Preferential Identification of Agonistic OX40 Antibodies by Using Cell Lysate to Pan Natively Paired, Humanized Mouse-Derived Yeast Surface Display Libraries

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Immunization and Sample Preparation
2.2. Generating Paired Heavy and Light Chain Libraries
2.3. Yeast Library Screening
2.4. Sequence Analysis
2.5. Monoclonal Antibody Expression and Characterization
3. Results
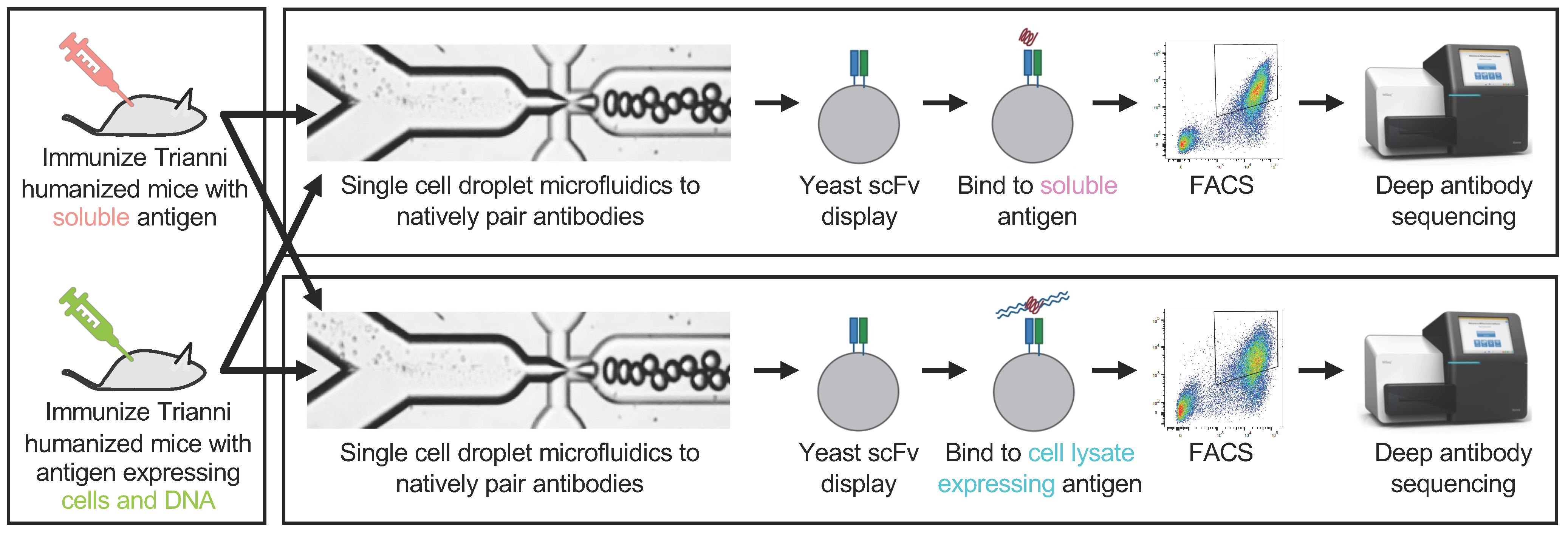
3.1. Overview of the Experimental Approach
3.2. Analysis of Serum Titers
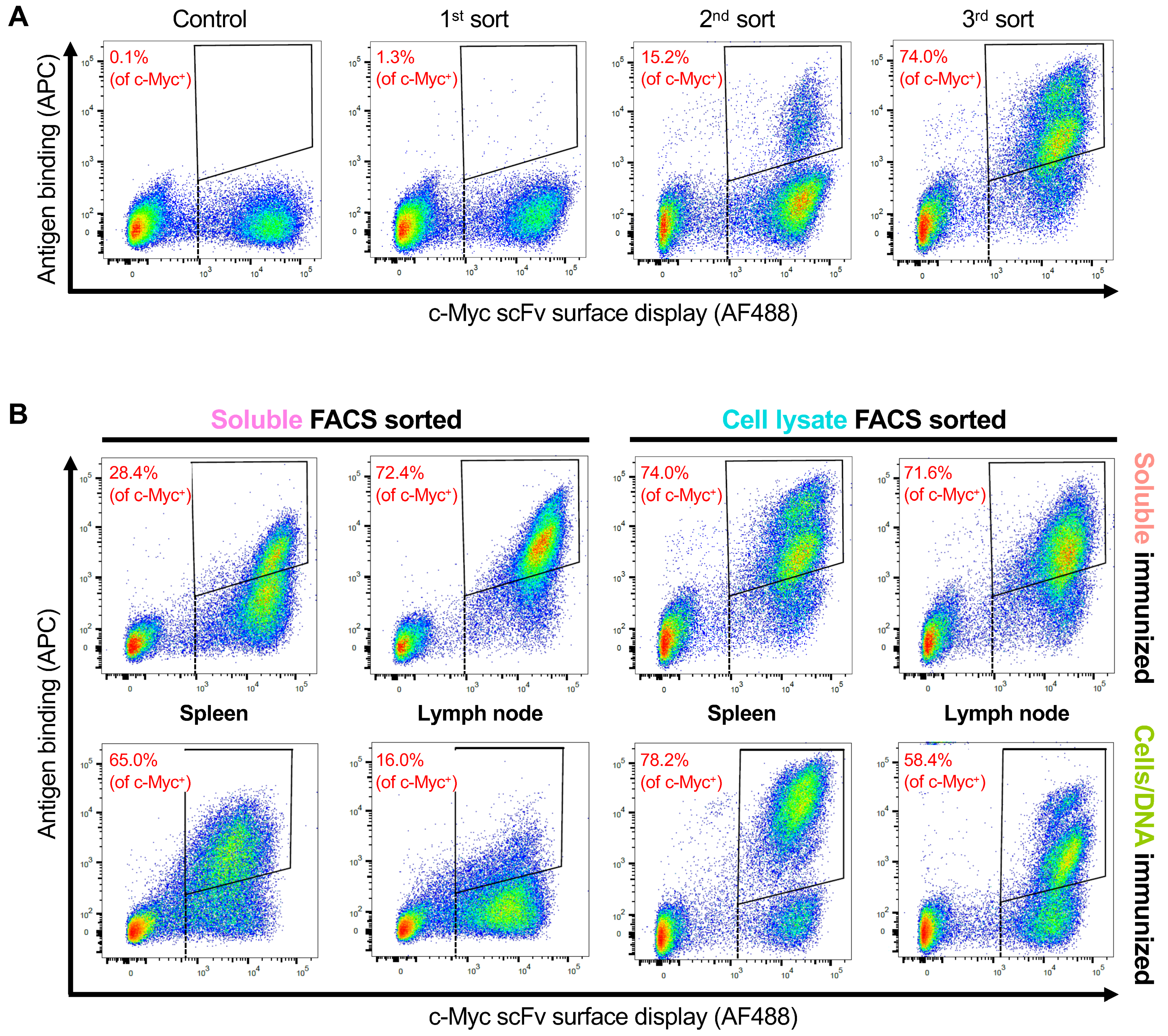
3.3. Selection of OX40 scFv Binders with FACS
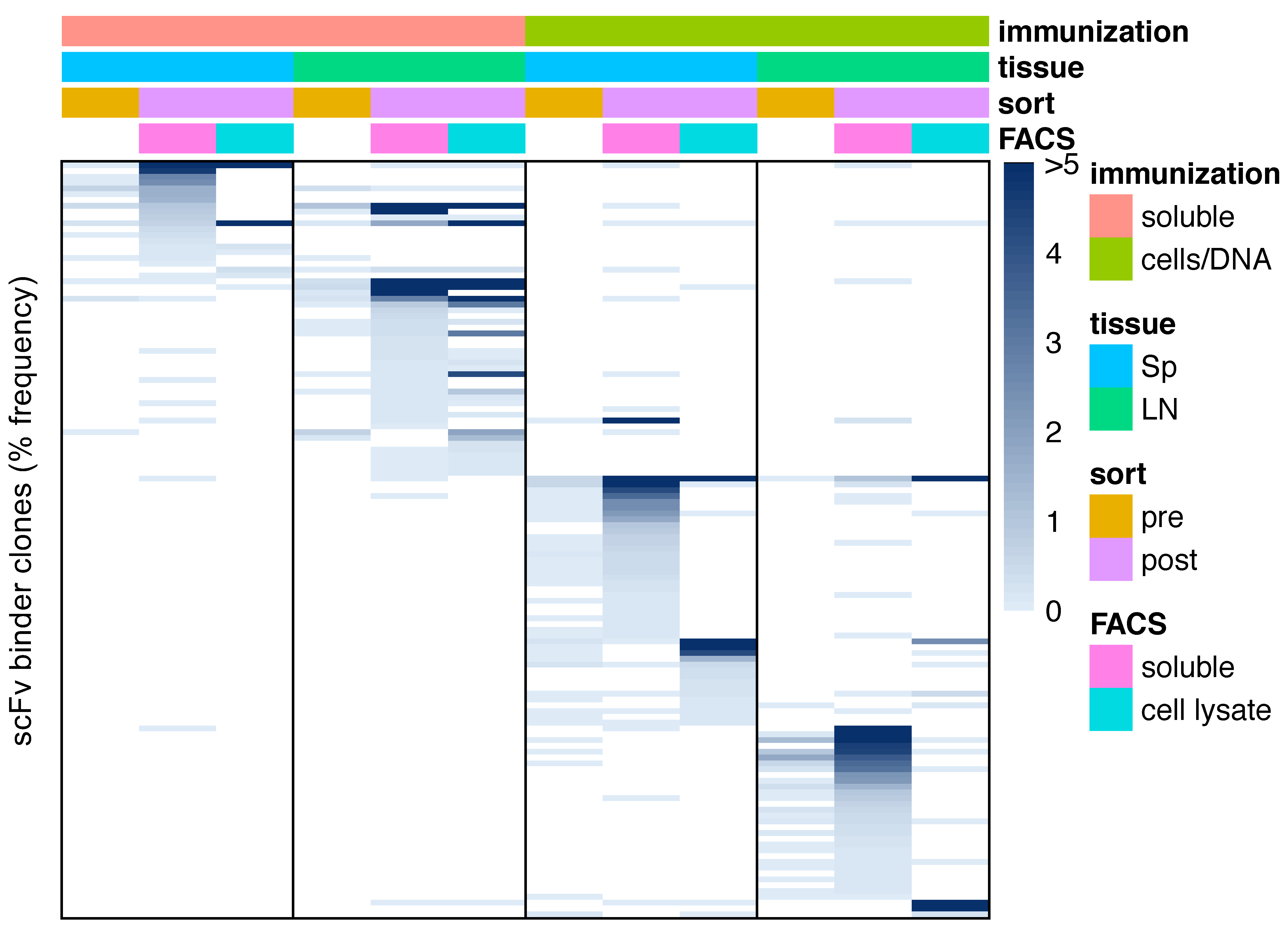
3.4. Sequence Characteristics of OX40 scFv Binders
3.5. Functional Characteristics of Monoclonal Antibody Binders
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 2011, 256, 495–497. [Google Scholar]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Kohl, T.O.; Ascoli, C.A. Direct and Indirect Cell-Based Enzyme-Linked Immunosorbent Assay. Cold Spring Harb. Protoc. 2017. [Google Scholar] [CrossRef] [PubMed]
- Even-Desrumeaux, K.; Chames, P. Phage Display and Selections on Cells. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 225–235. [Google Scholar]
- Nelson, A.L.; Dhimolea, E.; Reichert, J.M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 2010, 9, 767–774. [Google Scholar] [CrossRef]
- Spencer, S.; Bethea, D.; Raju, T.S.; Giles-Komar, J.; Feng, Y. Solubility evaluation of murine hybridoma antibodies. mAbs 2012, 4, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Jain, T.; Sun, T.; Durand, S.; Hall, A.; Houston, N.R.; Sharkey, B.; Bobrowicz, B.; Caffry, I.; Yu, Y.; Cao, Y.; et al. Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. USA 2017, 114, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; Mizrahi, R.A.; Spindler, M.J.; Adams, M.S.; Asensio, M.A.; Edgar, R.C.; Leong, J.; Leong, R.; Roalfe, L.; White, R.; et al. Rare, high-affinity anti-pathogen antibodies from human repertoires, discovered using microfluidics and molecular genomics. mAbs 2017, 9, 1282–1296. [Google Scholar] [CrossRef]
- Adler, A.S.; Mizrahi, R.A.; Spindler, M.J.; Adams, M.S.; Asensio, M.A.; Edgar, R.C.; Leong, J.; Leong, R.; Johnson, D.S. Rare, high-affinity mouse anti-PD-1 antibodies that function in checkpoint blockade, discovered using microfluidics and molecular genomics. mAbs 2017, 9, 1270–1281. [Google Scholar] [CrossRef]
- Adler, A.S.; Bedinger, D.; Adams, M.S.; Asensio, M.A.; Edgar, R.C.; Leong, R.; Leong, J.; Mizrahi, R.A.; Spindler, M.J.; Bandi, S.R.; et al. A natively paired antibody library yields drug leads with higher sensitivity and specificity than a randomly paired antibody library. mAbs 2018, 10, 431–443. [Google Scholar] [CrossRef]
- Rajan, S.; Kierny, M.R.; Mercer, A.; Wu, J.; Tovchigrechko, A.; Wu, H.; Xiao, X.; Chowdhury, P.S.; Dall′acqua, W.F.; Acqua, W.F.D.; et al. Recombinant human B cell repertoires enable screening for rare, specific, and natively paired antibodies. Commun. Biol. 2018, 1, 5. [Google Scholar] [CrossRef]
- Wang, B.; DeKosky, B.J.; Timm, M.R.; Lee, J.; Normandin, E.; Misasi, J.; Kong, R.; McDaniel, J.R.; Delidakis, G.; E Leigh, K.; et al. Functional interrogation and mining of natively paired human VH:VL antibody repertoires. Nat. Biotechnol. 2018, 36, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Linch, S.N.; McNamara, M.J.; Redmond, W.L. OX40 Agonists and Combination Immunotherapy: Putting the Pedal to the Metal. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, J.; Griffiths, J.; Tews, I.; Cragg, M.S. OX40: Structure and function—What questions remain? Mol. Immunol. 2017, 83, 13–22. [Google Scholar] [CrossRef]
- Compaan, D.M.; Hymowitz, S.G. The Crystal Structure of the Costimulatory OX40-OX40L Complex. Structure 2006, 14, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Tillotson, B.J.; De Larrinoa, I.F.; Skinner, C.A.; Klavas, D.M.; Shusta, E.V. Antibody affinity maturation using yeast display with detergent-solubilized membrane proteins as antigen sources. Protein. Eng. Des. Sel. 2012, 26, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.K.; Shusta, E.V. Antibody library screens using detergent-solubilized mammalian cell lysates as antigen sources. Protein. Eng. Des. Sel. 2010, 23, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 2015, 31, 3476–3482. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, M.-P.; Giudicelli, V.; Ginestoux, C.; Jabado-Michaloud, J.; Folch, G.; Bellahcene, F.; Wu, Y.; Gemrot, E.; Brochet, X.; Lane, J. IMGT(R), the international ImMunoGeneTics information system(R). Nucleic Acids Res. 2009, 37, D1006–D1012. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Compl. Syst. 2006, 1695, 1–9. [Google Scholar]
- Dreyer, A.M.; Beauchamp, J.; Matile, H.; Pluschke, G. An efficient system to generate monoclonal antibodies against membrane-associated proteins by immunisation with antigen-expressing mammalian cells. BMC Biotechnol. 2010, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, M.; Ghaderi, A. Production of a Mouse Monoclonal Antibody Against Mortalin by Whole Cell Immunization. Monoclon. Antib. Immunodiagn. Immunother. 2017, 36, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Chiba, J. Production of Antibodies against Multipass Membrane Proteins Expressed in Human Tumor Cells Using Dendritic Cell Immunization. J. Biomed. Biotechnol. 2009, 2009, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; Ge, X.; E Miklos, A.; A Hughes, R.; Kang, S.H.; Hoi, K.H.; Chrysostomou, C.; Hunicke-Smith, S.P.; Iverson, B.L.; Tucker, P.W.; et al. Monoclonal antibodies isolated without screening by analyzing the variable-gene repertoire of plasma cells. Nat. Biotechnol. 2010, 28, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Saggy, I.; Wine, Y.; Shefet-Carasso, L.; Nahary, L.; Georgiou, G.; Benhar, I. Antibody isolation from immunized animals: Comparison of phage display and antibody discovery via V gene repertoire mining. Protein. Eng. Des. Sel. 2012, 25, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.R.; Tzeng, W.-P.; Spesock, A.; Music, N.; Guo, Z.; Barrington, R.; Stevens, J.; Donis, R.O.; Katz, J.M.; York, I.A.; et al. Diversity of the murine antibody response targeting influenza A(H1N1pdm09) hemagglutinin. Virology 2014, 458, 114–124. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mAb ID | Enriched? Soluble Immunized, Soluble Sorted | Enriched? Soluble Immunized, Lysate Sorted | Enriched? Cells/DNA Immunized, Soluble Sorted | Enriched? Cells/DNA Immunized, Lysate Sorted | Binds Cells? | Promega In Vitro Assay EC50 (ug/mL) (Agonist) | KD (nM) [Octet, Global Fit] |
|---|---|---|---|---|---|---|---|
| tOX40.2 | Yes | No | No | No | No | not tested | not tested |
| tOX40.4 | Yes | Yes | No | No | Yes (2 peaks) | 0.044 | 9 |
| tOX40.15 | Yes | No | No | No | Yes (1 peak) | 1.276 | 6 |
| tOX40.19 | Yes | Yes | No | No | Yes (1 peak) | 0.811 | 7.7 |
| tOX40.20 | Yes | No | No | No | No | not tested | not tested |
| tOX40.21 | Yes | Yes | No | No | Yes (1 peak) | does not agonize | 7 |
| tOX40.22 | Yes | Yes | No | No | Yes (2 peaks) | 0.087 | 7.3 |
| tOX40.23 | Yes | Yes | No | No | Yes (2 peaks) | 0.419 | 5.2 |
| tOX40.24 | Yes | Yes | No | No | Yes (2 peaks) | 0.024 | 22.9 |
| tOX40.28 | Yes | No | No | No | No | not tested | not tested |
| tOX40.31 | Yes | Yes | No | No | Yes (2 peaks) | 0.043 | 22.4 |
| tOX40.33 | Yes | No | No | No | No | not tested | not tested |
| tOX40.34 | Yes | No | No | No | No | not tested | not tested |
| tOX40.35 | Yes | No | No | No | Yes (1 peak) | 4.316 | 2.7 |
| tOX40.36 | Yes | No | No | No | No | not tested | not tested |
| tOX40.37 | Yes | Yes | No | No | Yes (2 peaks) | 0.195 | 12.5 |
| tOX40.38 | Yes | No | No | No | No | not tested | not tested |
| tOX40.39 | Yes | Yes | No | No | Yes (2 peaks) | 0.199 | 90 |
| tOX40.40 | Yes | Yes | No | No | Yes (2 peaks) | 0.426 | 151 |
| tOX40.41 | No | Yes | No | No | Yes (2 peaks) | 0.068 | 58.4 |
| tOX40.42 | No | No | Yes | No | Yes (1 peak) | does not agonize | no binding |
| tOX40.43 | No | No | Yes | No | No | not tested | not tested |
| tOX40.44 | No | No | Yes | No | No | not tested | not tested |
| tOX40.45 | No | No | Yes | No | No | not tested | not tested |
| tOX40.46 | No | No | Yes | No | No | not tested | not tested |
| tOX40.47 | No | No | Yes | No | No | not tested | not tested |
| tOX40.48 | No | No | Yes | No | No | not tested | not tested |
| tOX40.49 | No | No | Yes | No | No | not tested | not tested |
| tOX40.50 | No | No | Yes | Yes | Yes (2 peaks) | 0.091 | 29.8 |
| tOX40.51 | No | No | No | Yes | No | not tested | not tested |
| tOX40.52 | No | No | No | Yes | No | not tested | not tested |
| tOX40.54 | No | No | No | Yes | Yes (2 peaks) | 0.321 | 184 |
| tOX40.55 | No | No | No | Yes | No | not tested | not tested |
| tOX40.56 | No | No | Yes | No | No | not tested | not tested |
| tOX40.57 | No | No | Yes | No | No | not tested | not tested |
| tOX40.58 | No | No | Yes | No | No | not tested | not tested |
| tOX40.59 | No | No | Yes | No | No | not tested | not tested |
| tOX40.60 | No | No | Yes | No | No | not tested | not tested |
| tOX40.61 | No | No | No | Yes | No | not tested | not tested |
| tOX40.62 | No | No | No | Yes | No | not tested | not tested |
| tOX40.63 | No | No | No | Yes | No | not tested | not tested |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medina-Cucurella, A.V.; Mizrahi, R.A.; Asensio, M.A.; Edgar, R.C.; Leong, J.; Leong, R.; Lim, Y.W.; Nelson, A.; Niedecken, A.R.; Simons, J.F.; et al. Preferential Identification of Agonistic OX40 Antibodies by Using Cell Lysate to Pan Natively Paired, Humanized Mouse-Derived Yeast Surface Display Libraries. Antibodies 2019, 8, 17. https://doi.org/10.3390/antib8010017
Medina-Cucurella AV, Mizrahi RA, Asensio MA, Edgar RC, Leong J, Leong R, Lim YW, Nelson A, Niedecken AR, Simons JF, et al. Preferential Identification of Agonistic OX40 Antibodies by Using Cell Lysate to Pan Natively Paired, Humanized Mouse-Derived Yeast Surface Display Libraries. Antibodies. 2019; 8(1):17. https://doi.org/10.3390/antib8010017
Chicago/Turabian StyleMedina-Cucurella, Angélica V., Rena A. Mizrahi, Michael A. Asensio, Robert C. Edgar, Jackson Leong, Renee Leong, Yoong Wearn Lim, Ayla Nelson, Ariel R. Niedecken, Jan Fredrik Simons, and et al. 2019. "Preferential Identification of Agonistic OX40 Antibodies by Using Cell Lysate to Pan Natively Paired, Humanized Mouse-Derived Yeast Surface Display Libraries" Antibodies 8, no. 1: 17. https://doi.org/10.3390/antib8010017
APA StyleMedina-Cucurella, A. V., Mizrahi, R. A., Asensio, M. A., Edgar, R. C., Leong, J., Leong, R., Lim, Y. W., Nelson, A., Niedecken, A. R., Simons, J. F., Spindler, M. J., Stadtmiller, K., Wayham, N., Adler, A. S., & Johnson, D. S. (2019). Preferential Identification of Agonistic OX40 Antibodies by Using Cell Lysate to Pan Natively Paired, Humanized Mouse-Derived Yeast Surface Display Libraries. Antibodies, 8(1), 17. https://doi.org/10.3390/antib8010017