Yun, W.-C.; Lin, K.-Y.A.; Tong, W.-C.; Lin, Y.-F.; Du, Y. Enhanced degradation of paracetamol in water using sulfate radical-based advanced oxidation processes catalyzed by 3-dimensional Co3O4 nanoflower. Chem. Eng. J. 2019, 373, 1329–1337. [Google Scholar] [CrossRef]
- Sun, H.; Ullah, R.; Chong, S.; Ang, H.M.; Tadé, M.O.; Wang, S. Room-light-induced indoor air purification using an efficient Pt/N-TiO2 photocatalyst. Appl. Catal. B 2011, 108, 127–133. [Google Scholar] [CrossRef]
- Matalkeh, M.; Nasrallah, G.K.; Shurrab, F.M.; Al-Absi, E.S.; Mohammed, W.; Elzatahry, A.; Saoud, K.M. Visible Light Photocatalytic Activity of Ag/WO3 Nanoparticles and its Antibacterial Activity Under Ambient Light and in The Dark. Results Eng. 2022, 13, 100313. [Google Scholar] [CrossRef]
- Rahimi-Nasrabadi, M.; Ghaderi, A.; Banafshe, H.R.; Eghbali-Arani, M.; Akbari, M.; Ahmadi, F.; Pourmasoud, S.; Sobhani-Nasab, A. Preparation of Co2TiO4/CoTiO3/Polyaniline ternary nano-hybrids for enhanced destruction of agriculture poison and organic dyes under visible-light irradiation. J. Mater. Sci. Mater. Electron. 2019, 30, 15854–15868. [Google Scholar] [CrossRef]
- Ramezani, M.; Hosseinpour-Mashkani, S.M. Controlled Synthesis, Characterization, and Photocatalytic Application of Co2TiO4 Nanoparticles. J. Electron. Mater. 2017, 46, 1371–1377. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, J.C.; Yip, H.Y.; Li, Q.; Kwong, K.W.; Xu, A.-W.; Wong, P.K. Ambient Light Reduction Strategy to Synthesize Silver Nanoparticles and Silver-Coated TiO2 with Enhanced Photocatalytic and Bactericidal Activities. Langmuir 2003, 19, 10372–10380. [Google Scholar] [CrossRef]
- Andrew Lin, K.-Y.; Zhang, Z.-Y. α-Sulfur as a metal-free catalyst to activate peroxymonosulfate under visible light irradiation for decolorization. RSC Adv. 2016, 6, 15027–15034. [Google Scholar] [CrossRef]
- Lin, K.-Y.A.; Zhang, Z.-Y. Metal-free activation of Oxone using one-step prepared sulfur-doped carbon nitride under visible light irradiation. Sep. Purif. Technol. 2017, 173, 72–79. [Google Scholar] [CrossRef]
- Zhang, M.-W.; Lin, K.-Y.A.; Huang, C.-F.; Tong, S. Enhanced degradation of toxic azo dye, amaranth, in water using Oxone catalyzed by MIL-101-NH2 under visible light irradiation. Sep. Purif. Technol. 2019, 227, 115632. [Google Scholar] [CrossRef]
- Guo, X.; Liang, J.; Wang, L.; Feng, Z.; Yu, T.; Zhang, Z.; Shao, Y.; Hao, C.; Li, G. Synthesis of Cobalt–Glycerate hierarchical structure and their conversion into hierarchical CoP nanospheres for the hydrogen evolution reaction. Int. J. Hydrogen Energy 2018, 43, 2034–2042. [Google Scholar] [CrossRef]
- Yang, Z.K.; Song, L.X.; Teng, Y.; Xia, J. Ethylenediamine-modulated synthesis of highly monodisperse copper sulfide microflowers with excellent photocatalytic performance. J. Mater. Chem. A 2014, 2, 20004–20009. [Google Scholar] [CrossRef]
- Lai, H.-K.; Chou, Y.-Z.; Lee, M.-H.; Lin, K.-Y.A. Coordination polymer-derived cobalt nanoparticle-embedded carbon nanocomposite as a magnetic multi-functional catalyst for energy generation and biomass conversion. Chem. Eng. J. 2018, 332, 717–726. [Google Scholar] [CrossRef]
- Lin, K.-Y.A.; Chang, H.-A.; Chen, R.-C. MOF-derived magnetic carbonaceous nanocomposite as a heterogeneous catalyst to activate oxone for decolorization of Rhodamine B in water. Chemosphere 2015, 130, 66–72. [Google Scholar] [CrossRef]
- Khiem, T.C.; Tuan, D.D.; Kwon, E.; Thanh, B.X.; Tsang, Y.F.; Munagapati, V.S.; Wen, J.-C.; Hu, C.; Lin, K.-Y. Hollow and Oval-Configured Ultrafine Co3O4 as a Highly-Efficient Activator of Monopersulfate for Catalytic Elimination of Azorubin S. Sustain. Environ. Res. 2022. [Google Scholar] [CrossRef]
- Liu, W.-J.; Kwon, E.; Huy, N.N.; Khiem, T.C.; Lisak, G.; Wi-Afedzi, T.; Wu, C.-C.; Ghanbari, F.; Lin, K.-Y.A. Facilely-prepared sulfide-doped Co3O4 nanocomposite as a boosted catalyst for activating Oxone to degrade a sunscreen agent. J. Taiwan Inst. Chem. Eng. 2022, 133, 104253. [Google Scholar] [CrossRef]
- Shafiee, M.R.M.; Parhizkar, J.; Radfar, S. Removal of Rhodamine B by g-C3N4/Co3O4/MWCNT composite stabilized in hydrogel via the synergy of adsorption and photocatalysis under visible light. J. Mater. Sci. Mater. Electron. 2019, 30, 12475–12486. [Google Scholar] [CrossRef]
- Tai, J.Y.; Leong, K.H.; Saravanan, P.; Aziz, A.A.; Sim, L.C. Dopant-free oxygen-rich titanium dioxide: LED light-induced photocatalysis and mechanism insight. J. Mater. Sci. 2017, 52, 11630–11642. [Google Scholar] [CrossRef] [Green Version]
- Kaewkam, P.; Kanchanapaetnukul, A.; Khamyan, J.; Phadmanee, N.; Lin, K.-Y.A.; Kobwittaya, K.; Sirivithayapakorn, S. UV-assisted TiO2 photocatalytic degradation of virgin LDPE films: Effect of UV-A, UV-C, and TiO2. J. Environ. Chem. Eng. 2022, 10, 108131. [Google Scholar] [CrossRef]
- Ningsih, L.A.; Yoshida, M.; Sakai, A.; Lin, K.-Y.A.; Wu, K.C.; Catherine, H.N.; Ahamad, T.; Hu, C. Ag-modified TiO2/SiO2/Fe3O4 sphere with core-shell structure for photo-assisted reduction of 4-nitrophenol. Environ. Res. 2022, 214, 113690. [Google Scholar] [CrossRef]
- Khanahmadzadeh, S.; Enhessari, M.; Solati, Z.; Mohebalizadeh, A.; Alipouramjad, A. Synthesis, characterization and optical band gap of the Co2TiO4 nanoparticles. Mater. Sci. Semicond. Process. 2015, 31, 599–603. [Google Scholar] [CrossRef]
- Jiang, Z.; Lu, W.; Li, Z.; HO, K.H.; Li, X.; Jiao, X.; Chen, D. Synthesis of amorphous cobalt sulfide polyhedral nanocages for high performance supercapacitors. J. Mater. Chem. A 2014, 2, 8603–8606. [Google Scholar] [CrossRef]
- Li, Y.; Li, F.-M.; Meng, X.-Y.; Li, S.-N.; Zeng, J.-H.; Chen, Y. Ultrathin Co3O4 Nanomeshes for the Oxygen Evolution Reaction. ACS Catal. 2018, 8, 1913–1920. [Google Scholar] [CrossRef]
- Hu, P.D.; Long, M.C. Cobalt-catalyzed sulfate radical-based advanced oxidation: A review on heterogeneous catalysts and applications. Appl. Catal. B-Environ. 2016, 181, 103–117. [Google Scholar] [CrossRef]
- Thota, S.; Reehuis, M.; Maljuk, A.; Hoser, A.; Hoffmann, J.U.; Weise, B.; Waske, A.; Krautz, M.; Joshi, D.; Nayak, S.; et al. Neutron diffraction study of the inverse spinels Co2 TiO4 and Co2 SnO4. Phys. Rev. B 2017, 96, 144104. [Google Scholar] [CrossRef]
- Yan, S.C.; Li, Z.S.; Zou, Z.G. Photodegradation of Rhodamine B and Methyl Orange over Boron-Doped g-C3N4 under Visible Light Irradiation. Langmuir 2010, 26, 3894–3901. [Google Scholar] [CrossRef]
- Chen, X.; Wang, W.; Xiao, H.; Hong, C.; Zhu, F.; Yao, Y.; Xue, Z. Accelerated TiO2 photocatalytic degradation of Acid Orange 7 under visible light mediated by peroxymonosulfate. Chem. Eng. J. 2012, 193, 290–295. [Google Scholar] [CrossRef]
- Othman, I.; Hisham Zain, J.; Abu Haija, M.; Banat, F. Catalytic activation of peroxymonosulfate using CeVO4 for phenol degradation: An insight into the reaction pathway. Appl. Catal. B 2020, 266, 118601. [Google Scholar] [CrossRef]
- Tang, W.; Zhang, Y.; Guo, H.; Liu, Y. Heterogeneous activation of peroxymonosulfate for bisphenol AF degradation with BiOI0.5Cl0.5. RSC Adv. 2019, 9, 14060–14071. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Zhang, L.; Chen, M.; Ahmad, F.; Fida, H.; Zhang, H. Heterogeneous Activation of Peroxymonosulfate by a Spinel CoAl2O4 Catalyst for the Degradation of Organic Pollutants. Catalysts 2022, 12, 847. [Google Scholar] [CrossRef]
- Liu, W.-J.; Park, Y.-K.; Chen, W.-H.; Bui, H.M.; Munagapati, V.S.; Tuan, D.D.; Wen, J.-C.; You, S.; Da Oh, W.; Lin, K.-Y.A. Highly-efficient degradation of ensulizole using monopersulfate activated by nanostructured cobalt oxide: A comparative study on effects of different nanostructures. J. Environ. Chem. Eng. 2022, 10, 107137. [Google Scholar] [CrossRef]
- Yin, J.-Y.; Oh, W.D.; Kwon, E.; Thanh, B.X.; You, S.; Wang, H.; Lin, K.-Y.A. Cobalt sulfide nanofilm-assembled cube as an efficient catalyst for activating monopersulfate to degrade UV filter, 4,4′-dihydroxybenzophenone, in water. Colloids Surf. A Physicochem. Eng. Asp. 2021, 625, 126891. [Google Scholar] [CrossRef]
- Lin, K.-Y.A.; Lin, J.-T.; Jochems, A.P. Oxidation of amaranth dye by persulfate and peroxymonosulfate activated by ferrocene. J. Chem. Technol. Biotechnol. 2017, 92, 163–172. [Google Scholar] [CrossRef]
- Lin, K.-Y.A.; Lin, T.-Y. Degradation of Acid Azo Dyes Using Oxone Activated by Cobalt Titanate Perovskite. Water Air Soil Pollut. 2017, 229, 10. [Google Scholar] [CrossRef]
- Lin, K.-Y.A.; Lin, J.-T.; Lin, Y.-F. Heterogeneous catalytic activation of percarbonate by ferrocene for degradation of toxic amaranth dye in water. J. Taiwan Inst. Chem. Eng. 2017, 78, 144–149. [Google Scholar] [CrossRef]
- Tan, C.; Gao, N.; Deng, Y.; Deng, J.; Zhou, S.; Li, J.; Xin, X. Radical induced degradation of acetaminophen with Fe3O4 magnetic nanoparticles as heterogeneous activator of peroxymonosulfate. J. Hazard. Mater. 2014, 276, 452–460. [Google Scholar] [CrossRef]
- Imamović, B.; Trebše, P.; Omeragić, E.; Bečić, E.; Pečet, A.; Dedić, M. Stability and Removal of Benzophenone-Type UV Filters from Water Matrices by Advanced Oxidation Processes. Molecules 2022, 27, 1874. [Google Scholar] [CrossRef]
- Kang, S.; Hwang, J. CoMn2O4 embedded hollow activated carbon nanofibers as a novel peroxymonosulfate activator. Chem. Eng. J. 2021, 406, 127158. [Google Scholar] [CrossRef]
- Guo, W.; Su, S.; Yi, C.; Ma, Z. Degradation of antibiotics amoxicillin by Co3O4-catalyzed peroxymonosulfate system. Environ. Prog. Sustain. Energy 2013, 32, 193–197. [Google Scholar] [CrossRef]
- Xu, L.J.; Chu, W.; Gan, L. Environmental application of graphene-based CoFe2O4 as an activator of peroxymonosulfate for the degradation of a plasticizer. Chem. Eng. J. 2015, 263, 435–443. [Google Scholar] [CrossRef]
- Liang, P.; Zhang, C.; Duan, X.; Sun, H.; Liu, S.; Tade, M.O.; Wang, S. An insight into metal organic framework derived N-doped graphene for the oxidative degradation of persistent contaminants: Formation mechanism and generation of singlet oxygen from peroxymonosulfate. Environ. Sci. Nano 2017, 4, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Li, M.; Wang, C.; Zhang, M.; Nasir Khan, M.A.; Sun, X.; Shen, J.; Han, W.; Wang, L.; Li, J. Singlet oxygen-dominated non-radical oxidation process for efficient degradation of bisphenol A under high salinity condition. Water Res. 2019, 148, 416–424. [Google Scholar] [CrossRef]
- Yang, S.; Wu, P.; Liu, J.; Chen, M.; Ahmed, Z.; Zhu, N. Efficient removal of bisphenol A by superoxide radical and singlet oxygen generated from peroxymonosulfate activated with Fe0-montmorillonite. Chem. Eng. J. 2018, 350, 484–495. [Google Scholar] [CrossRef]
- Hailili, R.; Wang, C.; Lichtfouse, E. Perovskite nanostructures assembled in molten salt based on halogen anions KX (X = F, Cl and Br): Regulated morphology and defect-mediated photocatalytic activity. Appl. Catal. B 2018, 232, 531–543. [Google Scholar] [CrossRef]
- Latch, D.E.; McNeill, K. Microheterogeneity of singlet oxygen distributions in irradiated humic acid solutions. Science 2006, 311, 1743–1747. [Google Scholar] [CrossRef]
- Waclawek, S.; Grubel, K.; Cernik, M. Simple spectrophotometric determination of monopersulfate. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 149, 928–933. [Google Scholar] [CrossRef]
- Tuan, D.D.; Kwon, E.; Phattarapattamawong, S.; Thanh, B.X.; Khiem, T.C.; Lisak, G.; Wang, H.; Lin, K.-Y.A. Nitrogen-containing carbon hollow nanocube-confined cobalt nanoparticle as a magnetic and efficient catalyst for activating monopersulfate to degrade a UV filter in water. J. Environ. Chem. Eng. 2022, 10, 106989. [Google Scholar] [CrossRef]
- Ma, J.; Feng, Y.; Yang, X.; Wu, Y.; Wang, S.; Zhang, C.; Shi, Q. Sulphate radical oxidation of benzophenone: Kinetics, mechanisms and influence of water matrix anions. Environ. Technol. 2021, 42, 4324–4332. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, P.-H.; Khiem, T.C.; Kwon, E.; Chang, H.-C.; Bui, H.M.; Duan, X.; Yang, H.; Ghotekar, S.; Chen, W.-H.; Tsai, Y.-C.; et al. Ambient-Visible-Light-Mediated Enhanced Degradation of UV Stabilizer Bis(4-hydroxyphenyl)methanone by Nanosheet-Assembled Cobalt Titanium Oxide: A Comparative and DFT-Assisted Investigation. Water 2022, 14, 3318. https://doi.org/10.3390/w14203318
Mao P-H, Khiem TC, Kwon E, Chang H-C, Bui HM, Duan X, Yang H, Ghotekar S, Chen W-H, Tsai Y-C, et al. Ambient-Visible-Light-Mediated Enhanced Degradation of UV Stabilizer Bis(4-hydroxyphenyl)methanone by Nanosheet-Assembled Cobalt Titanium Oxide: A Comparative and DFT-Assisted Investigation. Water. 2022; 14(20):3318. https://doi.org/10.3390/w14203318
Chicago/Turabian StyleMao, Po-Hsin, Ta Cong Khiem, Eilhann Kwon, Hou-Chien Chang, Ha Manh Bui, Xiaoguang Duan, Hongta Yang, Suresh Ghotekar, Wei-Hsin Chen, Yu-Chih Tsai, and et al. 2022. "Ambient-Visible-Light-Mediated Enhanced Degradation of UV Stabilizer Bis(4-hydroxyphenyl)methanone by Nanosheet-Assembled Cobalt Titanium Oxide: A Comparative and DFT-Assisted Investigation" Water 14, no. 20: 3318. https://doi.org/10.3390/w14203318
APA StyleMao, P. -H., Khiem, T. C., Kwon, E., Chang, H. -C., Bui, H. M., Duan, X., Yang, H., Ghotekar, S., Chen, W. -H., Tsai, Y. -C., & Lin, K. -Y. A. (2022). Ambient-Visible-Light-Mediated Enhanced Degradation of UV Stabilizer Bis(4-hydroxyphenyl)methanone by Nanosheet-Assembled Cobalt Titanium Oxide: A Comparative and DFT-Assisted Investigation. Water, 14(20), 3318. https://doi.org/10.3390/w14203318