
TLR2 and TLR9 Blockade Using Specific Intrabodies Inhibits Inflammation-Mediated Pancreatic Cancer Cell Growth

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.1.1. Chemicals
2.1.2. Antibodies
2.2. Cell Lines
2.3. Methods
2.3.1. Transfection
2.3.2. Generation of Stable Cells
2.3.3. Cell Proliferation Assay
2.3.4. Live/Dead Cell Staining Using Propidium Iodide (PI)
2.3.5. Brefeldin A Treatment
2.3.6. Immunofluorescence
2.3.7. Quantitation of Immunostaining
2.3.8. Counting of Ki67-Positive Cells and Plotting the Graphs
2.3.9. Colocalization Analysis
2.3.10. Phase Contrast Imaging
2.3.11. Western Blotting
2.3.12. RNA Isolation, cDNA Synthesis, and Quantitative Real-Time PCR
| Component | Amount |
| RNA | x µL |
| 10 mM dNTP | 1 µL |
| Random hexamers (50 ng/µL) | 2 µL |
| DEPC water | to make 10 µL |
| Component | Amount |
| 10× RT buffer | 2 µL |
| 25 mM MgCl2 | 4 µL |
| 0.1 M DTT | 2 µL |
| RNaseOUT™ (ribonuclease inhibitor) | 1 µL |
2.3.13. TaqMan RT-PCR
| Component | Amount |
| TaqMan™ Fast Advanced Master Mix (2×) | 5 µL |
| TaqMan™ Assay (20×) | 0.5 µL |
| Nuclease-free water | 2.5 µL |
| cDNA | 2 µL |
2.3.14. Immunoprecipitation
2.4. Statistical Analyses
3. Results
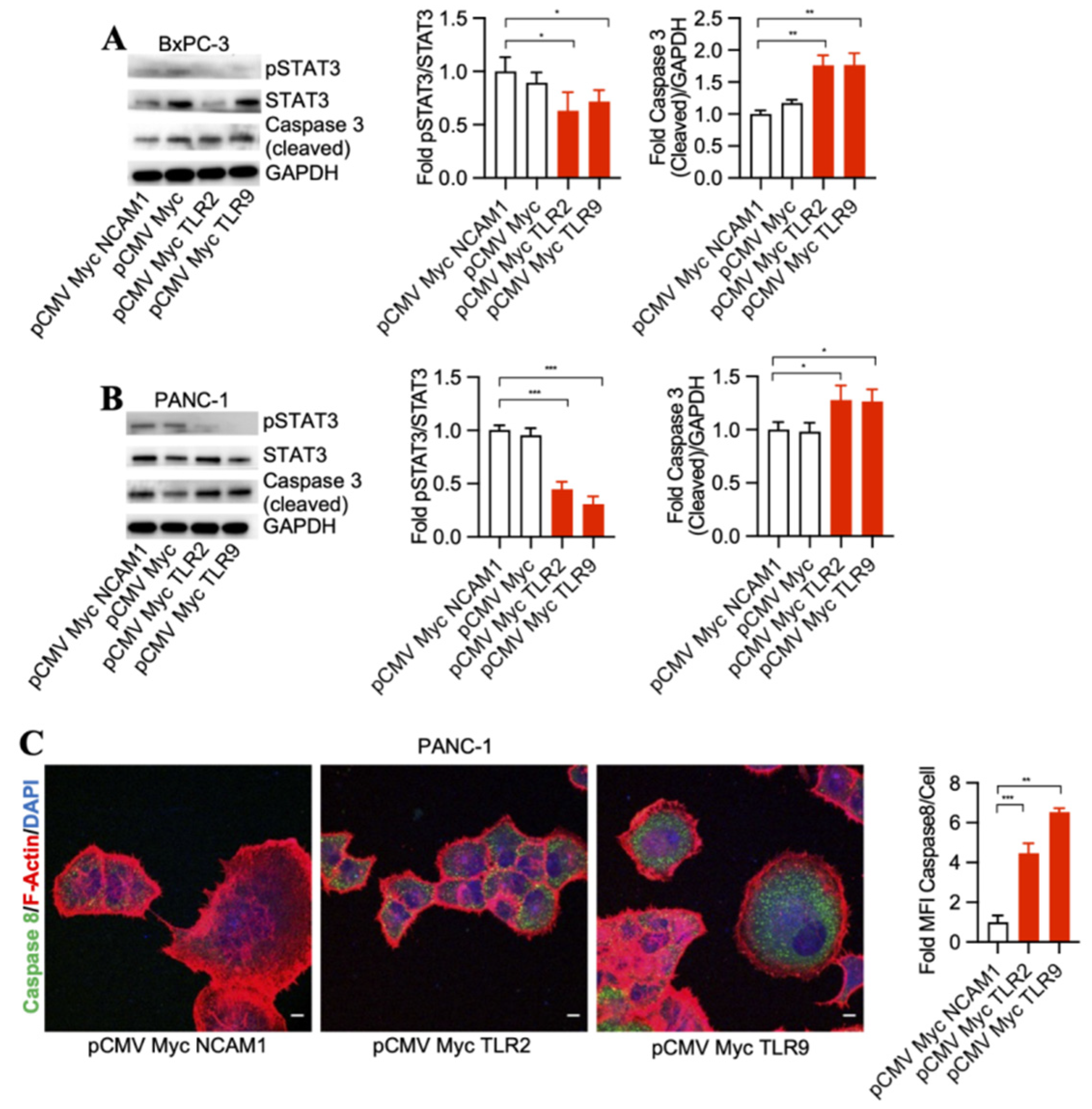
3.1. TLR2 and TLR9 Intrabodies Cause Pancreatic Cancer Cell Death
3.2. TLR2 and TLR9 Intrabody-Mediated Pancreatic Cancer Cell Death through Inhibition of the STAT3 Phosphorylation Pathway
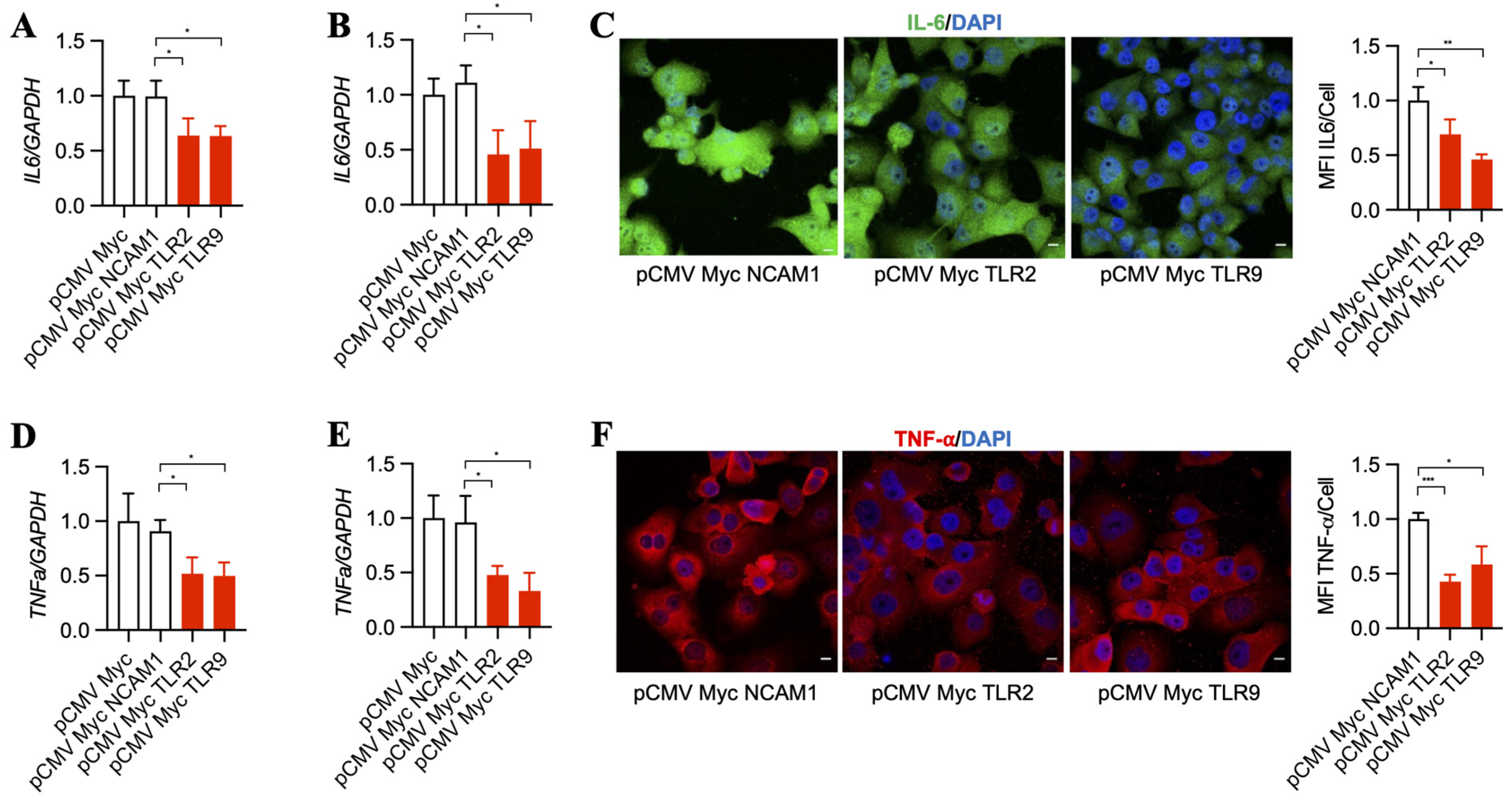
3.3. Inhibition of TLR2 and TLR9 by Its Specific Intrabodies Blocks Inflammatory Mediators IL-6 and TNF-α
3.4. TLR2 and TLR9 Intrabodies Bind to TLR2 and TLR9 Proteins to Retain These Proteins in the Endoplasmic Reticulum
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Katz, M.H.G.; Shi, Q.; Meyers, J.; Herman, J.M.; Chuong, M.; Wolpin, B.M.; Ahmad, S.; Marsh, R.; Schwartz, L.; Behr, S.; et al. Efficacy of Preoperative mFOLFIRINOX vs. mFOLFIRINOX Plus Hypofractionated Radiotherapy for Borderline Resectable Adenocarcinoma of the Pancreas: The A021501 Phase 2 Randomized Clinical Trial. JAMA Oncol. 2022, 8, 1263–1270. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rizzato, C.; Kraft, P.; Kooperberg, C.; Petersen, G.M.; Wang, Z.; Arslan, A.A.; Beane-Freeman, L.; Bracci, P.M.; Buring, J.; et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat. Genet. 2014, 46, 994–1000. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e113. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef]
- West, A.C.; Jenkins, B.J. Inflammatory and non-inflammatory roles for Toll-like receptors in gastrointestinal cancer. Curr. Pharm. Des. 2015, 21, 2968–2977. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 2018, 8, 5072–5087. [Google Scholar] [CrossRef]
- Man, S.M.; Jenkins, B.J. Context-dependent functions of pattern recognition receptors in cancer. Nat. Rev. Cancer 2022, 22, 397–413. [Google Scholar] [CrossRef]
- Vaz, J.; Andersson, R. Intervention on toll-like receptors in pancreatic cancer. World J. Gastroenterol. 2014, 20, 5808–5817. [Google Scholar] [CrossRef]
- Ochi, A.; Graffeo, C.S.; Zambirinis, C.P.; Rehman, A.; Hackman, M.; Fallon, N.; Barilla, R.M.; Henning, J.R.; Jamal, M.; Rao, R.; et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J. Clin. Investig. 2012, 122, 4118–4129. [Google Scholar] [CrossRef]
- Zambirinis, C.P.; Levie, E.; Nguy, S.; Avanzi, A.; Barilla, R.; Xu, Y.; Seifert, L.; Daley, D.; Greco, S.H.; Deutsch, M.; et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J. Exp. Med. 2015, 212, 2077–2094. [Google Scholar] [CrossRef]
- Pratesi, G.; Petrangolini, G.; Tortoreto, M.; Addis, A.; Belluco, S.; Rossini, A.; Selleri, S.; Rumio, C.; Menard, S.; Balsari, A. Therapeutic synergism of gemcitabine and CpG-oligodeoxynucleotides in an orthotopic human pancreatic carcinoma xenograft. Cancer Res. 2005, 65, 6388–6393. [Google Scholar] [CrossRef]
- Hamzah, J.; Altin, J.G.; Herringson, T.; Parish, C.R.; Hammerling, G.J.; O’Donoghue, H.; Ganss, R. Targeted liposomal delivery of TLR9 ligands activates spontaneous antitumor immunity in an autochthonous cancer model. J. Immunol. 2009, 183, 1091–1098. [Google Scholar] [CrossRef]
- Rosa, R.; Melisi, D.; Damiano, V.; Bianco, R.; Garofalo, S.; Gelardi, T.; Agrawal, S.; Di Nicolantonio, F.; Scarpa, A.; Bardelli, A.; et al. Toll-like receptor 9 agonist IMO cooperates with cetuximab in K-ras mutant colorectal and pancreatic cancers. Clin. Cancer Res. 2011, 17, 6531–6541. [Google Scholar] [CrossRef]
- Ochi, A.; Nguyen, A.H.; Bedrosian, A.S.; Mushlin, H.M.; Zarbakhsh, S.; Barilla, R.; Zambirinis, C.P.; Fallon, N.C.; Rehman, A.; Pylayeva-Gupta, Y.; et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J. Exp. Med. 2012, 209, 1671–1687. [Google Scholar] [CrossRef]
- Pu, N.; Chen, Q.; Gao, S.; Liu, G.; Zhu, Y.; Yin, L.; Hu, H.; Wei, L.; Wu, Y.; Maeda, S.; et al. Genetic landscape of prognostic value in pancreatic ductal adenocarcinoma microenvironment. Ann. Transl. Med. 2019, 7, 645. [Google Scholar] [CrossRef]
- Xu, C.; Sui, S.; Shang, Y.; Yu, Z.; Han, J.; Zhang, G.; Ntim, M.; Hu, M.; Gong, P.; Chen, H.; et al. The landscape of immune cell infiltration and its clinical implications of pancreatic ductal adenocarcinoma. J. Adv. Res. 2020, 24, 139–148. [Google Scholar] [CrossRef]
- Grimmig, T.; Moench, R.; Kreckel, J.; Haack, S.; Rueckert, F.; Rehder, R.; Tripathi, S.; Ribas, C.; Chandraker, A.; Germer, C.T.; et al. Toll Like Receptor 2, 4, and 9 Signaling Promotes Autoregulative Tumor Cell Growth and VEGF/PDGF Expression in Human Pancreatic Cancer. Int. J. Mol. Sci. 2016, 17, 2060. [Google Scholar] [CrossRef]
- Human Protein Atlas. Electronic Address. 2023. Available online: https://www.proteinatlas.org (accessed on 1 June 2023).
- Kirschning, C.J.; Dreher, S.; Maass, B.; Fichte, S.; Schade, J.; Koster, M.; Noack, A.; Lindenmaier, W.; Wagner, H.; Boldicke, T. Generation of anti-TLR2 intrabody mediating inhibition of macrophage surface TLR2 expression and TLR2-driven cell activation. BMC Biotechnol. 2010, 10, 31. [Google Scholar] [CrossRef]
- Reimer, E.; Somplatzki, S.; Zegenhagen, D.; Hanel, S.; Fels, A.; Bollhorst, T.; Hovest, L.G.; Bauer, S.; Kirschning, C.J.; Boldicke, T. Molecular cloning and characterization of a novel anti-TLR9 intrabody. Cell Mol. Biol. Lett. 2013, 18, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Popkov, M.; Jendreyko, N.; McGavern, D.B.; Rader, C.; Barbas, C.F. Targeting tumor angiogenesis with adenovirus-delivered anti-Tie-2 intrabody. Cancer Res. 2005, 65, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Jendreyko, N.; Popkov, M.; Rader, C.; Barbas, C.F., 3rd. Phenotypic knockout of VEGF-R2 and Tie-2 with an intradiabody reduces tumor growth and angiogenesis in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 8293–8298. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, H.; Jiang, H.; Namusamba, M.; Wang, C.; Lan, T.; Wang, T.; Wang, B. A BAP31 intrabody induces gastric cancer cell death by inhibiting p27(kip1) proteasome degradation. Int. J. Cancer 2019, 144, 2051–2062. [Google Scholar] [CrossRef] [PubMed]
- Boldicke, T. Therapeutic Potential of Intrabodies for Cancer Immunotherapy: Current Status and Future Directions. Antibodies 2022, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Denecke, J.; De Rycke, R.; Botterman, J. Plant and mammalian sorting signals for protein retention in the endoplasmic reticulum contain a conserved epitope. EMBO J. 1992, 11, 2345–2355. [Google Scholar] [CrossRef] [PubMed]
- Napier, R.M.; Fowke, L.C.; Hawes, C.; Lewis, M.; Pelham, H.R. Immunological evidence that plants use both HDEL and KDEL for targeting proteins to the endoplasmic reticulum. J. Cell Sci. 1992, 102 Pt 2, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Marschall, A.L.; Dubel, S.; Boldicke, T. Specific in vivo knockdown of protein function by intrabodies. mAbs 2015, 7, 1010–1035. [Google Scholar] [CrossRef] [PubMed]
- Ajay, A.K.; Saikumar, J.; Bijol, V.; Vaidya, V.S. Heterozygosity for fibrinogen results in efficient resolution of kidney ischemia reperfusion injury. PLoS ONE 2012, 7, e45628. [Google Scholar] [CrossRef] [PubMed]
- Ajay, A.K.; Kim, T.M.; Ramirez-Gonzalez, V.; Park, P.J.; Frank, D.A.; Vaidya, V.S. A bioinformatics approach identifies signal transducer and activator of transcription-3 and checkpoint kinase 1 as upstream regulators of kidney injury molecule-1 after kidney injury. J. Am. Soc. Nephrol. 2014, 25, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Craciun, F.L.; Ajay, A.K.; Hoffmann, D.; Saikumar, J.; Fabian, S.L.; Bijol, V.; Humphreys, B.D.; Vaidya, V.S. Pharmacological and genetic depletion of fibrinogen protects from kidney fibrosis. Am. J. Physiol. Renal Physiol. 2014, 307, F471–F484. [Google Scholar] [CrossRef] [PubMed]
- Ajay, A.K.; Zhao, L.; Vig, S.; Fujiwara, M.; Thakurela, S.; Jadhav, S.; Cho, A.; Chiu, I.J.; Ding, Y.; Ramachandran, K.; et al. Deletion of STAT3 from Foxd1 cell population protects mice from kidney fibrosis by inhibiting pericytes trans-differentiation and migration. Cell Rep. 2022, 38, 110473. [Google Scholar] [CrossRef]
- Foster, S.L.; Hargreaves, D.C.; Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007, 447, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Gupta, S. TLR1/2, TLR7, and TLR9 signals directly activate human peripheral blood naive and memory B cell subsets to produce cytokines, chemokines, and hematopoietic growth factors. J. Clin. Immunol. 2011, 31, 89–98. [Google Scholar] [CrossRef]
- Yan, J.; Hua, F.; Liu, H.Z.; Yang, H.Z.; Hu, Z.W. Simultaneous TLR2 inhibition and TLR9 activation synergistically suppress tumor metastasis in mice. Acta Pharmacol. Sin. 2012, 33, 503–512. [Google Scholar] [CrossRef]
- Woo, S.R.; Corrales, L.; Gajewski, T.F. Innate immune recognition of cancer. Annu. Rev. Immunol. 2015, 33, 445–474. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Casolino, R.; Braconi, C.; Malleo, G.; Paiella, S.; Bassi, C.; Milella, M.; Dreyer, S.B.; Froeling, F.E.M.; Chang, D.K.; Biankin, A.V.; et al. Reshaping preoperative treatment of pancreatic cancer in the era of precision medicine. Ann. Oncol. 2021, 32, 183–196. [Google Scholar] [CrossRef]
- Connor, A.A.; Gallinger, S. Pancreatic cancer evolution and heterogeneity: Integrating omics and clinical data. Nat. Rev. Cancer 2022, 22, 131–142. [Google Scholar] [CrossRef]
- Di Federico, A.; Mosca, M.; Pagani, R.; Carloni, R.; Frega, G.; De Giglio, A.; Rizzo, A.; Ricci, D.; Tavolari, S.; Di Marco, M.; et al. Immunotherapy in Pancreatic Cancer: Why Do We Keep Failing? A Focus on Tumor Immune Microenvironment, Predictive Biomarkers and Treatment Outcomes. Cancers 2022, 14, 2429. [Google Scholar] [CrossRef]
- Rangelova, E.; Kaipe, H. Immunotherapy in pancreatic cancer-an emerging role: A narrative review. Chin. Clin. Oncol. 2022, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Hsiue, E.H.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef] [PubMed]
- Aslan, M.; Shahbazi, R.; Ulubayram, K.; Ozpolat, B. Targeted Therapies for Pancreatic Cancer and Hurdles Ahead. Anticancer Res. 2018, 38, 6591–6606. [Google Scholar] [CrossRef] [PubMed]
- Hester, R.; Mazur, P.K.; McAllister, F. Immunotherapy in Pancreatic Adenocarcinoma: Beyond “Copy/Paste”. Clin. Cancer Res. 2021, 27, 6287–6297. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, P.; Jugnarain, V.; Vaugrente, O.; Vallet, A.; Boulo, T.; Gauthier, C.; Inoue, A.; Sibille, N.; Gauthier, C.; Jean-Alphonse, F.; et al. A single-domain intrabody targeting the follicle-stimulating hormone receptor impacts FSH-induced G protein-dependent signaling. FEBS Lett. 2023. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Q.; Wang, J.; Wang, C.; Lan, T.; Wang, T.; Wang, B. Knockdown of BAP31 Downregulates Galectin-3 to Inhibit the Wnt/β-Catenin Signaling Pathway to Modulate 5-FU Chemosensitivity and Cancer Stemness in Colorectal Cancer. Int. J. Mol. Sci. 2023, 24, 14402. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wu, Y.; Xiao, T.; Cheng, C.; Zhang, T.; Gao, Z.; Hu, S.; Ren, Z.; Yu, X.; Yang, F.; et al. A specifc anti-cyclin D1 intrabody represses breast cancer cell proliferation by interrupting the cyclin D1–CDK4 interaction. Breast Cancer Res. Treat. 2023, 198, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.; Hu, M.; Ren, H.; Wang, J.; Cheng, X.; Li, R.; Yuan, B.; Balan, Y.; Bai, Z.; Huang, H. VHH212 nanobody targeting the hypoxia-inducible factor 1α suppresses angiogenesis and potentiates gemcitabine therapy in pancreatic cancer in vivo. Cancer Biol. Med. 2021, 18, 772. [Google Scholar] [CrossRef]
- Schmitt, L.C.; Rau, A.; Seifert, O.; Honer, J.; Hutt, M.; Schmid, S.; Zantow, J.; Hust, M.; Dübel, S.; Olayioye, M.A.; et al. Inhibition of HER3 activation and tumor growth with a human antibody binding to a conserved epitope formed by domain III and IV. Mabs 2017, 9, 831–843. [Google Scholar] [CrossRef]
- Russo, G.; Unkauf, T.; Meier, D.; Wenzel, E.V.; Langreder, N.; Kai-Thomas Schneider, K.-T.; Wiesner, R.; Bischoff, R.; Stadler, V.; Dübel, S. In vitro evolution of myc-tag antibodies: In-depth specificity and affinity analysis of Myc1-9E10 and Hyper-Myc. Biol. Chem. 2022, 403, 479–494. [Google Scholar] [CrossRef]
- Yang, J.L.; Pan, X.Y.; Zhao, W.X.; Hu, Q.C.; Ding, F.; Feng, Q.; Li, G.Y.; Luo, Y. The antitumor efficacy of a novel adenovirus-mediated anti-p21Ras single chain fragment variable antibody on human cancers in vitro and in vivo. Int. J. Oncol. 2016, 48, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.M.; Choi, D.K.; Jung, K.; Bae, J.; Kim, J.S.; Park, S.W.; Song, K.H.; Kim, Y.S. Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration. Nat. Commun. 2017, 8, 15090. [Google Scholar] [CrossRef] [PubMed]
- Somplatzki, S.; Muhlenhoff, M.; Kroger, A.; Gerardy-Schahn, R.; Boldicke, T. Intrabodies against the Polysialyltransferases ST8SiaII and ST8SiaIV inhibit Polysialylation of NCAM in rhabdomyosarcoma tumor cells. BMC Biotechnol. 2017, 17, 42. [Google Scholar] [CrossRef]
- Shin, S.M.; Kim, J.S.; Park, S.W.; Jun, S.Y.; Kweon, H.J.; Choi, D.K.; Lee, D.; Cho, Y.B.; Kim, Y.S. Direct targeting of oncogenic RAS mutants with a tumor-specific cytosol-penetrating antibody inhibits RAS mutant-driven tumor growth. Sci. Adv. 2020, 6, eaay2174. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Sikorski, A.F. Editorial focus: Understanding off-target effects as the key to successful RNAi therapy. Cell Mol. Biol. Lett. 2019, 24, 69. [Google Scholar] [CrossRef]
- Vaghari-Tabari, M.; Hassanpour, P.; Sadeghsoltani, F.; Malakoti, F.; Alemi, F.; Qujeq, D.; Asemi, Z.; Yousefi, B. CRISPR/Cas9 gene editing: A new approach for overcoming drug resistance in cancer. Cell Mol. Biol. Lett. 2022, 27, 49. [Google Scholar] [CrossRef]
- Guo, C.; Ma, X.; Gao, F.; Guo, Y. Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 2023, 11, 1143157. [Google Scholar] [CrossRef] [PubMed]
- Nurmi, A.M.; Hagstrom, J.; Mustonen, H.; Seppanen, H.; Haglund, C. The expression and prognostic value of toll-like receptors (TLRs) in pancreatic cancer patients treated with neoadjuvant therapy. PLoS ONE 2022, 17, e0267792. [Google Scholar] [CrossRef] [PubMed]
- Topcu, K.S.B.; Korucu, E.N.; Menevse, E.; Kocak, N.; Duran, T. Investigation of the effects of the toll-like receptor 4 pathway on immune checkpoint vista in pancreatic cancer. Investig. New Drugs 2022, 40, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Goumas, F.A.; Holmer, R.; Egberts, J.H.; Gontarewicz, A.; Heneweer, C.; Geisen, U.; Hauser, C.; Mende, M.M.; Legler, K.; Rocken, C.; et al. Inhibition of IL-6 signaling significantly reduces primary tumor growth and recurrencies in orthotopic xenograft models of pancreatic cancer. Int. J. Cancer 2015, 137, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Okusaka, T.; Ishii, H.; Kyogoku, A.; Yoshimori, M.; Kajimura, N.; Yamaguchi, K.; Kakizoe, T. Elevated serum interleukin-6 levels in patients with pancreatic cancer. Jpn. J. Clin. Oncol. 1998, 28, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Fan, W.; Xu, Z.; Chen, H.; He, Y.; Yang, G.; Yang, G.; Hu, H.; Tang, S.; Wang, P.; et al. Inhibiting tumor necrosis factor-alpha diminishes desmoplasia and inflammation to overcome chemoresistance in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 81110–81122. [Google Scholar] [CrossRef] [PubMed]
- Ikebe, M.; Kitaura, Y.; Nakamura, M.; Tanaka, H.; Yamasaki, A.; Nagai, S.; Wada, J.; Yanai, K.; Koga, K.; Sato, N.; et al. Lipopolysaccharide (LPS) increases the invasive ability of pancreatic cancer cells through the TLR4/MyD88 signaling pathway. J. Surg. Oncol. 2009, 100, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wu, C.; Ma, J.; Yang, Y.; Man, X.; Wu, H.; Li, S. Toll-like receptor 4 promotes angiogenesis in pancreatic cancer via PI3K/AKT signaling. Exp. Cell Res. 2016, 347, 274–282. [Google Scholar] [CrossRef]
- Guo, H.Y.; Cui, Z.J. Extracellular Histones Activate Plasma Membrane Toll-Like Receptor 9 to Trigger Calcium Oscillations in Rat Pancreatic Acinar Tumor Cell AR4-2J. Cells 2018, 8. [Google Scholar] [CrossRef]
- Meng, S.; Li, Y.; Zang, X.; Jiang, Z.; Ning, H.; Li, J. Effect of TLR2 on the proliferation of inflammation-related colorectal cancer and sporadic colorectal cancer. Cancer Cell Int. 2020, 20, 95. [Google Scholar] [CrossRef] [PubMed]
- Brignole, C.; Marimpietri, D.; Di Paolo, D.; Perri, P.; Morandi, F.; Pastorino, F.; Zorzoli, A.; Pagnan, G.; Loi, M.; Caffa, I.; et al. Therapeutic targeting of TLR9 inhibits cell growth and induces apoptosis in neuroblastoma. Cancer Res. 2010, 70, 9816–9826. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Q.; Ma, A.; Li, Y.; Li, R.; Wang, Y. Functional expression of TLR9 in esophageal cancer. Oncol. Rep. 2014, 31, 2298–2304. [Google Scholar] [CrossRef]
- Xie, X.; Ma, L.; Zhou, Y.; Shen, W.; Xu, D.; Dou, J.; Shen, B.; Zhou, C. Polysaccharide enhanced NK cell cytotoxicity against pancreatic cancer via TLR4/MAPKs/NF-kappaB pathway in vitro/vivo. Carbohydr. Polym. 2019, 225, 115223. [Google Scholar] [CrossRef]
- Xu, C.; Zhu, M.; Wang, Q.; Cui, J.; Huang, Y.; Huang, X.; Huang, J.; Gai, J.; Li, G.; Qiao, P.; et al. TROP-2-directed nanobody-drug conjugate elicited potent antitumor effect in pancreatic cancer. J. Nanobiotechnol. 2023, 21, 410. [Google Scholar] [CrossRef]
- Bolesina, N.; Gatti, G.; López de Blanc, S.; Dhooge, S.; Rocha, D.; Fernandez, E.; Ferreyra, R.; Palla, V.; Grupe, V.; Morelatto, R.; et al. Oral squamous cell carcinoma (OSCC) tumors from heavy alcohol consumers are associated with higher levels of TLR9 and a particular immunophenotype: Impact on patient survival. Front. Immunol. 2022, 13, 941667. [Google Scholar] [CrossRef] [PubMed]
- Eskuri, M.; Kemi, N.; Helminen, O.; Huhta, H.; Kauppila, J.H. Toll-like receptors 1, 2, 4, 5, and 6 in gastric cancer. Virchows Arch. 2023, 1–10. [Google Scholar] [CrossRef]
- Mukherjee, S.; Patra, R.; Behzadi, P.; Masotti, A.; Paolini, A.; Sarshar, M. Toll-like receptor-guided therapeutic intervention of human cancers: Molecular and immunological perspectives. Front. Immunol. 2023, 14, 1244345. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, Z.; Zou, S.; Li, F.; Chen, H.; Peng, C.; Deng, X.; Wen, C.; Shen, B.; Zhan, Q. A Novel c-MET-Targeting Antibody-Drug Conjugate for Pancreatic Cancer. Front. Oncol. 2021, 11, 634881. [Google Scholar] [CrossRef]
- Zhou, C.; Yi, C.; Yi, Y.; Qin, W.; Yan, Y.; Dong, X.; Zhang, X.; Huang, Y.; Zhang, R.; Wei, J.; et al. LncRNA PVT1 promotes gemcitabine resistance of pancreatic cancer via activating Wnt/β-catenin and autophagy pathway through modulating the miR-619-5p/Pygo2 and miR-619-Sp ATG 14 axes. Mol. Cancer 2020, 19, 118. [Google Scholar] [CrossRef]
- Jung, J.Y.; Ryu, H.J.; Lee, S.H.; Kim, D.Y.; Kim, M.J.; Lee, E.J.; Ryu, Y.M.; Kim, S.Y.; Kim, K.P.; Choi, E.Y.; et al. Tumor Immunity and SiRNA Nanoparticle Targeting PD-L1 Activates Abrogates Pancreatic Cancer Growth in Humanized Preclinical Model. Cells 2021, 10, 2734. [Google Scholar] [CrossRef] [PubMed]
- Chanda, N.; Kattumuri, V.; Shukla, R.; Zambre, A.; Katti, K.; Upendran, A.; Kulkarni, R.R.; Kan, P.; Fent, G.M.; Casteel, S.W.; et al. Bombesin functionalized gold nanoparticles show in vitro and in vivo cancer receptor specificity. Proc. Natl. Acad. Sci. USA 2010, 107, 8760–8765. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, P.P.G.; Gaglione, S.; Sewastianik, T.; Carrasco, R.D.; Langer, R.; Mitchell, M.J. Nanoparticles for Immune Cytokine TRAIL-Based Cancer therapy. ACS Nano 2018, 12, 912–931. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Assay ID |
|---|---|
| IL-6 | Hs00174131_m1 |
| TNFα1 | Hs00855471_g1 |
| GAPDH | Hs99999905_m1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajay, A.K.; Gasser, M.; Hsiao, L.-L.; Böldicke, T.; Waaga-Gasser, A.M. TLR2 and TLR9 Blockade Using Specific Intrabodies Inhibits Inflammation-Mediated Pancreatic Cancer Cell Growth. Antibodies 2024, 13, 11. https://doi.org/10.3390/antib13010011
Ajay AK, Gasser M, Hsiao L-L, Böldicke T, Waaga-Gasser AM. TLR2 and TLR9 Blockade Using Specific Intrabodies Inhibits Inflammation-Mediated Pancreatic Cancer Cell Growth. Antibodies. 2024; 13(1):11. https://doi.org/10.3390/antib13010011
Chicago/Turabian StyleAjay, Amrendra K., Martin Gasser, Li-Li Hsiao, Thomas Böldicke, and Ana Maria Waaga-Gasser. 2024. "TLR2 and TLR9 Blockade Using Specific Intrabodies Inhibits Inflammation-Mediated Pancreatic Cancer Cell Growth" Antibodies 13, no. 1: 11. https://doi.org/10.3390/antib13010011
APA StyleAjay, A. K., Gasser, M., Hsiao, L. -L., Böldicke, T., & Waaga-Gasser, A. M. (2024). TLR2 and TLR9 Blockade Using Specific Intrabodies Inhibits Inflammation-Mediated Pancreatic Cancer Cell Growth. Antibodies, 13(1), 11. https://doi.org/10.3390/antib13010011