Molecular Engineering of Therapeutic Cytokines

Abstract
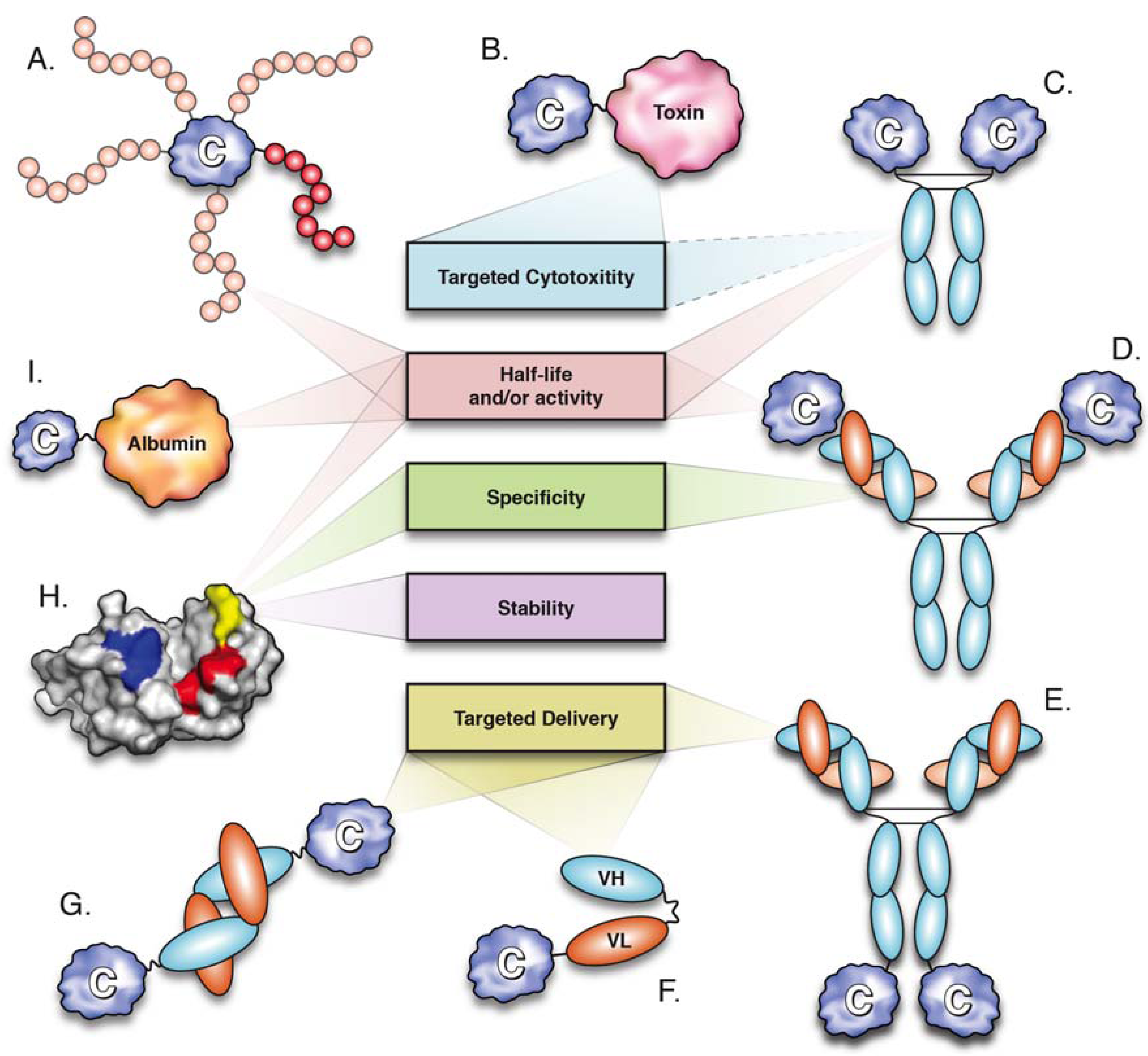
:1. Introduction
2. Cytokine Toxicity

{kind=link}
| Cytokine | Main biological functions | Indications | Status | Brand name, ClinicalTrials.gov ID or EudraCT No. |
|---|---|---|---|---|
| IL-1Ra | Binds IL-1 receptor without inducing signalling, natural IL-1 antagonist | Rheumatoid arthritis, neonatal-onset multisystem inflammatory disease | Approved | Kineret (Anakinra) |
| IL-2 | T-cell generation, homeostasis and proliferation; stimulates NK cells. | Metastatic melanoma, renal cell carcinoma | Approved | Proleukin (Aldesleukin) |
| IL-7 | Development of pre-B cells and pre-T-cells | Solid tumours, HIV-induced CD4 lymphopenia, HIV-related PML, T-cell lymphopenia after stem cell transplant | Phase II | NCT00062049, NCT01190111, NCT01190111, NCT00684008, 2010-019773-15, 2012-000725-41 |
| IL-10 | Inhibition of pro-inflammatory cytokine production by macrophages | Psoriasis, scar reduction, Wegener’s granulomatosis | Phase I, II | NCT00001761, NCT00001761, NCT00001761, 2004-001455-11 |
| IL-11 | Promotes haematopoiesis, synergises with IL-3 and IL-4 | Prevention of severe thrombocytopenia after myelosuppressive chemotherapy | Approved | Neumega (Oprelvekin) |
| IL-12 | NK cell activation, polarisation towards development of TH1 CD4+ cells | Ovarian cancer, metastatic melanoma, cutaneous T-cell non-Hodgkin lymphoma and other cancers; HIV-induced CD4 lymphopenia | Phase I, II | NCT00016289, NCT00026143 NCT00052377, NCT00000857 |
| IL-15 | Similar to IL-2, stimulates T-cells and NK cells | Metastatic melanoma, renal cell carcinoma and other cancers | Phase I, I/II | NCT00001761, NCT01021059, NCT01572493, NCT01369888 |
| IL-21 | Induction of B-cells, T-cells and NK cells | Metastatic melanoma, metastatic renal cancer, ovarian cancer, solid tumours | Phase I, II | NCT00336986, NCT00095108, NCT00523380, NCT01629758, 2006-005350-79 |
| IFN-α | Leukocyte interferon, anti-proliferative and anti-viral cytotoxic activity, increased expression of MHC class I antigen | Hairy cell leukaemia, AIDS-related Kaposi's sarcoma, hepatitis B/C, follicular lymphoma, malignant melanoma | Approved | Infergen, Intron-A, Roferon A |
| IFN-β | Fibroblast interferon, similar activity to IFN-α | Relapsing multiple sclerosis | Approved | Avonex, Betaseron, Extavia, Rebif |
| IFN-γ | Modulates T-cell growth and differentiation; promotes development of TH1 CD4+ cells | Chronic granulomatous disease, malignant osteoporosis | Approved | Actinmmune, Imukin |
| G-CSF | Stimulates neutrophil development and differentiation | Chemotherapy-induced neutropenia | Approved | Granocyte (Lenograstim), Neupogen (Filgrastim) |
| GM-CSF | Growth and development of macrophage and granulocyte precursors | Neutropenia under chemotherapy and bone-marrow transplant | Approved | Leucomax (Molgramostim), Leukine (Sargramostim) |
| TNF-α | Promotes inflammation, inhibition of tumorigenesis | Soft tissue sarcoma of the limb—administered via isolated limb perfusion | Approved | Beromun (Tasonermin) |
| TRAP | Induction of T-cell dependent B-cell activation, promotes antibody class-switching, T-cell priming | X-linked hyper IgM syndrome, metastatic melanoma, metastatic kidney cancer | Phase I, II | NCT00001145, NCT00053391, NCT00020540, 2010-023103-94 |
| TRAIL | Apoptosis of tumour cells and activated T-cells | Metastatic colorectal cancer, non-Hodgkin lymphoma | Phase I | NCT00873756, NCT00400764 |
| Modification /cytokine | Property | Indications | Status | Drug name(s), ClinicaTrials.gov or EudraCT ID | |
|---|---|---|---|---|---|
| PEGylation | |||||
| IFN-α2a | Chronic hepatitis B/C | Approved | Pegasys (peginterferon alfa-2a) | ||
| IFN-α2b | Extended half-life | Chronic hepatitis C | Approved | Pegintron, Sylatron (peginterferon alfa-2b) | |
| G-CSF | Chemotherapy-induced neutropenia | Approved | Neulasta (pegfilgrastim) | ||
| IL-29 (IFN-λ1) | Chronic hepatitis C | Phase I | NCT00565539 | ||
| Fusion to albumin | |||||
| IFN-α2b | Extended half-life | Chronic hepatitis C | Phase III | Albinterferon Alfa-2b | |
| Fusion to toxins | |||||
| IL-2-DT | Targeted cytotoxicity | Cutaneous T-cell lymphoma | Approved | Ontak (denileukin diftitox) | |
| IL-13-PE | Glioblastoma multiform, malignant glioma | Phase I, III | Cintredekin besudotox—NCT00880061, NCT00076986 | ||
| Fusion to Abs (Immunocytokines) | |||||
| F16-IL2 | Targets A1 domain of tenascin C | Breast cancer, solid tumours | Phase II | NCT01134250 | |
| L19-12 | Targets fibronectin EDB splice variant | Metastatic melanoma, renal cell carcinoma, pancreatic cancer and solid tumours | Phase I, II | NCT01058538, NCT01198522, NCT01253096, NCT01055522 | |
| hu14.18-IL2 | Targets GD2 disialoganglioside | Metastatic melanoma, neuroblastoma | Phase II | NCT00590824, NCT00082758, NCT00109863 | |
| KS-IL2 | Targets EpCAM | Various cancers | Phase I | Tucotuzumab celmoleukin - NCT00132522 | |
| NHS-IL2LT | Targets DNA/ histones in necroses | Lung cancer, non-Hodgkin lymphoma | Phase I | Selectikine - NCT00879866, NCT01032681 | |
| NHS-IL12 | Targets DNA/ histones in necroses | Epithelial and mesenchymal solid tumours | Phase I | NCT01417546 | |
| BC1-IL12 | Targets fibronectin EDB splice variant | Metastatic melanoma and renal cell carcinoma | Phase I | NCT00625768 | |
| L19-TNFα | Targets fibronectin EDB splice variant | Solid tumours, colorectal cancer, melanoma of the lower limb | Phase I, II | NCT01253837, NCT01213732 | |
| F8-IL10 | Targets fibronectin EDA splice variant | Rheumatoid arthritis | Phase I | 2008-008729-31 (PH-F8IL10-02/08) | |
| Mutagenesis | |||||
| IL-2 | Mutation of free-cysteine to reduce aggregation | Metastatic melanoma and renal cell carcinoma | Approved | Proleukin (aldesleukin) | |
| IL-4 | Disruption of receptor signalling-subunit binding to generate antagonist | Asthma and atopic eczema | Phase II | Aerovant (pitrakinra) - NCT00801853, NCT00535431, NCT00676884 | |
3. Cytokine PEGylation

4. Antibody-IL-2 Immune Complexes
5. Cytokine Fusion Proteins
5.1. Fusion of Cytokines to the Fc Domain
5.2. Fusion to Serum Albumin
5.3. Fusion to Human Transferrin
5.4. Fusion to Agonistic Receptors
5.6. Fusion to Toxins
5.7. Fusion to Antibodies and Antibody Fragments (Immunocytokines)
5.7.1. Target Antigens
5.7.2. Antibody Format
5.7.3. Cytokine Partners
6. Cytokine Mutagenesis
6.1. Stability
6.2. Half-Life Extension
6.3. Modulation of Specificity
6.4. Development of Cytokine Antagonists
7. Concluding Remarks
Acknowledgements
Conflict of Interest
References
- Dinarello, C.A. IL-1: Discoveries, controversies and future directions. Eur. J. Immunol. 2010, 40, 599–606. [Google Scholar] [CrossRef]
- Beeson, P.B. Temperature-elevating effect of a substance obtained from polymorphonuclear leucocytes. J. Clin. Invest. 1948, 27, 524–524. [Google Scholar]
- Morgan, D.A.; Ruscetti, F.W.; Gallo, R. Selective in vitro growth of T-lymphocytes from normal human bone marrows. Science 1976, 193, 1007–1008. [Google Scholar]
- Gillis, S.; Smith, K.A. Long-term culture of tumor-specific cytotoxic T-cells. Nature 1977, 268, 154–156. [Google Scholar] [CrossRef]
- Gordon, J.; Maclean, L.D. A lymphocyte-stimulating factor produced in vitro. Nature 1965, 208, 795–796. [Google Scholar] [CrossRef]
- Isaacs, A.; Lindenmann, J. Virus interference. 1. The interferon. Proc. Royal Soc. B 1957, 147, 258–267. [Google Scholar] [CrossRef]
- Pestka, S. The interferons: 50 years after their discovery, there is much more to learn. J. Biol. Chem. 2007, 282, 20047–20051. [Google Scholar] [CrossRef]
- Cantell, K.; Hirvonen, S.; Kauppinen, H.L.; Myllyla, G. Production of interferon in human leukocytes from normal donors with the use of Sendai virus. Methods Enzymol. 1981, 78, 29–38. [Google Scholar] [CrossRef]
- Goeddel, D.V.; Yelverton, E.; Ullrich, A.; Heyneker, H.L.; Miozzari, G.; Holmes, W.; Seeburg, P.H.; Dull, T.; May, L.; Stebbing, N.; et al. Human-leukocyte interferon produced by Escherichia-coli is biologically-active. Nature 1980, 287, 411–416. [Google Scholar] [CrossRef]
- Nagata, S.; Taira, H.; Hall, A.; Johnsrud, L.; Streuli, M.; Ecsodi, J.; Boll, W.; Cantell, K.; Weissmann, C. Synthesis in Escherichia-coli of a polypeptide with human-leukocyte interferon activity. Nature 1980, 284, 316–320. [Google Scholar] [CrossRef]
- Devos, R.; Plaetinck, G.; Cheroutre, H.; Simons, G.; Degrave, W.; Tavernier, J.; Remaut, E.; Fiers, W. Molecular-cloning of human interleukin-2 carrier DNA and its expression in Escherichia-coli. Nucleic Acids Res. 1983, 11, 4307–4323. [Google Scholar] [CrossRef]
- Taniguchi, T.; Matsui, H.; Fujita, T.; Takaoka, C.; Kashima, N.; Yoshimoto, R.; Hamuro, J. Structure and expression of a cloned cDNA for human interleukin-2. Nature 1983, 302, 305–310. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef]
- Boyman, O.; Surh, C.D.; Sprent, J. Potential use of IL-2/anti-IL-2 antibody immune complexes for the treatment of cancer and autoimmune disease. Expert Opin. Biol. Ther. 2006, 6, 1323–1331. [Google Scholar] [CrossRef]
- Atkins, M.B.; Lotze, M.T.; Dutcher, J.P.; Fisher, R.I.; Weiss, G.; Margolin, K.; Abrams, J.; Sznol, M.; Parkinson, D.; Hawkins, M.; et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999, 17, 2105–2116. [Google Scholar]
- Rosenberg, S.A.; Yang, J.C.; White, D.E.; Steinberg, S.M. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2—Identification of the antigens mediating response. Ann. Surg. 1998, 228, 307–317. [Google Scholar] [CrossRef]
- Jevševar, S.; Kunstelj, M.; Porekar, V.G. PEGylation of therapeutic proteins. Biotechnol. J. 2010, 5, 113–128. [Google Scholar]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef]
- Maack, T.; Johnson, V.; Kau, S.T.; Figueiredo, J.; Sigulem, D. Renal filtration, transport, and metabolism of low-molecular-weight proteins - Review. Kidney Int. 1979, 16, 251–270. [Google Scholar] [CrossRef]
- Crawford, J. Once-per-cycle pegfilgrastim (Neulasta) for the management of chemotherapy-induced neutropenia. Semin. Oncol. 2003, 30, 24–30. [Google Scholar] [CrossRef]
- Bukowski, R.M.; Tendler, C.; Cutler, D.; Rose, E.; Laughlin, M.M.; Statkevich, P. Treating cancer with PEG intron—Pharmacokinetic profile and dosing guidelines for an improved interferon-alpha-2b formulation. Cancer 2002, 95, 389–396. [Google Scholar] [CrossRef]
- Perry, C.M.; Jarvis, B. Peginterferon-alpha-2a (40kD)—A review of its use in the management of chronic hepatitis C. Drugs 2001, 61, 2263–2288. [Google Scholar] [CrossRef]
- Glue, P.; Fang, J.W.S.; Rouzier-Panis, R.; Raffanel, C.; Sabo, R.; Gupta, S.K.; Salfi, M.; Jacobs, S.; Hepatitis, C.I.T.G. Pegylated interferon-alpha 2b: Pharmacokinetics, pharmacodynamics, safety, and preliminary efficacy data. Clin. Pharmacol. Ther. 2000, 68, 556–567. [Google Scholar] [CrossRef]
- Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Parkinson, D.R.; Marincola, F.M.; Weber, J.S.; Seipp, C.A.; White, D.E.; Rosenberg, S.A. The use of polyethylene glycol-modified interleukin-2 (PEG-IL-2) in the treatment of patients with metastatic renal-cell carcinoma and melanoma—A phase-I study and a randomised prospective study comparing IL-2 alone versus IL-2 combined with PEG-IL-2. Cancer 1995, 76, 687–694. [Google Scholar]
- Katre, N.V.; Knauf, M.J.; Laird, W.J. Chemical modification of recombinant interleukin-2 by polyethilene-glycol increases its potency in the murine Meth-A sarcoma model. Proc. Natl. Acad. Sci. USA 1987, 84, 1487–1491. [Google Scholar] [CrossRef]
- Klein, B.; Brailly, H. Cytokine-binding proteins—Stimulating antagonists. Immunol. Today 1995, 16, 216–220. [Google Scholar] [CrossRef]
- Sato, J.; Hamaguchi, N.; Doken, K.; Gotoh, K.; Ootsu, K.; Iwasa, S.; Ogawa, Y.; Toguchi, H. Enhancement of antitumor-activity of recombinant interleukin-2 (rIL-2) by immunocomplexing with a monoclonal-antibody against rIL-2. Biotherapy 1993, 6, 225–231. [Google Scholar] [CrossRef]
- Rosenblum, M.G.; Unger, B.W.; Gutterman, J.U.; Hersh, E.M.; David, G.S.; Frincke, J.M. Modification of human-leukocyte interferon pharmacology with a monoclonal-antibody. Cancer Res. 1985, 45, 2421–2424. [Google Scholar]
- Finkelman, F.D.; Madden, K.B.; Morris, S.C.; Holmes, J.M.; Boiani, N.; Katona, I.M.; Maliszewski, C.R. Anti-cytokine antibodies as carrier proteins—Prolongation of in-vivo effects of exogenous cytokines by injection of cytokine anti-cytokine antibody complexes. J. Immunol. 1993, 151, 1235–1244. [Google Scholar]
- Boyman, O.; Kovar, M.; Rubinstein, M.P.; Surh, C.D.; Sprent, J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006, 311, 1924–1927. [Google Scholar] [CrossRef]
- Schein, C.H. The shape of the messenger: Using protein structure information to design novel cytokine-based therapeutics. Curr. Pharm. Des. 2002, 8, 2113–2129. [Google Scholar] [CrossRef]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar]
- Krieg, C.; Letourneau, S.; Pantaleo, G.; Boyman, O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11906–11911. [Google Scholar] [CrossRef]
- Tomala, J.; Chmelova, H.; Mrkvan, T.; Rihova, B.; Kovar, M. In Vivo Expansion of Activated Naive CD8(+) T Cells and NK Cells Driven by Complexes of IL-2 and Anti-IL-2 Monoclonal Antibody As Novel Approach of Cancer Immunotherapy. J. Immunol. 2009, 183, 4904–4912. [Google Scholar] [CrossRef]
- Kamimura, D.; Sawa, Y.; Sato, M.; Agung, E.; Hirano, T.; Murakami, M. IL-2 in vivo activities and antitumor efficacy enhanced by an anti-IL-2 mAb. J. Immunol. 2006, 177, 306–314. [Google Scholar]
- Webster, K.E.; Walters, S.; Kohler, R.E.; Mrkvan, T.; Boyman, O.; Surh, C.D.; Grey, S.T.; Sprent, J. In vivo expansion of T reg cells with IL-2-mAb complexes: Induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J. Exp. Med. 2009, 206, 751–760. [Google Scholar] [CrossRef]
- Tang, Q.; Adams, J.Y.; Penaranda, C.; Melli, K.; Piaggio, E.; Sgouroudis, E.; Piccirillo, C.A.; Salomon, B.L.; Bluestone, J.A. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 2008, 28, 687–697. [Google Scholar] [CrossRef]
- Liu, R.; Zhou, Q.; La Cava, A.; Campagnolo, D.I.; Van Kaer, L.; Shi, F.-D. Expansion of regulatory T cells via IL-2/anti-IL-2 mAb complexes suppresses experimental myasthenia. Eur. J. Immunol. 2010, 40, 1577–1589. [Google Scholar] [CrossRef]
- Wilson, M.S.; Pesce, J.T.; Ramalingam, T.R.; Thompson, R.W.; Cheever, A.; Wynn, T.A. Suppression of Murine Allergic Airway Disease by IL-2:Anti-IL-2 Monoclonal Antibody-Induced Regulatory T Cells. J. Immunol. 2008, 181, 6942–6954. [Google Scholar]
- Tam Nguyen, D.; Kyaw, T.S.; Kanellakis, P.; To, K.; Tipping, P.; Toh, B.-H.; Bobik, A.; Agrotis, A. Cytokine Therapy With Interleukin-2/Anti-Interleukin-2 Monoclonal Antibody Complexes Expands CD4+CD25+Foxp3+ Regulatory T Cells and Attenuates Development and Progression of Atherosclerosis. Circulation 2012, 126, 1256–1266. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Letourneau, S.; van Leeuwen, E.M.; Krieg, C.; Martin, C.; Pantaleo, G.; Sprent, J.; Surh, C.D.; Boyman, O. IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor alpha subunit CD25. Proc. Natl. Acad. Sci. USA 2010, 107, 2171–2176. [Google Scholar] [CrossRef]
- Carter, P.J. Introduction to current and future protein therapeutics: A protein engineering perspective. Exp. Cell Res. 2011, 317, 1261–1269. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Hu, J.; Shao, Z.; Pleass, R.J. Fc-fusion proteins: New developments and future perspectives. EMBO Mol. Med. 2012, 4, 1015–1028. [Google Scholar] [CrossRef]
- Bitonti, A.J.; Dumont, J.A.; Low, S.C.; Peters, R.T.; Kropp, K.E.; Palombella, V.J.; Stattel, J.M.; Lu, Y.C.; Tan, C.A.; Song, J.J.; et al. Pulmonary delivery of an erythropoietin Fc fusion protein in non-human primates through an immunoglobulin transport pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 9763–9768. [Google Scholar] [CrossRef]
- Dumont, J.A.; Bitonti, A.J.; Clark, D.; Evans, S.; Pickford, M.; Newman, S.P. Delivery of an erythropoietin-Fc fusion protein by inhalation in humans through an immunoglobulin transport pathway. J. Aerosol Med. 2005, 18, 294–303. [Google Scholar] [CrossRef]
- Chang, C.H.; Gupta, P.; Goldenberg, D.M. Advances and challenges in developing cytokine fusion proteins as improved therapeutics. Expert Opin. Drug Discov. 2009, 4, 181–194. [Google Scholar] [CrossRef]
- Strohl, W.R. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr. Opin. Biotechnol. 2009, 20, 685–691. [Google Scholar] [CrossRef]
- Shoji-Hosaka, E.; Kobayashi, Y.; Wakitani, M.; Uchida, K.; Niwa, R.; Nakamura, K.; Shitara, K. Enhanced Fc-dependent cellular cytotoxicity of Fc fusion proteins derived from TNF receptor II and LFA-3 by fucose removal from Asn-linked oligosaccharides. J. Biochem. (Tokyo) 2006, 140, 777–783. [Google Scholar] [CrossRef]
- Davis, P.M.; Abraham, R.; Xu, L.; Nadler, S.G.; Suchard, S.J. Abatacept binds to the fc receptor CD64 but does not mediate complement-dependent cytotoxicity or antibody-dependent cellular cytotoxicity. J. Rheumatol. 2007, 34, 2204–2210. [Google Scholar]
- McGuire, H.M.; Walters, S.; Vogelzang, A.; Lee, C.M.Y.; Webster, K.E.; Sprent, J.; Christ, D.; Grey, S.; King, C. Interleukin-21 Is Critically Required in Autoimmune and Allogeneic Responses to Islet Tissue in Murine Models. Diabetes 2011, 60, 867–875. [Google Scholar] [CrossRef]
- Bubier, J.A.; Bennett, S.M.; Sproule, T.J.; Lyons, B.L.; Olland, S.; Young, D.A.; Roopenian, D.C. Treatment of BXSB-Yaa mice with IL-21R-Fc fusion protein minimally attenuates systemic lupus erythematosus. Ann. NY Acad. Sci. 2007, 1110, 590–601. [Google Scholar]
- Young, D.A.; Hegen, M.; Margery Ma, H.L.; Whitters, M.J.; Albert, L.M.; Lowe, L.; Senices, M.; Wu, P.W.; Sibley, B.; Leathurby, Y.; et al. Blockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum. 2007, 56, 1152–1163. [Google Scholar] [CrossRef]
- Feng, X.M.; Zheng, X.X.; Yi, S.N.; Lehnert, A.M.; Strom, T.B.; O'Connell, P.J. IL-10/Fc inhibits macrophage function and prolongs pancreatic islet xenograft survival. Transplantation 1999, 68, 1775–1783. [Google Scholar] [CrossRef]
- FloresVillanueva, P.O.; Zheng, X.X.; Strom, T.B.; Stadecker, M.J. Recombinant IL-10 and IL-10/Fc treatment down-regulate egg antigen-specific delayed hypersensitivity reactions and egg granuloma formation in schistosomiasis. J. Immunol. 1996, 156, 3315–3320. [Google Scholar]
- Zheng, X.X.; Steele, A.W.; Hancock, W.W.; Stevens, A.C.; Nickerson, P.W.; RoyChaudhury, P.; Tian, Y.; Strom, T.B. A noncytolytic IL-10/Fc fusion protein prevents diabetes, blocks autoimmunity, and promotes suppressor phenomena in NOD mice. J. Immunol. 1997, 158, 4507–4513. [Google Scholar]
- Zheng, X.X.; Steele, A.W.; Nickerson, P.W.; Steurer, W.; Steiger, J.; Strom, T.B. Administration of noncytolytic IL-10/Fc in murine models of lipopolysaccharide-induced septic shock and allogeneic islet transplantation. J. Immunol. 1995, 154, 5590–5600. [Google Scholar]
- Schwager, K.; Kaspar, M.; Bootz, F.; Marcolongo, R.; Paresce, E.; Neri, D.; Trachsel, E. Preclinical characterization of Dekavil (F8-IL10), a novel clinical-stage immunocytokine which inhibits the progression of collagen-induced arthritis. Arthritis Res. Ther. 2009, 11. Article No. R142. [Google Scholar]
- Zheng, X.X.; Steele, A.W.; Hancock, W.W.; Kawamoto, K.; Li, X.C.; Nickerson, P.W.; Li, Y.S.; Tian, Y.; Strom, T.B. IL-2 receptor-targeted cytolytic IL-2/Fc fusion protein treatment blocks diabetogenic autoimmunity in nonobese diabetic mice. J. Immunol. 1999, 163, 4041–4048. [Google Scholar]
- Nam, H.J.; Song, M.-Y.; Choi, D.-H.; Yang, S.-H.; Jin, H.-T.; Sung, Y.-C. Marked enhancement of antigen-specific T-cell responses by IL-7-fused nonlytic, but not lytic, Fc as a genetic adjuvant. Eur. J. Immunol. 2010, 40, 351–358. [Google Scholar] [CrossRef]
- Anderson, C.L.; Chaudhury, C.; Kim, J.; Bronson, C.L.; Wani, M.A.; Mohanty, S. Perspective - FcRn transports albumin: Relevance to immunology and medicine. Trends Immunol. 2006, 27, 343–348. [Google Scholar] [CrossRef]
- Osborn, B.L.; Gu, M.; Grzegorzewski, K.J.; Logan, T.F.; Crowder, K.; Weiss, G.R.; Syed, S.; Rowensky, E.; Tolcher, A.; Agarwala, S.S. Preliminary pharmacokinetic evaluation of Albuleukin; an interleukin-2 human serum albumin fusion protein, in solid tumor patients. Proc. Amer. Assoc. Cancer Res. 2004, 2004, 1099. [Google Scholar]
- Halpern, W.; Riccobene, T.A.; Agostini, H.; Baker, K.; Stolow, D.; Gu, M.L.; Hirsch, J.; Mahoney, A.; Carrell, J.; Boyd, E.; et al. Albugranin (TM), a recombinant human granulocyte colony stimulating factor (G-CSF) genetically fused to recombinant human albumin induces prolonged myelopoietic effects in mice and monkeys. Pharm. Res. 2002, 19, 1720–1729. [Google Scholar] [CrossRef]
- Nelson, D.R.; Benhamou, Y.; Chuang, W.-L.; Lawitz, E.J.; Rodriguez-Torres, M.; Flisiak, R.; Rasenack, J.W.F.; Kryczka, W.; Lee, C.-M.; Bain, V.G.; et al. Albinterferon Alfa-2b Was Not Inferior to Pegylated Interferon-alpha in a Randomized Trial of Patients With Chronic Hepatitis C Virus Genotype 2 or 3. Gastroenterology 2010, 139, 1267–1276. [Google Scholar] [CrossRef]
- Subramanian, G.M.; Fiscella, M.; Lamouse-Smith, A.; Zeuzem, S.; McHutchison, J.G. Albinterferon alpha-2b: A genetic fusion protein for the treatment of chronic hepatitis C. Nat. Biotechnol. 2007, 25, 1411–1419. [Google Scholar] [CrossRef]
- Zeuzem, S.; Sulkowski, M.S.; Lawitz, E.J.; Rustgi, V.K.; Rodriguez-Torres, M.; Bacon, B.R.; Grigorescu, M.; Tice, A.D.; Lurie, Y.; Cianciara, J.; et al. Albinterferon Alfa-2b Was Not Inferior to Pegylated Interferon-alpha in a Randomized Trial of Patients With Chronic Hepatitis C Virus Genotype 1. Gastroenterology 2010, 139, 1257–1266. [Google Scholar] [CrossRef]
- Sung, C.; Nardelli, B.; LaFleur, D.W.; Blatter, E.; Corcoran, M.; Olsen, H.S.; Birse, C.E.; Pickeral, O.K.; Zhang, J.; Shah, D. An IFN-β-albumin fusion protein that displays improved pharmacokinetic and pharmacodynamic properties in nonhuman primates. J. Int. Cytokine Res. 2003, 23, 25–36. [Google Scholar] [CrossRef]
- Ward, E.S.; Gussow, D.; Griffiths, A.D.; Jones, P.T.; Winter, G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia-coli. Nature 1989, 341, 544–546. [Google Scholar] [CrossRef]
- Holt, L.J.; Basran, A.; Jones, K.; Chorlton, J.; Jespers, L.S.; Brewis, N.D.; Tomlinson, I.M. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng. Des. Sel. 2008, 21, 283–288. [Google Scholar] [CrossRef]
- Walker, A.; Dunlevy, G.; Rycroft, D.; Topley, P.; Holt, L.J.; Herbert, T.; Davies, M.; Cook, F.; Holmes, S.; Jespers, L.; et al. Anti-serum albumin domain antibodies in the development of highly potent, efficacious and long-acting interferon. Protein Eng. Des. Sel. 2010, 23, 271–278. [Google Scholar] [CrossRef]
- Qian, Z.M.; Li, H.Y.; Sun, H.Z.; Ho, K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol. Rev. 2002, 54, 561–587. [Google Scholar] [CrossRef]
- Daniels, T.R.; Delgado, T.; Helguera, G.; Penichet, M.L. The transferrin receptor part II: Targeted delivery of therapeutic agents into cancer cells. Clin. Immunol. 2006, 121, 159–176. [Google Scholar] [CrossRef]
- Daniels, T.R.; Delgado, T.; Rodriguez, J.A.; Helguera, G.; Penichet, M.L. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin. Immunol. 2006, 121, 144–158. [Google Scholar] [CrossRef]
- Kim, B.-J.; Zhou, J.; Martin, B.; Carlson, O.D.; Maudsley, S.; Greig, N.H.; Mattson, M.P.; Ladenheim, E.E.; Wustner, J.; Turner, A.; et al. Transferrin Fusion Technology: A Novel Approach to Prolonging Biological Half-Life of Insulinotropic Peptides. J. Pharmacol. Exp. Ther. 2010, 334, 682–692. [Google Scholar] [CrossRef]
- Banerjee, D.; Flanagan, P.R.; Cluett, J.; Valberg, L.S. Transferrin receptors in the human gastrointestinal-tract - relationship to body iron stores. Gastroenterology 1986, 91, 861–869. [Google Scholar]
- Bai, Y.; Ann, D.K.; Shen, W.C. Recombinant granulocyte colony-stimulating factor-transferrin fusion protein as an oral myelopoietic agent. Proc. Natl. Acad. Sci. USA 2005, 102, 7292–7296. [Google Scholar]
- Bai, Y.; Shen, W.-C. Improving the oral efficacy of recombinant granulocyte colony-stimulating factor and transferrin fusion protein by spacer optimization. Pharm. Res. 2006, 23, 2116–2121. [Google Scholar] [CrossRef]
- Jones, S.A.; Rose-John, S. The role of soluble receptors in cytokine biology: The agonistic properties of the sIL-6R/IL-6 complex. BBA-Mol. Cell Res. 2002, 1592, 251–263. [Google Scholar]
- Rubinstein, M.P.; Kovar, M.; Purton, J.F.; Cho, J.H.; Boyman, O.; Surh, C.D.; Sprent, J. Converting IL-15 to a superagonist by binding to soluble IL-15R alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 9166–9171. [Google Scholar]
- Dubois, S.; Mariner, J.; Waldmann, T.A.; Tagaya, Y. IL-15R alpha recycles and presents IL-15 in trans to neighboring cells. Immunity 2002, 17, 537–547. [Google Scholar] [CrossRef]
- Waldmann, T.A. The biology of interleukin-2 and interleukin-15: Implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 2006, 6, 595–601. [Google Scholar] [CrossRef]
- Mortier, E.; Quemener, A.; Vusio, P.; Lorenzen, I.; Boublik, Y.; Grotzinger, J.; Plet, A.; Jacques, Y. Soluble interleukin-15 receptor alpha (IL-15R alpha)-sushi as a selective and potent agonist of IL-15 action through IL-15R beta/gamma. Hyperagonist IL-15 x IL-15R alpha fusion proteins. J. Biol. Chem. 2006, 281, 1612–1619. [Google Scholar]
- Fischer, M.; Goldschmitt, J.; Peschel, C.; Brakenhoff, J.P.; Kallen, K.J.; Wollmer, A.; Grotzinger, J.; Rose-John, S.I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat. Biotechnol. 1997, 15, 142–145. [Google Scholar] [CrossRef]
- Pflanz, S.; Tacken, I.; Grotzinger, J.; Jacques, Y.; Dahmen, H.; Heinrich, P.C.; Muller-Newen, G. A fusion protein of interleukin-11 and soluble interleukin-11 receptor acts as a superagonist on cells expressing gp130. FEBS Lett. 1999, 450, 117–122. [Google Scholar] [CrossRef]
- Peters, M.; Blinn, G.; Jostock, T.; Schirmacher, P.; Zum Buschenfelde, K.H.M.; Galle, P.R.; Rose-John, S. Combined interleukin 6 and soluble interleukin 6 receptor accelerates murine liver regeneration. Gastroenterology 2000, 119, 1663–1671. [Google Scholar] [CrossRef]
- Manoukian, G.; Hagemeister, F. Denileukin diftitox: A novel immunotoxin. Expert Opin. Biol. Ther. 2009, 9, 1445–1451. [Google Scholar] [CrossRef]
- Shimamura, T.; Husain, S.R.; Puri, R.K. The IL-4 and IL-13 pseudomonas exotoxins: New hope for brain tumor therapy. Neurosurg. Focus 2006, 20, E11–E11. [Google Scholar]
- Foss, F. Clinical experience with denileukin diftitox (ONTAK). Semin. Oncol. 2006, 33, S11–S16. [Google Scholar] [CrossRef]
- Waters, C.A.; Schimke, P.A.; Snider, C.E.; Itoh, K.; Smith, K.A.; Nichols, J.C.; Strom, T.B.; Murphy, J.R. Interleukin-2 receptor-targeted cytotoxicity—Receptor-binding requirements for entry of a diphtheria toxin-related interleukin-2 fusion protein into cells. Eur. J. Immunol. 1990, 20, 785–791. [Google Scholar] [CrossRef]
- Bacha, P.; Williams, D.P.; Waters, C.; Williams, J.M.; Murphy, J.R.; Strom, T.B. Interleukin-2 receptor targeted cyto-toxicity interleukin-2 receptor mediated action of a diphtheria-toxin related interleukin-2 fusion protein. J. Exp. Med. 1988, 167, 612–622. [Google Scholar] [CrossRef]
- Olsen, E.; Duvic, M.; Frankel, A.; Kim, Y.; Martin, A.; Vonderheid, E.; Jegasothy, B.; Wood, G.; Gordon, M.; Heald, P.; et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J. Clin. Oncol. 2001, 19, 376–388. [Google Scholar]
- Ruddle, J.B.; Harper, C.A.; Honemann, D.; Seymour, J.F.; Prince, H.M.; Harper, C.A. A denileukin diftitox (Ontak) associated retinopathy? Br. J. Ophthalmol. 2006, 90, 1070–1071. [Google Scholar]
- Ruddle, J.B.; Prince, H.M. Denileukin diftitox and vision loss. Leuk. Lymphoma 2007, 48, 655–656. [Google Scholar] [CrossRef]
- Takeuchi, M.; Keino, H.; Kezuka, T.; Usui, M.; Taguchi, O. Immune responses to retinal self-antigens in CD25(+)CD4(+) regulatory T-Cell-depleted mice. Invest. Ophthalmol. Visual Sci. 2004, 45, 1879–1886. [Google Scholar] [CrossRef]
- Iwamoto, F.M.; Lamborn, K.R.; Robins, H.I.; Mehta, M.P.; Chang, S.M.; Butowski, N.A.; DeAngelis, L.M.; Abrey, L.E.; Zhang, W.-T.; Prados, M.D.; et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro-Oncology 2010, 12, 855–861. [Google Scholar] [CrossRef]
- Mut, M.; Sherman, J.H.; Shaffrey, M.E.; Schiff, D. Cintredekin besudotox in treatment of malignant glioma. Expert Opin. Biol. Ther. 2008, 8, 805–812. [Google Scholar] [CrossRef]
- Jackaman, C.; Bundell, C.S.; Kinnear, B.F.; Smith, A.M.; Filion, P.; van Hagen, D.; Robinson, B.W.S.; Nelson, D.J. IL-2 intratumoral immunotherapy enhances CD8(+) T cells that mediate destruction of tumor cells and tumor-associated vasculature: A novel mechanism for IL-2(1). J. Immunol. 2003, 171, 5051–5063. [Google Scholar]
- Gutbrodt, K.L.; Neri, D. Immunocytokines. Antibodies 2012, 1, 70–87. [Google Scholar] [CrossRef]
- van Horssen, R.; ten Hagen, T.L.M.; Eggermont, A.M.M. TNF-alpha in cancer treatment: Molecular insights, antitumor effects, and clinical utility. Oncologist 2006, 11, 397–408. [Google Scholar] [CrossRef]
- Sondel, P.M.; Gillies, S.D. Current and Potential Uses of Immunocytokines as Cancer Immunotherapy. Antibodies 2012, 1, 149–171. [Google Scholar] [CrossRef]
- Kontermann, R.E. Antibody-cytokine fusion proteins. Arch. Biochem. Biophys. 2012, 526, 194–205. [Google Scholar] [CrossRef]
- Pasche, N.; Neri, D. Immunocytokines: A novel class of potent armed antibodies. Drug Discov. Today 2012, 17, 583–590. [Google Scholar] [CrossRef]
- Connor, J.P.; Cristea, M.C.; Lewis, N.L.; Lewis, L.D.; Komarnitsky, P.B.; Mattiacci, M.R.; Felder, M.; Stewart, S.; Harter, J.; Henslee-Downey, J.; et al. A phase 1b study of humanized KS-interleukin-2 (huKS-IL2) immunocytokine with cyclophosphamide in patients with EpCAM-positive advanced solid tumors. BMC Cancer 2013, 13. Article No. 20. [Google Scholar]
- Patriarca, C.; Macchi, R.M.; Marschner, A.K.; Mellstedt, H. Epithelial cell adhesion molecule expression (CD326) in cancer: A short review. Cancer Treat. Rev. 2012, 38, 68–75. [Google Scholar] [CrossRef]
- Albertini, M.R.; Hank, J.A.; Gadbaw, B.; Kostlevy, J.; Haldeman, J.; Schalch, H.; Gan, J.; Kim, K.; Eickhoff, J.; Gillies, S.D.; et al. Phase II trial of hu14.18-IL2 for patients with metastatic melanoma. Cancer Immunol. Immunother. 2012, 61, 2261–2271. [Google Scholar] [CrossRef]
- Mujoo, K.; Cheresh, D.A.; Yang, H.M.; Reisfeld, R.A. Disialoganglioside-GD2 on human neuroblastoma-cells—Target antigen for monoclonal antibody-mediated cytolysis and suppression of tumor-growth. Cancer Res. 1987, 47, 1098–1104. [Google Scholar]
- Yamane, B.H.; Hank, J.A.; Albertini, M.R.; Sondel, P.M. The development of antibody-IL-2 based immunotherapy with hu14.18-IL2 (EMD-273063) in melanoma and neuroblastoma. Expert Opin. Investig. Drugs 2009, 18, 991–1000. [Google Scholar] [CrossRef]
- Helguera, G.; Rodriguez, J.A.; Penichet, M.L. Cytokines fused to antibodies and their combinations as therapeutic agents against different peritoneal HER2/neu expressing tumors. Mol. Cancer Ther. 2006, 5, 1029–1040. [Google Scholar] [CrossRef]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; Press, M.F. Studies of the HER-2/neu proto-oncogene in human-breast and ovarian-cancer. Science 1989, 244, 707–712. [Google Scholar]
- Gillies, S.D.; Lan, Y.; Williams, S.; Carr, F.; Forman, S.; Raubitschek, A.; Lo, K.M. An anti-CD20-IL-2 immunocytokine is highly efficacious in a SCID mouse model of established human B lymphoma. Blood 2005, 105, 3972–3978. [Google Scholar] [CrossRef]
- Rossi, E.A.; Goldenberg, D.M.; Cardillo, T.M.; Stein, R.; Chang, C.-H. CD20-targeted tetrameric interferon-alpha, a novel and potent immunocytokine for the therapy of B-cell lymphomas. Blood 2009, 114, 3864–3871. [Google Scholar] [CrossRef]
- Hirsch, B.; Brauer, J.; Fischdick, M.; Loddenkemper, C.; Bulfone-Paus, S.; Stein, H.; Duerkop, H. Anti-CD30 Human IL-2 Fusion Proteins Display Strong and Specific Cytotoxicity In Vivo. Curr. Drug Targets 2009, 10, 110–117. [Google Scholar] [CrossRef]
- Frey, K.; Fiechter, M.; Schwager, K.; Belloni, B.; Barysch, M.J.; Neri, D.; Dummer, R. Different patterns of fibronectin and tenascin-C splice variants expression in primary and metastatic melanoma lesions. Exp. Dermatol. 2011, 20, 685–688. [Google Scholar] [CrossRef]
- Kriegsmann, J.; Berndt, A.; Hansen, T.; Borsi, L.; Zardi, L.; Brauer, R.; Petrow, P.K.; Otto, M.; Kirkpatrick, C.J.; Gay, S.; et al. Expression of fibronectin splice variants and oncofetal glycosylated fibronectin in the synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Rheumatol. Int. 2004, 24, 25–33. [Google Scholar] [CrossRef]
- Schliemann, C.; Wiedmer, A.; Pedretti, M.; Szczepanowski, M.; Klapper, W.; Neri, D. Three clinical-stage tumor targeting antibodies reveal differential expression of oncofetal fibronectin and tenascin-C isoforms in human lymphoma. Leuk. Res. 2009, 33, 1718–1722. [Google Scholar] [CrossRef]
- Gillessen, S.; Gnad-Vogt, U.S.; Gallerani, E.; Beck, J.; Sessa, C.; Omlin, A.; Mattiacci, M.R.; Liedert, B.; Kramer, D.; Laurent, J.; et al. A phase I dose-escalation study of the immunocytokine EMD 521873 (Selectikine) in patients with advanced solid tumours. Eur. J. Cancer 2013, 49, 35–44. [Google Scholar] [CrossRef]
- Epstein, A.L.; Chen, F.M.; Taylor, C.R. A novel method for the detection of necrotic lesions in human cancers. Cancer Res. 1988, 48, 5842–5848. [Google Scholar]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Naramura, M.; Gillies, S.D.; Mendelsohn, J.; Reisfeld, R.A.; Mueller, B.M. Mechanisms of cellular cytotoxicity mediated by a recombinant antibody IL2 fusion protein against human-melanoma cells. Immunol. Lett. 1993, 39, 91–99. [Google Scholar] [CrossRef]
- Jain, R.K.; Baxter, L.T. Mechanisms of heterogeneous distribution of monoclonal-antibodies and other macromolecules in tumors—Significance of elevated interstitial pressure. Cancer Res. 1988, 48, 7022–7032. [Google Scholar]
- Yokota, T.; Milenic, D.E.; Whitlow, M.; Schlom, J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992, 52, 3402–3408. [Google Scholar]
- Milenic, D.E.; Yokota, T.; Filpula, D.R.; Finkelman, M.A.J.; Dodd, S.W.; Wood, J.F.; Whitlow, M.; Snoy, P.; Schlom, J. Construction, binding-properties, metabolism, and tumor targeting of a single-chain Fv derived from the pancarcinoma monoclonal-antibody CC49. Cancer Res. 1991, 51, 6363–6371. [Google Scholar]
- Adams, G.P.; Schier, R.; McCall, A.M.; Crawford, R.S.; Wolf, E.J.; Weiner, L.M.; Marks, J.D. Prolonged in vivo tumour retention of a human diabody targeting the extracellular domain of human HER2/neu. Br. J. Cancer 1998, 77, 1405–1412. [Google Scholar] [CrossRef]
- Rossi, E.A.; Rossi, D.L.; Stein, R.; Goldenberg, D.M.; Chang, C.-H. A Bispecific Antibody-IFN alpha 2b Immunocytokine Targeting CD20 and HLA-DR Is Highly Toxic to Human Lymphoma and Multiple Myeloma Cells. Cancer Res. 2010, 70, 7600–7609. [Google Scholar] [CrossRef]
- Kontermann, R.E. Dual targeting strategies with bispecific antibodies. mAbs 2012, 4, 182–197. [Google Scholar] [CrossRef]
- Bremer, E.; de Bruyn, M.; Wajant, H.; Helfrich, W. Targeted Cancer Immunotherapy Using Ligands of the Tumor Necrosis Factor Super-Family. Curr. Drug Targets 2009, 10, 94–103. [Google Scholar] [CrossRef]
- Galeazzi, M.; Baldi, C.; Prisco, E.; Bardelli, M.; Neri, D.; Giovannoni, L.; Selvi, E.; Caporali, R. A Phase Ib Clinical Trial with F8-IL10, an Anti-Inflammatory Immunocytokine for the Treatment of Rheumatoid Arthritis (RA), Used in Combination with Methotrexate (MTX). Arthritis Rheum. 2012, 64, S553–S554. [Google Scholar]
- ReidhaarOlson, J.F.; DeSouzaHart, J.A.; Selick, H.E. Identification of residues critical to the activity of human granulocyte colony-stimulating factor. Biochemistry (Mosc.) 1996, 35, 9034–9041. [Google Scholar] [CrossRef]
- Young, D.C.; Zhan, H.J.; Cheng, Q.L.; Hou, J.Z.; Matthews, D.J. Characterization of the receptor binding determinants of granulocyte colony stimulating factor. Protein Sci. 1997, 6, 1228–1236. [Google Scholar] [CrossRef]
- Collins, L.; Tsien, W.H.; Seals, C.; Hakimi, J.; Weber, D.; Bailon, P.; Hoskings, J.; Greene, W.C.; Toome, V.; Ju, G. Identification of specific residues of human interleukin-2 that affect binding to the 70-kDa subunit (p70) of the interleukin-2 receptor. Proc. Natl. Acad. Sci. USA 1988, 85, 7709–7713. [Google Scholar] [CrossRef]
- Ju, G.; Collins, L.; Kaffka, K.L.; Tsien, W.H.; Chizzonite, R.; Crowl, R.; Bhatt, R.; Kilian, P.L. Structure-function analysis of human interleukin-2—Identification of amino-acid-residues required for biological-activity. J. Biol. Chem. 1987, 262, 5723–5731. [Google Scholar]
- Weir, M.P.; Chaplin, M.A.; Wallace, D.M.; Dykes, C.W.; Hobden, A.N. Structure activity relationships of recombinant human interleukin-2. Biochemistry (Mosc.) 1988, 27, 6883–6892. [Google Scholar]
- Lee, C.M.Y.; McGuire, H.; Basten, A.; King, C.; Christ, D. Expression, purification and characterization of recombinant interleukin-21. J. Immunol. Methods 2010, 362, 185–189. [Google Scholar] [CrossRef]
- Marshall, S.A.; Lazar, G.A.; Chirino, A.J.; Desjarlais, J.R. Rational design and engineering of therapeutic proteins. Drug Discov. Today 2003, 8, 212–221. [Google Scholar] [CrossRef]
- Lowe, D.; Dudgeon, K.; Rouet, R.; Schofield, P.; Jermutus, L.; Christ, D. Aggregation, stability, and formulation of human antibody therapeutics. Adv. Protein Chem. Struct. Biol. 2011, 84, 41–61. [Google Scholar] [CrossRef]
- Dudgeon, K.; Rouet, R.; Kokmeijer, I.; Schofield, P.; Stolp, J.; Langley, D.; Stock, D.; Christ, D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 10879–10884. [Google Scholar]
- Arakawa, T.; Prestrelski, S.J.; Narhi, L.O.; Boone, T.C.; Kenney, W.C. Cysteine-17 of recombinant human granulocyte-colony-stimulating factor is partially solvent-exposed. J. Protein Chem. 1993, 12, 525–531. [Google Scholar] [CrossRef]
- Doyle, M.V.; Lee, M.T.; Fong, S. Comparison of the biological-activities of human recombinant interleukin-2125 and native interleukin-2. J. Biol. Response Mod. 1985, 4, 96–109. [Google Scholar]
- Lin, L. Betaseron. Dev. Biol. Stand. 1998, 96, 97–104. [Google Scholar]
- Bishop, B.; Koay, D.C.; Sartorelli, A.C.; Regan, L. Reengineering granulocyte colony-stimulating factor for enhanced stability. J. Biol. Chem. 2001, 276, 33465–33470. [Google Scholar]
- Luo, P.; Hayes, R.J.; Chan, C.; Stark, D.M.; Hwang, M.Y.; Jacinto, J.M.; Juvvadi, P.; Chung, H.S.; Kundu, A.; Ary, M.L.; et al. Development of a cytokine analog with enhanced stability using computational ultrahigh throughput screening. Protein Sci. 2002, 11, 1218–1226. [Google Scholar] [CrossRef]
- Tang, L.; Persky, A.M.; Hochhaus, G.; Meibohm, B. Pharmacokinetic aspects of biotechnology products. J. Pharm. Sci. 2004, 93, 2184–2204. [Google Scholar] [CrossRef]
- French, A.R.; Lauffenburger, D.A. Controlling receptor/ligand trafficking: Effects of cellular and molecular properties on endosomal sorting. Ann. Biomed. Eng. 1997, 25, 690–707. [Google Scholar] [CrossRef]
- Lauffenburger, D.A.; Fallon, E.M.; Haugh, J.M. Scratching the (cell) surface: Cytokine engineering for improved ligand/receptor trafficking dynamics. Chem. Biol. 1998, 5, R257–R263. [Google Scholar] [CrossRef]
- Fallon, E.M.; Liparoto, S.F.; Lee, K.J.; Ciardelli, T.L.; Lauffenburger, D.A. Increased endosomal sorting of ligand to recycling enhances potency of an interleukin-2 analog. J. Biol. Chem. 2000, 275, 6790–6797. [Google Scholar]
- Sarkar, C.A.; Lowenhaupt, K.; Horan, T.; Boone, T.C.; Tidor, B.; Lauffenburger, D.A. Rational cytokine design for increased lifetime and enhanced potency using pH-activated "histidine switching". Nat. Biotechnol. 2002, 20, 908–913. [Google Scholar] [CrossRef]
- Rao, B.M.; Driver, I.; Lauffenburger, D.A.; Wittrup, K.D. High-affinity CD25-binding IL-2 mutants potently stimulate persistent T cell growth. Biochemistry (Mosc.) 2005, 44, 10696–10701. [Google Scholar] [CrossRef]
- Rao, B.M.; Girvin, A.T.; Ciardelli, T.; Lauffenburger, D.A.; Wittrup, K.D. Interleukin-2 mutants with enhanced alpha-receptor subunit binding affinity. Protein Eng. 2003, 16, 1081–1087. [Google Scholar] [CrossRef]
- Liu, D.V.; Maier, L.M.; Hafler, D.A.; Wittrup, K.D. Engineered Interleukin-2 Antagonists for the Inhibition of Regulatory T Cells. J. Immunother. 2009, 32, 887–894. [Google Scholar] [CrossRef]
- Levin, A.M.; Bates, D.L.; Ring, A.M.; Krieg, C.; Lin, J.T.; Su, L.; Moraga, I.; Raeber, M.E.; Bowman, G.R.; Novick, P.; et al. Exploiting a natural conformational switch to engineer an interleukin-2 'superkine'. Nature 2012, 484, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Junttila, I.S.; Creusot, R.J.; Moraga, I.; Bates, D.L.; Wong, M.T.; Alonso, M.N.; Suhoski, M.M.; Lupardus, P.; Meier-Schellersheim, M.; Engleman, E.G.; et al. Redirecting cell-type specific cytokine responses with engineered interleukin-4 superkines. Nat. Chem. Biol. 2012, 8, 990–998. [Google Scholar] [CrossRef]
- Hershey, G.K.K. IL-13 receptors and signaling pathways: An evolving web. J. Allergy Clin. Immunol. 2003, 111, 677–690. [Google Scholar]
- Antoniu, S.A. Pitrakinra, a dual IL-4/IL-13 antagonist for the potential treatment of asthma and eczema. Curr. Opin. Investig. Drugs 2010, 11, 1286–1294. [Google Scholar]
- Antoniu, S.A.; Cojocaru, I. Pitrakinra for asthma. Expert Opin. Biol. Ther. 2010, 10, 1609–1615. [Google Scholar] [CrossRef]
- Wenzel, S.; Wilbraham, D.; Fuller, R.; Burmeister Getz, E.; Lonphre, M. Effect of an interieukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: Results of two phase 2a studies. Lancet 2007, 370, 1422–1431. [Google Scholar] [CrossRef]
- Tony, H.P.; Shen, B.J.; Reusch, P.; Sebald, W. Design of human interleukin-4 antagonists inhibiting interleukin-4-dependent and interleukin-13-dependent responses in T-cells and B-cells with high-efficiency. Eur. J. Biochem. 1994, 225, 659–665. [Google Scholar] [CrossRef]
- Malara, N.; Foca, D.; Casadonte, F.; Sesto, M.; Paolino, D.; Tassone, P.; Venuta, S.; Savino, R. Treatment with an adenoviral vector expressing the IL-6 receptor superantagonist sant7 compared with the treatment with the recombinant sant7 protein in MM cell lines. J. Immunother. 2006, 29, 674–675. [Google Scholar]
- Tassone, P.; Neri, P.; Burger, R.; Savino, R.; Shammas, M.; Catley, L.; Podar, K.; Chauhan, D.; Masciari, S.; Gozzini, A.; et al. Combination therapy with interleukin-6 receptor superantagonist Sant7 and dexamethasone induces antitumor effects in a novel SCID-hu In vivo model of human multiple myeloma. Clin. Cancer Res. 2005, 11, 4251–4258. [Google Scholar] [CrossRef]
- Gallelli, L.; Falcone, D.; Pelaia, G.; Renda, T.; Terracciano, R.; Malara, N.; Vatrella, A.; Sanduzzi, A.; D'Agostino, B.; Rossi, F.; et al. Interleukin-6 receptor superantagonist Sant7 inhibits TGF-beta-induced proliferation of human lung fibroblasts. Cell Prolif. 2008, 41, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Savino, R.; Ciapponi, L.; Lahm, A.; Demartis, A.; Cabibbo, A.; Toniatti, C.; Delmastro, P.; Altamura, S.; Ciliberto, G. Rational design of a receptor super-antagonist of human interleukin-6. EMBO J. 1994, 13, 5863–5870. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vazquez-Lombardi, R.; Roome, B.; Christ, D. Molecular Engineering of Therapeutic Cytokines. Antibodies 2013, 2, 426-451. https://doi.org/10.3390/antib2030426
Vazquez-Lombardi R, Roome B, Christ D. Molecular Engineering of Therapeutic Cytokines. Antibodies. 2013; 2(3):426-451. https://doi.org/10.3390/antib2030426
Chicago/Turabian StyleVazquez-Lombardi, Rodrigo, Brendan Roome, and Daniel Christ. 2013. "Molecular Engineering of Therapeutic Cytokines" Antibodies 2, no. 3: 426-451. https://doi.org/10.3390/antib2030426
APA StyleVazquez-Lombardi, R., Roome, B., & Christ, D. (2013). Molecular Engineering of Therapeutic Cytokines. Antibodies, 2(3), 426-451. https://doi.org/10.3390/antib2030426