IgE Antibodies: From Structure to Function and Clinical Translation




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
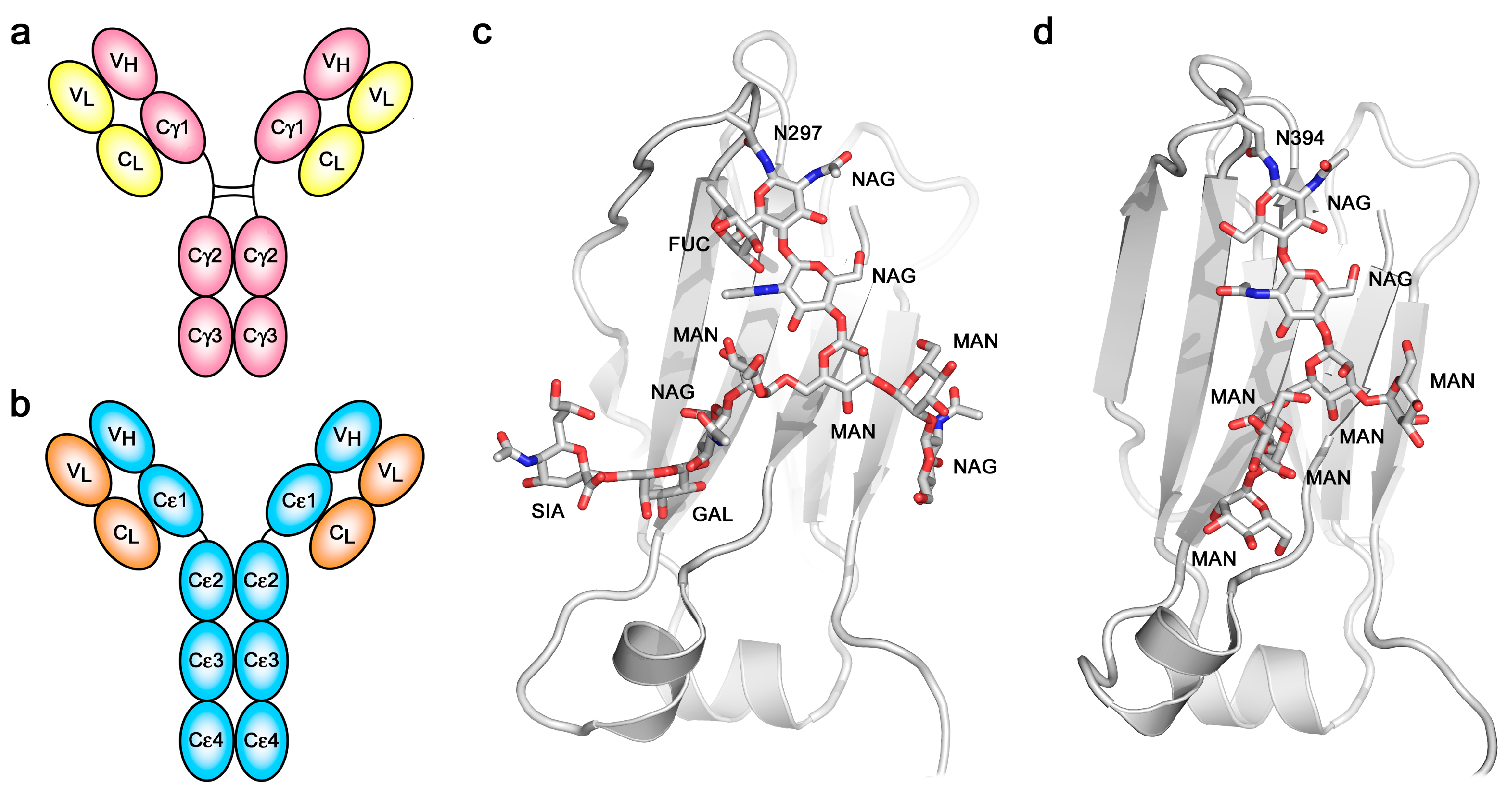
2. The Structure of IgE
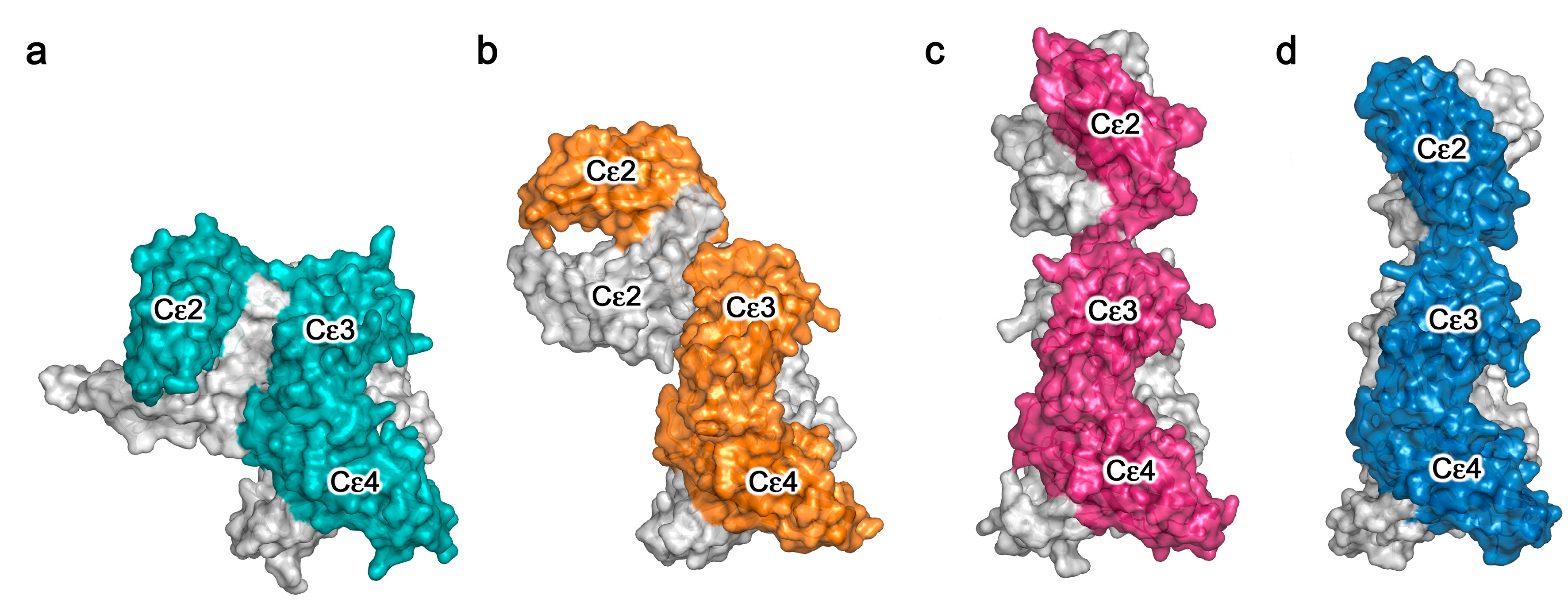
3. Conformational Dynamics in IgE-Fc
4. IgE-Receptor Interactions
5. IgE—An Allosteric Antibody
6. Antigen (Allergen) Binding
7. Rationale for Harnessing IgE-Mediated Functions against Cancer
7.1. Epidemiological Links between IgE, Allergy and Cancer
7.2. Features of IgE that may Translate to Immune Protective Functions against Tumours
8. Pre-Clinical Studies of IgE Antibodies Targeting Cancer Antigens: The Advent of Allergo Oncology
8.1. Engineering Platforms for Production of IgE Antibodies for Research and Clinical Translation
8.2. Functional Evaluations of Anti-Tumour IgEs
8.2.1. In Vitro and In Vivo Functional Profiles of Engineered IgEs Targeting Several Cancer Antigens
8.2.2. MOv18 IgE, the First Anti-Tumour IgE to Reach Clinical Testing: Evaluation of In Vitro Effector Functions
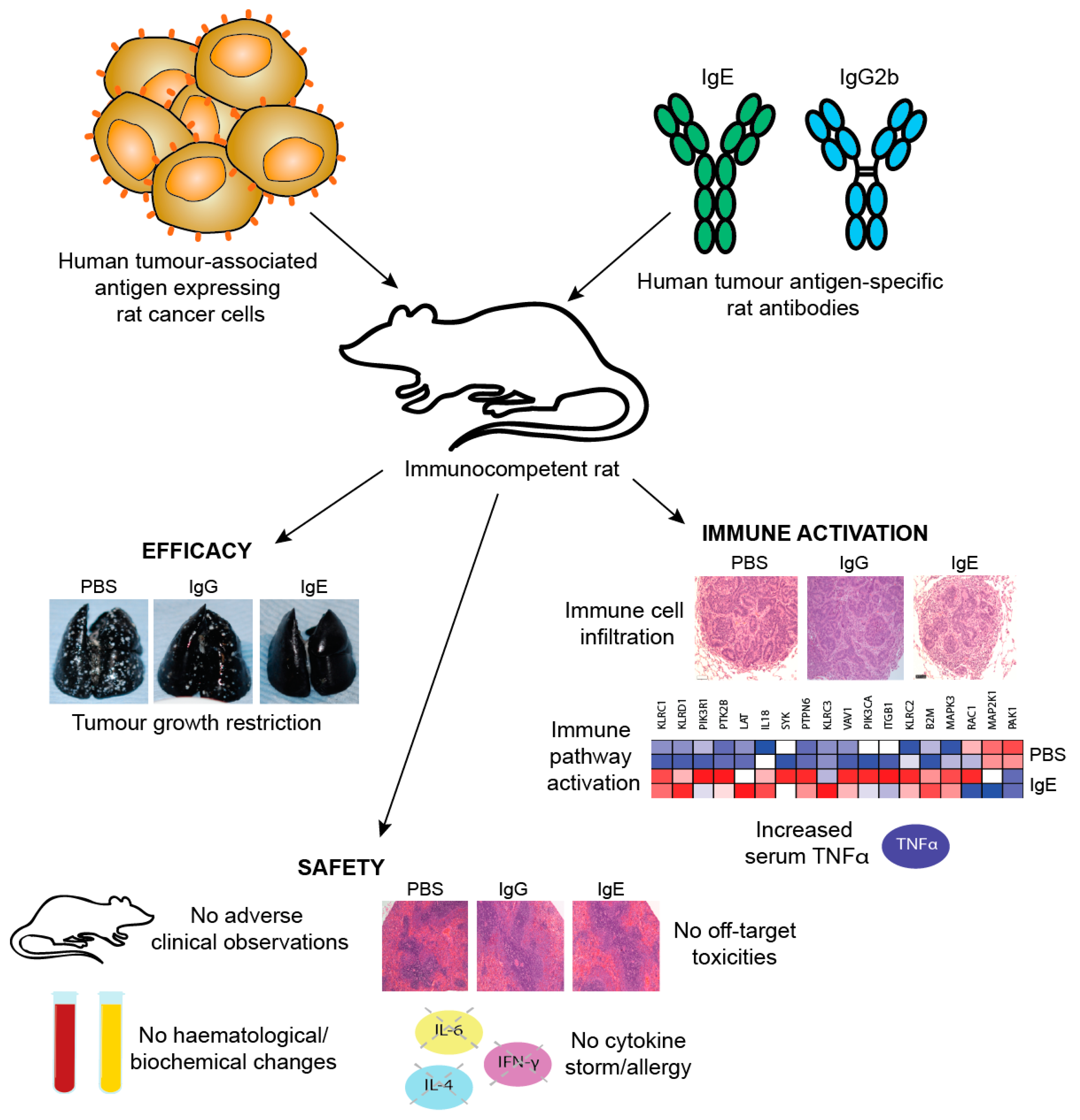
8.2.3. In vivo efficacy studies of MOv18 IgE
8.3. Evidence for IgE Activating Monocytes and Macrophages against Cancer
8.3.1. Monocytes and Macrophages as Key Effector Cells in MOv18 IgE-Potentiated Anti-Tumour Functions
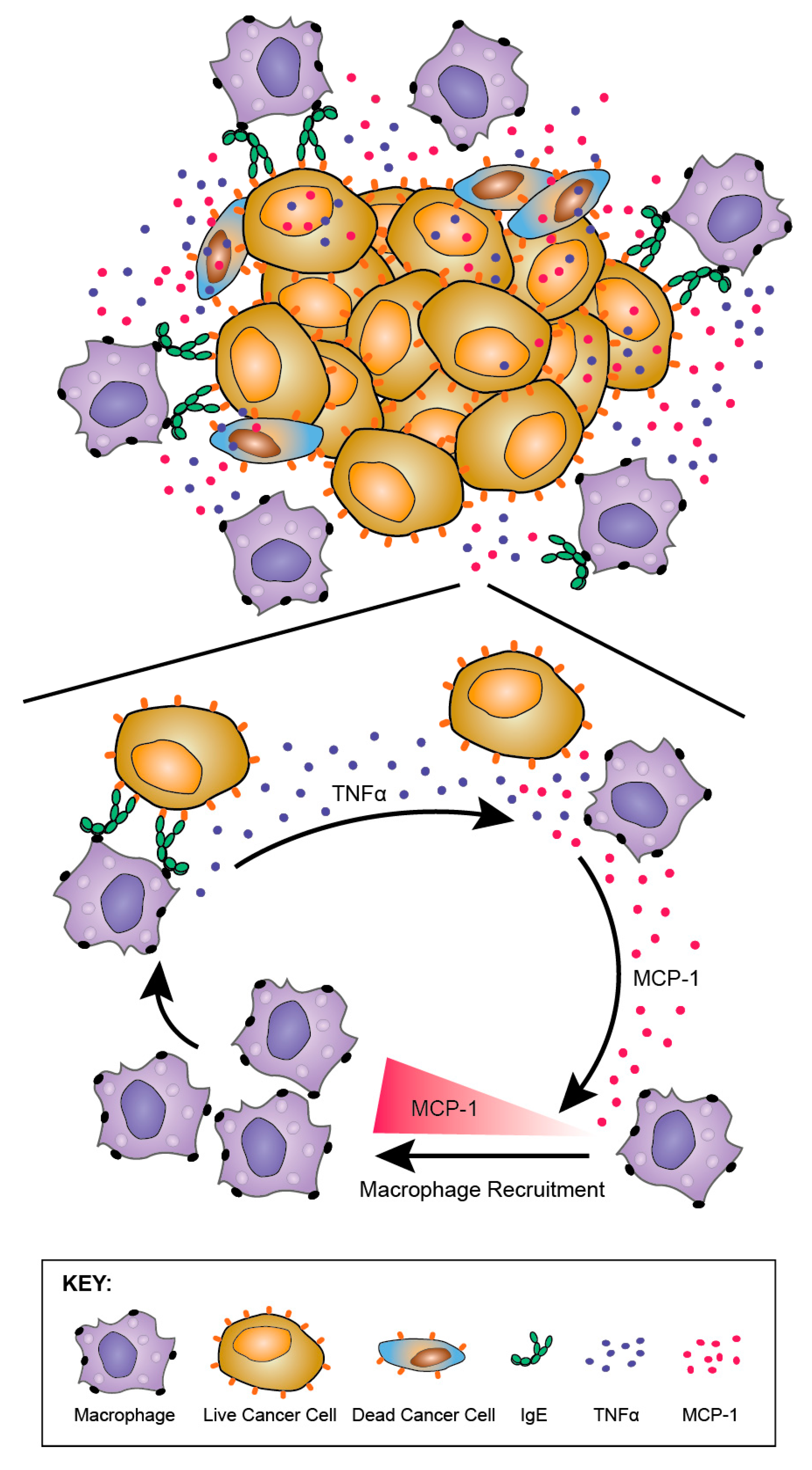
8.3.2. Anti-Tumour IgE Directs Monocytes and Macrophages
8.3.3. TNFα/MCP-1 Axis as a Mechanism of MOv18 IgE-Mediated Activation of Human Monocytes
9. Towards Clinical Translation of First-In-Class IgE to a First-In-Man Clinical Trial
9.1. Predicting Safety of IgE: Using Ex Vivo Functional Assays Adapted from Allergy Diagnosis
9.2. Predicting Safety of IgE: In Vivo Models
9.3. Monitoring Antibody Safety in Trials
10. Thoughts for the Design of New IgE-Based Therapeutic Agents
10.1. Expression Systems and IgE Glyco-Profiling
10.2. Selecting Tumour Targets and Malignant Indications for IgE Therapeutic Agents
10.3. Challenges for IgE-Based Therapies
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADCC | antibody-dependent cell-mediated cytotoxicity |
| ADCP | antibody-dependent cell-mediated phagocytosis |
| APC | antigen presenting cell |
| BAL | broncho-alveolar lavage |
| BAT | basophil activation test |
| CCA | colorectal cancer antigen |
| CDR | complementarity-determining region |
| CTCs | circulating tumour cells |
| CTL | cytotoxic T lymphocyte |
| DCs | dendritic cells |
| EGFR | epidermal growth factor receptor |
| EM | electron microscopy |
| FR | framework region |
| FRα | folate receptor alpha |
| FRET | fluorescence (Förster) resonance energy transfer |
| GMP | Good Manufacturing Practice |
| IHC | immunohistochemical/immunohistochemistry |
| i.p. | intraperitoneal |
| i.v. | intravenous |
| MCP-1 | macrophage chemoattractant protein-1 |
| MD | molecular dynamics |
| MMTV | mammary tumour virus |
| NIP | 4-hydroxy-3-nitro-phenacetyl |
| NK | Natural Killer |
| PBMCs | peripheral blood mononuclear cells |
| PDX | patient-derived xenograft |
| PIPE | Polymerase Incomplete Primer Extension |
| PSA | prostate specific antigen |
| RBL | rat basophil leukaemia |
| SAXS | small-angle X-ray scattering |
| s.c. | subcutaneous |
| Th | T helper |
| TME | tumour microenvironment |
| TNFα | tumour necrosis factor |
| UCOE | Ubiquitous Chromatin Opening Elements |
| WAG | Wistar Albino Glaxo |
References
- Platts-Mills, T.A.; Heymann, P.W.; Commins, S.P.; Woodfolk, J.A. The discovery of IgE 50 years later. Ann. Allergy Asthma Immunol. 2016, 116, 179–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennich, H.H.; Ishizaka, K.; Johansson, S.G.O.; Rowe, D.S.; Stanworth, D.R.; Terry, W.D. Immunoglobulin E, a new class of human immunoglobulin. Bull. World Health Organ. 1968, 38, 151–152. [Google Scholar] [CrossRef]
- Ishizaka, K.; Ishizaka, T.; Hornbrook, M.M. Physicochemical properties of reaginic antibody. V. Correlation of reaginic activity with γE globulin antibody. J. Immunol. 1966, 97, 840–853. [Google Scholar] [PubMed]
- Gould, H.J.; Sutton, B.J. IgE in allergy and asthma today. Nat. Rev. Immunol. 2008, 8, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Tsai, M.; Starkl, P.; Marichal, T.; Galli, S.J. IgE and mast cells in host defense against parasites and venoms. Semin. Immunopathol. 2016, 38, 581–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, B.J.; Davies, A.M. Structure and dynamics of IgE-receptor interactions: FcεRI and CD23/FcεRII. Immunol. Rev. 2015, 268, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Kraft, S.; Kinet, J.-P. New developments in FcεRI regulation, function and inhibition. Nat. Rev. Immunol. 2007, 7, 365–378. [Google Scholar] [CrossRef]
- Kinet, J.P. The high-affinity IgE receptor (FcεRI): From physiology to pathology. Annu. Rev. Immunol. 1999, 17, 931–972. [Google Scholar] [CrossRef]
- Gounni, A.S.; Wellemans, V.; Yang, J.; Bellesort, F.; Kassiri, K.; Gangloff, S.; Guenounou, M.; Halayko, A.J.; Hamid, Q.; Lamkhioued, B. Human airway smooth muscle cells express the high affinity receptor for IgE (FcεRI): A critical role of FcεRI in human airway smooth muscle cell function. J. Immunol. 2005, 175, 2613–2621. [Google Scholar] [CrossRef]
- Campbell, A.M.; Vachier, I.; Chanez, P.; Vignola, A.M.; Lebel, B.; Kochan, J.; Godard, P.; Bousquet, J. Expression of the high-affinity receptor for IgE on bronchial epithelial cells of asthmatics. Am. J. Respir. Cell Mol. Biol. 1998, 19, 92–97. [Google Scholar] [CrossRef]
- Untersmayr, E.; Bises, G.; Starkl, P.; Bevins, C.L.; Scheiner, O.; Boltz-Nitulescu, G.; Wrba, F.; Jensen-Jarolim, E. The high affinity IgE receptor FcεRI is expressed by human intestinal epithelial cells. PLoS ONE 2010, 5, e9023. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, P.M.; Pietersz, G.A. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat. Rev. Drug Discov. 2012, 11, 311–331. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.H.; Ford, J.W.; Sturgill, J.L.; Gibb, D.R. CD23: An overlooked regulator of allergic disease. Curr. Allergy Asthma Rep. 2007, 7, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Yukawa, K.; Kikutani, H.; Owaki, H.; Yamasaki, K.; Yokota, A.; Nakamura, H.; Barsumian, E.L.; Hardy, R.R.; Suemura, M.; Kishimoto, T. A B cell-specific differentiation antigen, CD23, is a receptor for IgE (Fc epsilon R) on lymphocytes. J. Immunol. 1987, 138, 2576–2580. [Google Scholar]
- Bonnefoy, J.Y.; Aubry, J.P.; Peronne, C.; Wijdenes, J.; Banchereau, J. Production and characterization of a monoclonal antibody specific for the human lymphocyte low affinity receptor for IgE: CD 23 is a low affinity receptor for IgE. J. Immunol. 1987, 138, 2970–2978. [Google Scholar] [PubMed]
- Palaniyandi, S.; Tomei, E.; Li, Z.; Conrad, D.H.; Zhu, X. CD23-dependent transcytosis of IgE and immune complex across the polarized human respiratory epithelial cells. J. Immunol. 2011, 186, 3484–3496. [Google Scholar] [CrossRef]
- Tu, Y.; Salim, S.; Bourgeois, J.; Di Leo, V.; Irvine, E.J.; Marshall, J.K.; Perdue, M.H. CD23-mediated IgE transport across human intestinal epithelium: Inhibition by blocking sites of translation or binding. Gastroenterology 2005, 129, 928–940. [Google Scholar] [CrossRef]
- Li, H.; Nowak-Wegrzyn, A.; Charlop-Powers, Z.; Shreffler, W.; Chehade, M.; Thomas, S.; Roda, G.; Dahan, S.; Sperber, K.; Berin, M.C. Transcytosis of IgE-antigen complexes by CD23a in human intestinal epithelial cells and its role in food allergy. Gastroenterology 2006, 131, 47–58. [Google Scholar] [CrossRef]
- McCloskey, N.; Hunt, J.; Beavil, R.L.; Jutton, M.R.; Grundy, G.J.; Girardi, E.; Fabiane, S.M.; Fear, D.J.; Conrad, D.H.; Sutton, B.J.; et al. Soluble CD23 monomers inhibit and oligomers stimulate IgE synthesis in human B cells. J. Biol. Chem. 2007, 282, 24083–24091. [Google Scholar] [CrossRef]
- Gould, H.J.; Beavil, R.L.; Reljić, R.; Shi, J.; Ma, C.W.; Sutton, B.J.; Ghirlando, R. IgE Homeostasis: Is CD23 the safety switch? In IgE Regulation: Molecular Mechanisms; Vercelli, D., Ed.; Wiley: Chichester, UK, 1997; pp. 37–59. [Google Scholar]
- Cooper, A.M.; Hobson, P.S.; Jutton, M.R.; Kao, M.W.; Drung, B.; Schmidt, B.; Fear, D.J.; Beavil, A.J.; McDonnell, J.M.; Sutton, B.J.; et al. Soluble CD23 controls IgE synthesis and homeostasis in human B cells. J. Immunol. 2012, 188, 3199–3207. [Google Scholar] [CrossRef]
- Palaniyandi, S.; Liu, X.; Periasamy, S.; Ma, A.; Tang, J.; Jenkins, M.; Tuo, W.; Song, W.; Keegan, A.D.; Conrad, D.H.; et al. Inhibition of CD23-mediated IgE transcytosis suppresses the initiation and development of allergic airway inflammation. Mucosal Immunol. 2015, 8, 1262–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitropoulou, A.N.; Bowen, H.; Dodev, T.; Davies, A.M.; Bax, H.; Beavil, R.L.; Beavil, A.J.; Gould, H.J.; James, L.K.; Sutton, B.J. Structure of a patient-derived antibody in complex with allergen reveals simultaneous conventional and superantigen-like recognition. Proc. Natl. Acad. Sci. USA 2018, 115, E8707–E8716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Calvert, R.A.; Sutton, B.J.; Doré, K.A. IgY: A key isotype in antibody evolution. Biol. Rev. Camb. Philos. Soc. 2017, 92, 2144–2156. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, A.; Munn, E.A. Conformation of the free and antigen-bound IgM antibody molecules. Nature 1969, 224, 1307–1309. [Google Scholar] [CrossRef] [PubMed]
- Crispin, M.; Yu, X.; Bowden, T.A. Crystal structure of sialylated IgG Fc: Implications for the mechanism of intravenous immunoglobulin therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3544–E3546. [Google Scholar] [CrossRef] [PubMed]
- Doré, K.A.; Davies, A.M.; Drinkwater, N.; Beavil, A.J.; McDonnell, J.M.; Sutton, B.J. Thermal sensitivity and flexibility of the Cε3 domains in immunoglobulin E. Biochim. Biophys. Acta 2017, 1865, 1336–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padlan, E.A.; Davies, D.R. A model of the Fc of Immunoglobulin-E. Mol. Immunol. 1986, 23, 1063–1075. [Google Scholar] [CrossRef]
- Holowka, D.; Baird, B. Structural studies on the membrane-bound immunoglobulin E (IgE)-receptor complex. 2. Mapping of distances between sites on IgE and the membrane surface. Biochemistry 1983, 22, 3475–3484. [Google Scholar] [CrossRef]
- Holowka, D.; Conrad, D.H.; Baird, B. Structural mapping of membrane-bound immunoglobulin-E receptor complexes: Use of monoclonal anti-IgE antibodies to probe the conformation of receptor-bound IgE. Biochemistry 1985, 24, 6260–6267. [Google Scholar] [CrossRef]
- Zheng, Y.; Shopes, B.; Holowka, D.; Baird, B. Conformations of IgE bound to its receptor FcεRI and in solution. Biochemistry 1991, 30, 9125–9132. [Google Scholar] [CrossRef]
- Zheng, Y.; Shopes, B.; Holowka, D.; Baird, B. Dynamic conformations compared for IgE and IgG1 in solution and bound to receptors. Biochemistry 1992, 31, 7446–7456. [Google Scholar] [CrossRef] [PubMed]
- Beavil, A.J.; Young, R.J.; Sutton, B.J.; Perkins, S.J. Bent domain structure of recombinant human IgE-Fc in solution by X-ray and neutron scattering in conjunction with an automated curve fitting procedure. Biochemistry 1995, 34, 14449–14461. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Beavil, R.L.; Fabiane, S.M.; Beavil, A.J.; Sohi, M.K.; Keown, M.; Young, R.J.; Henry, A.J.; Owens, R.J.; Gould, H.J.; et al. The crystal structure of IgE Fc reveals an asymmetrically bent conformation. Nat. Immunol. 2002, 3, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.; Keeble, A.H.; Dale, R.E.; Corbett, M.K.; Beavil, R.L.; Levitt, J.; Swann, M.J.; Suhling, K.; Ameer-Beg, S.; Sutton, B.J.; et al. A fluorescent biosensor reveals conformational changes in human immunoglobulin E Fc: Implications for mechanisms of receptor binding, inhibition, and allergen recognition. J. Biol. Chem. 2012, 287, 17459–17470. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.M.; Allan, E.G.; Keeble, A.H.; Delgado, J.; Cossins, B.P.; Mitropoulou, A.N.; Pang, M.O.Y.; Ceska, T.; Beavil, A.J.; Craggs, G.; et al. Allosteric mechanism of action of the therapeutic anti-IgE antibody omalizumab. J. Biol. Chem. 2017, 292, 9975–9987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drinkwater, N.; Cossins, B.P.; Keeble, A.H.; Wright, M.; Cain, K.; Hailu, H.; Oxbrow, A.; Delgado, J.; Shuttleworth, L.K.; Kao, M.W.; et al. Human immunoglobulin E flexes between acutely bent and extended conformations. Nat. Struct. Mol. Biol. 2014, 21, 397–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-B.; Ramadani, F.; Pang, M.O.Y.; Beavil, R.L.; Holdom, M.D.; Mitropoulou, A.N.; Beavil, A.J.; Gould, H.J.; Chang, T.-W.; Sutton, B.J.; et al. Structural basis for selective inhibition of immunoglobulin E-receptor interactions by an anti-IgE antibody. Sci. Rep. 2018, 8, 11548. [Google Scholar] [CrossRef]
- Arnold, J.N.; Radcliffe, C.M.; Wormald, M.R.; Royle, L.; Harvey, D.J.; Crispin, M.; Dwek, R.A.; Sim, R.B.; Rudd, P.M. The glycosylation of human serum IgD and IgE and the accessibility of identified oligomannose structures for interaction with mannan-binding lectin. J. Immunol. 2004, 173, 6831–6840. [Google Scholar] [CrossRef]
- Plomp, R.; Hensbergen, P.J.; Rombouts, Y.; Zauner, G.; Dragan, I.; Koeleman, C.A.; Deelder, A.M.; Wuhrer, M. Site-specific N-glycosylation analysis of human immunoglobulin E. J. Proteome Res. 2014, 13, 536–546. [Google Scholar] [CrossRef]
- Shade, K.T.; Platzer, B.; Washburn, N.; Mani, V.; Bartsch, Y.C.; Conroy, M.; Pagan, J.D.; Bosques, C.; Mempel, T.R.; Fiebiger, E.; et al. A single glycan on IgE is indispensible for initiation of anaphylaxis. J. Exp. Med. 2015, 212, 457–467. [Google Scholar] [CrossRef]
- Fridriksson, E.K.; Beavil, A.; Holowka, D.; Gould, H.J.; Baird, B.; McLafferty, F.W. Heterogeneous glycosylation of immunoglobulin E constructs characterized by top-down high-resolution 2-D mass spectrometry. Biochemistry 2000, 39, 3369–3376. [Google Scholar] [CrossRef]
- Taylor, A.I.; Fabiane, S.M.; Sutton, B.J.; Calvert, R.A. The crystal structure of an avian IgY-Fc fragment reveals conservation with both mammalian IgG and IgE. Biochemistry 2009, 48, 558–562. [Google Scholar] [CrossRef]
- Holdom, M.D.; Davies, A.M.; Nettleship, J.E.; Bagby, S.C.; Dhaliwal, B.; Girardi, E.; Hunt, J.; Gould, H.J.; Beavil, A.J.; McDonnell, J.M.; et al. Conformational changes in IgE contribute to its uniquely slow dissociation rate from receptor FcεRI. Nat. Struct. Mol. Biol. 2011, 18, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu. Rev. Immunol. 2007, 25, 21–50. [Google Scholar] [CrossRef] [PubMed]
- Helm, B.; Marsh, P.; Vercelli, D.; Padlan, E.; Gould, H.; Geha, R. The mast cell binding site on human immunoglobulin E. Nature 1988, 331, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Vercelli, D.; Helm, B.; Marsh, P.; Padlan, E.; Geha, R.; Gould, H. The B-cell binding site on human immunoglobulin E. Nature 1989, 338, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Basu, M.; Hakimi, J.; Dharm, E.; Kondas, J.A.; Tsien, W.H.; Pilson, R.S.; Lin, P.; Gilfillan, A.; Haring, P.; Braswell, E.H.; et al. Purification and characterization of human recombinant IgE-Fc fragments that bind to the human high affinity IgE receptor. J. Biol. Chem. 1993, 268, 13118–13127. [Google Scholar]
- Hunt, J.; Beavil, R.L.; Calvert, R.A.; Gould, H.J.; Sutton, B.J.; Beavil, A.J. Disulfide linkage controls the affinity and stoichiometry of IgE Fcε3-4 binding to FcεRI. J. Biol. Chem. 2005, 280, 16808–16814. [Google Scholar] [CrossRef]
- Sayers, I.; Cain, S.A.; Swan, J.R.; Pickett, M.A.; Watt, P.J.; Holgate, S.T.; Padlan, E.A.; Schuck, P.; Helm, B.A. Amino acid residues that influence FcεRI-mediated effector functions of human immunoglobulin E. Biochemistry 1998, 37, 16152–16164. [Google Scholar] [CrossRef]
- Dhaliwal, B.; Yuan, D.; Pang, M.O.; Henry, A.J.; Cain, K.; Oxbrow, A.; Fabiane, S.M.; Beavil, A.J.; McDonnell, J.M.; Gould, H.J.; et al. Crystal structure of IgE bound to its B-cell receptor CD23 reveals a mechanism of reciprocal allosteric inhibition with high affinity receptor FcεRI. Proc. Natl. Acad. Sci. USA 2012, 109, 12686–12691. [Google Scholar] [CrossRef]
- Cohen, E.S.; Dobson, C.L.; Käck, H.; Wang, B.; Sims, D.A.; Lloyd, C.O.; England, E.; Rees, D.G.; Guo, H.; Karagiannis, S.N.; et al. A novel IgE-neutralizing antibody for the treatment of severe uncontrolled asthma. mAbs 2014, 6, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, B.; Pang, M.O.; Keeble, A.H.; James, L.K.; Gould, H.J.; McDonnell, J.M.; Sutton, B.J.; Beavil, A.J. IgE binds asymmetrically to its B cell receptor CD23. Sci. Rep. 2017, 7, 45533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, D.; Keeble, A.H.; Hibbert, R.G.; Fabiane, S.; Gould, H.J.; McDonnell, J.M.; Beavil, A.J.; Sutton, B.J.; Dhaliwal, B. Ca2+-dependent structural changes in the B-cell receptor CD23 increase its affinity for human immunoglobulin E. J. Biol. Chem. 2013, 288, 21667–21677. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, B.; Pang, M.O.Y.; Yuan, D.; Beavil, A.J.; Sutton, B.J. A range of Cε3-Cε4 interdomain angles in IgE Fc accommodate binding to its receptor CD23. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Garman, S.C.; Wurzburg, B.A.; Tarchevskaya, S.S.; Kinet, J.-P.; Jardetzky, T.S. Structure of the Fc fragment of human IgE bound to its high-affinity receptor FcεRIα. Nature 2000, 406, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Wurzburg, B.A.; Garman, S.C.; Jardetzky, T.S. Structure of the human IgE-Fc Cε3-Cε4 reveals conformational flexibility in the antibody effector domains. Immunity 2000, 13, 375–385. [Google Scholar] [CrossRef]
- Wurzburg, B.A.; Jardetzky, T.S. Conformational Flexibility in the IgE-Fc3-4 Revealed in Multiple Crystal Forms. J. Mol. Biol. 2009, 393, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Jabs, F.; Plum, M.; Laursen, N.S.; Jensen, R.K.; Mølgaard, B.; Miehe, M.; Mandolesi, M.; Rauber, M.M.; Pfützner, W.; Jakob, T.; et al. Trapping IgE in a closed conformation by mimicking CD23 binding prevents and disrupts FcεRI interaction. Nat. Commun. 2018, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Oi, V.T.; Vuong, T.M.; Hardy, R.; Reidler, J.; Dangl, J.; Herzenberg, L.A.; Stryer, L. Correlation between segmental flexibility and effector function of antibodies. Nature 1983, 307, 136–140. [Google Scholar] [CrossRef]
- Gould, H.J.; Sutton, B.J.; Beavil, A.J.; Beavil, R.L.; McCloskey, N.; Coker, H.A.; Fear, D.; Smurthwaite, L. The biology of IgE and the basis of allergic disease. Annu. Rev. Immunol. 2003, 21, 579–628. [Google Scholar] [CrossRef]
- Hibbert, R.G.; Teriete, P.; Grundy, G.J.; Beavil, R.L.; Reljic, R.; Holers, V.M.; Hannan, J.P.; Sutton, B.J.; Gould, H.J.; McDonnell, J.M. The structure of human CD23 and its interactions with IgE and CD21. J. Exp. Med. 2005, 202, 751–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubry, J.-P.; Pochon, S.; Graber, P.; Jansen, K.U.; Bonnefoy, J.-Y. CD21 is a ligand for CD23 and regulates IgE production. Nature 1992, 358, 505–507. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.L.; Katz, D.H. The binding of IgE to murine FcεRII is calcium-dependent but not inhibited by carbohydrate. J. Immunol. 1990, 144, 2638–2646. [Google Scholar] [PubMed]
- Karagiannis, S.N.; Warrack, J.K.; Jennings, K.H.; Murdock, P.R.; Christie, G.; Moulder, K.; Sutton, B.J.; Gould, H.J. Endocytosis and recycling of the complex between CD23 and HLA-DR in human B cells. Immunology 2001, 103, 319–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, C.B.F.; Moestrup, S.K. How calcium makes endocytic receptors attractive. Trends Biochem. Sci. 2014, 39, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.E.; Chen, B.-H.; Woodward, E.C.; Conrad, D.H. Production of a chimeric form of CD23 that is oligomeric and blocks IgE binding to the FcεRI. J. Immunol. 1998, 161, 6696–6704. [Google Scholar] [PubMed]
- Suemura, M.; Kikutani, H.; Sugiyama, K.; Uchibayashi, N.; Aitani, M.; Kuritani, T.; Barsumian, E.L.; Yamatodani, A.; Kishimoto, T. Significance of soluble Fcε receptor II (sFcεRII/CD23) in serum and possible application of sFcεRII for the prevention of allergic reactions. Allergy Proc. 1991, 12, 133–137. [Google Scholar] [CrossRef]
- Borthakur, S.; Hibbert, R.G.; Pang, M.O.; Yahya, N.; Bax, H.J.; Kao, M.W.; Cooper, A.M.; Beavil, A.J.; Sutton, B.J.; Gould, H.J.; et al. Mapping of the CD23 binding site on immunoglobulin E (IgE) and allosteric control of the IgE-FcεRI interaction. J. Biol. Chem. 2012, 287, 31457–31461. [Google Scholar] [CrossRef]
- Henry, A.J.; McDonnell, J.M.; Ghirlando, R.; Sutton, B.J.; Gould, H.J. Conformation of the isolated Cε3 domain of IgE and its complex with the high-affinity receptor, FcεRI. Biochemistry 2000, 39, 7406–7413. [Google Scholar] [CrossRef]
- Vangelista, L.; Laffer, S.; Turek, R.; Grönlund, H.; Sperr, W.R.; Valent, P.; Pastore, A.; Valenta, R. The immunoglobulin-like modules Cε3 and α2 are the minimal units necessary for human IgE-FεRI interaction. J. Clin. Investig. 1999, 103, 1571–1578. [Google Scholar] [CrossRef]
- Price, N.E.; Price, N.C.; Kelly, S.M.; McDonnell, J.M. The key role of protein flexibility in modulating IgE interactions. J. Biol. Chem. 2005, 280, 2324–2330. [Google Scholar] [CrossRef] [PubMed]
- Harwood, N.E.; McDonnell, J.M. The intrinsic flexibility of IgE and its role in binding FcεRI. Biomed. Pharmacother. 2007, 61, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, S.; Andrejeva, G.; McDonnell, J.M. Basis of the intrinsic flexibility of the Cε3 domain of IgE. Biochemistry 2011, 50, 4608–4614. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, B.; Pang, M.O.; Yuan, D.; Yahya, N.; Fabiane, S.M.; McDonnell, J.M.; Gould, H.J.; Beavil, A.J.; Sutton, B.J. Conformational plasticity at the IgE-binding site of the B-cell receptor CD23. Mol. Immunol. 2013, 56, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Dorrington, K.J.; Bennich, H. Thermally induced structural changes in immunoglobulin E. J. Biol. Chem. 1973, 248, 8378–8384. [Google Scholar]
- Eggel, A.; Baravalle, G.; Hobi, G.; Kim, B.; Buschor, P.; Forrer, P.; Shin, J.S.; Vogel, M.; Stadler, B.M.; Dahinden, C.A.; et al. Accelerated dissociation of IgE-FcεRI complexes by disruptive inhibitors actively desensitizes allergic effector cells. J. Allergy Clin. Immunol. 2014, 133, 1709–1719. [Google Scholar] [CrossRef]
- Pennington, L.F.; Tarchevskaya, S.; Brigger, D.; Sathiyamoorthy, K.; Graham, M.T.; Nadeau, K.C.; Eggel, A.; Jardetzky, T.S. Structural basis of omalizumab therapy and omalizumab-mediated IgE exchange. Nat. Commun. 2016, 7, 11610. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Eggel, A.; Tarchevskaya, S.S.; Vogel, M.; Prinz, H.; Jardetzky, T.S. Accelerated disassembly of IgE-receptor complexes by a disruptive macromolecular inhibitor. Nature 2012, 491, 613–617. [Google Scholar] [CrossRef]
- Roux, K.H.; Strelets, L.; Brekke, O.H.; Sandlie, I.; Michaelsen, T.E. Comparisons of the ability of human IgG3 hinge mutants, IgM, IgE, and IgA2, to form small immune complexes: A role for flexibility and geometry. J. Immunol. 1998, 161, 4083–4090. [Google Scholar]
- Gieras, A.; Linhart, B.; Roux, K.H.; Dutta, M.; Khodoun, M.; Zafred, D.; Cabauatan, C.R.; Lupinek, C.; Weber, M.; Focke-Tejkl, M.; et al. IgE epitope proximity determines immune complex shape and effector cell activation capacity. J. Allergy Clin. Immunol. 2016, 137, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Christensen, L.H.; Holm, J.; Lund, G.; Riise, E.; Lund, K. Several distinct properties of the IgE repertoire determine effector cell degranulation in response to allergen challenge. J. Allergy Clin. Immunol. 2008, 122, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Padlan, E.A.; Silverton, E.W.; Sheriff, S.; Cohen, G.H.; Smith-Gill, S.J.; Davies, D.R. Structure of an antibody-antigen complex: Crystal structure of the HyHEL-10 Fab-lysozyme complex. Proc. Natl. Acad. Sci USA 1989, 86, 5938–5942. [Google Scholar] [CrossRef] [PubMed]
- Mirza, O.; Henriksen, A.; Ipsen, H.; Larsen, J.N.; Wissenbach, M.; Spangfort, M.D.; Gajhede, M. Dominant epitopes and allergic cross-reactivity: Complex formation between a Fab fragment of a monoclonal murine IgG antibody and the major allergen from birch pollen Bet v 1. J. Immunol. 2000, 165, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Padavattan, S.; Schirmer, T.; Schmidt, M.; Akdis, C.; Valenta, R.; Mittermann, I.; Soldatova, L.; Slater, J.; Mueller, U.; Markovic-Housley, Z. Identification of a B-cell Epitope of Hyaluronidase, a Major Bee Venom Allergen, from its Crystal Structure in Complex with a Specific Fab. J. Mol. Biol. 2007, 368, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gustchina, A.; Alexandratos, J.; Wlodawer, A.; Wünschmann, S.; Kepley, C.L.; Chapman, M.D.; Pomés, A. Crystal structure of a dimerized cockroach allergen Bla g 2 complexed with a monoclonal antibody. J. Biol. Chem. 2008, 283, 22806–22814. [Google Scholar] [CrossRef] [PubMed]
- Chruszcz, M.; Pomés, A.; Glesner, J.; Vailes, L.D.; Osinski, T.; Porebski, P.J.; Majorek, K.A.; Heymann, P.W.; Platts-Mills, T.A.; Minor, W.; et al. Molecular determinants for antibody binding on group 1 house dust mite allergens. J. Biol. Chem. 2012, 287, 7388–7398. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gustchina, A.; Glesner, J.; Wünschmann, S.; Vailes, L.D.; Chapman, M.D.; Pomés, A.; Wlodawer, A. Carbohydrates Contribute to the Interactions between Cockroach Allergen Bla g 2 and a Monoclonal Antibody. J. Immunol. 2011, 186, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Osinski, O.; Pomés, A.; Majorek, K.A.; Glesner, J.; Offermann, L.R.; Vailes, L.D.; Chapman, M.D.; Minor, W.; Chruszcz, M. Structural Analysis of Der p 1–Antibody Complexes and Comparison with Complexes of Proteins or Peptides with Monoclonal Antibodies. J. Immunol. 2015, 195, 307–316. [Google Scholar] [CrossRef]
- Orengo, J.M.; Radin, A.R.; Kamat, V.; Badithe, A.; Ben, L.H.; Bennett, B.L.; Zhong, S.; Birchard, D.; Limnander, A.; Rafique, A.; et al. Treating cat allergy with monoclonal IgG antibodies that bind allergen and prevent IgE engagement. Nat. Commun. 2018, 9, 1421. [Google Scholar] [CrossRef] [Green Version]
- Niemi, M.; Jylhä, S.; Laukkanen, M.-L.; Söderlund, H.; Mäkinen-Kiljunen, S.; Kallio, J.M.; Hakulinen, N.; Haahtela, T.; Takkinen, K.; Rouvinen, J. Molecular Interactions between a Recombinant IgE Antibody and the β-Lactoglobulin Allergen. Structure 2007, 15, 1413–1421. [Google Scholar] [CrossRef] [Green Version]
- Padavattan, S.; Flicker, S.; Schirmer, T.; Madritsch, C.; Randow, S.; Reese, G.; Vieths, S.; Lupinek, C.; Ebner, C.; Valenta, R.; et al. High-affinity IgE recognition of a conformational epitope of the major respiratory allergen Phl p 2 as revealed by X-ray crystallography. J. Immunol. 2009, 182, 2141–2151. [Google Scholar] [CrossRef]
- Glesner, J.; Wünschmann, S.; Li, M.; Gustchina, A.; Wlodawer, A.; Himly, M.; Chapman, M.D.; Pomés, A. Mechanisms of allergen-antibody interaction of cockroach allergen Bla g 2 with monoclonal antibodies that inhibit IgE antibody binding. PLoS ONE 2011, 6, e22223. [Google Scholar] [CrossRef]
- Marone, G.; Rossi, F.W.; Detoraki, A.; Granata, F.; Marone, G.; Genovese, A.; Spadaro, G. Role of superallergens in allergic disorders. Chem. Immunol. Allergy 2007, 93, 195–213. [Google Scholar] [CrossRef]
- Zacharia, B.E.; Sherman, P. Atopy, helminths, and cancer. Med. Hypotheses 2003, 60, 1–5. [Google Scholar] [CrossRef]
- Finkelman, F.D.; Urban, J.F., Jr. The other side of the coin: The protective role of the TH2 cytokines. J. Allergy Clin. Immunol. 2001, 107, 772–780. [Google Scholar] [CrossRef]
- Gurish, M.F.; Bryce, P.J.; Tao, H.; Kisselgof, A.B.; Thornton, E.M.; Miller, H.R.; Friend, D.S.; Oettgen, H.C. IgE enhances parasite clearance and regulates mast cell responses in mice infected with Trichinella spiralis. J. Immunol. 2004, 172, 1139–1145. [Google Scholar] [CrossRef]
- Ure, D.M. Negative association between allergy and cancer. Scott. Med. J. 1969, 14, 51–54. [Google Scholar] [CrossRef]
- Schlitter, H.E. Is there an allergy against malignant tumor tissue and what can it signify in regard to the defense of the body against cancer? Strahlentherapie 1961, 114, 203–204. [Google Scholar]
- McCormick, D.P.; Ammann, A.J.; Ishizaka, K.; Miller, D.G.; Hong, R. A study of allergy in patients with malignant lymphoma and chronic lymphocytic leukemia. Cancer 1971, 27, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Augustin, R.; Chandradasa, K.D. IgE levels and allergic skin reactions in cancer and non-cancer patients. Int. Arch. Allergy Appl. Immunol. 1971, 41, 141–143. [Google Scholar] [CrossRef]
- Jacobs, D.; Landon, J.; Houri, M.; Merrett, T.G. Circulating levels of immunoglobulin E in patients with cancer. Lancet 1972, 2, 1059–1061. [Google Scholar] [CrossRef]
- Allegra, J.; Lipton, A.; Harvey, H.; Luderer, J.; Brenner, D.; Mortel, R.; Demers, L.; Gillin, M.; White, D.; Trautlein, J. Decreased prevalence of immediate hypersensitivity (atopy) in a cancer population. Cancer Res. 1976, 36, 3225–3226. [Google Scholar]
- Neuchrist, C.; Kornfehl, J.; Grasl, M.; Lassmann, H.; Kraft, D.; Ehrenberger, K.; Scheiner, O. Distribution of immunoglobulins in squamous cell carcinoma of the head and neck. Int. Arch. Allergy Immunol. 1994, 104, 97–100. [Google Scholar] [CrossRef]
- Fu, S.L.; Pierre, J.; Smith-Norowitz, T.A.; Hagler, M.; Bowne, W.; Pincus, M.R.; Mueller, C.M.; Zenilman, M.E.; Bluth, M.H. Immunoglobulin E antibodies from pancreatic cancer patients mediate antibody-dependent cell-mediated cytotoxicity against pancreatic cancer cells. Clin. Exp. Immunol. 2008, 153, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Crawford, G.; Hayes, M.D.; Seoane, R.C.; Ward, S.; Dalessandri, T.; Lai, C.; Healy, E.; Kipling, D.; Proby, C.; Moyes, C.; et al. Epithelial damage and tissue γδ T cells promote a unique tumor-protective IgE response. Nat. Immunol. 2018, 19, 859–870. [Google Scholar] [CrossRef]
- Disney-Hogg, L.; Cornish, A.J.; Sud, A.; Law, P.J.; Kinnersley, B.; Jacobs, D.I.; Ostrom, Q.T.; Labreche, K.; Eckel-Passow, J.E.; Armstrong, G.N.; et al. Impact of atopy on risk of glioma: A Mendelian randomisation study. BMC Med. 2018, 16, 42. [Google Scholar] [CrossRef]
- Helby, J.; Bojesen, S.E.; Nielsen, S.F.; Nordestgaard, B.G. IgE and risk of cancer in 37,747 individuals from the general population. Ann. Oncol. 2015, 26, 1784–1790. [Google Scholar] [CrossRef]
- Liao, H.C.; Wu, S.Y.; Ou, C.Y.; Hsiao, J.R.; Huang, J.S.; Tsai, S.T.; Huang, C.C.; Wong, T.Y.; Lee, W.T.; Chen, K.C.; et al. Allergy symptoms, serum total immunoglobulin E, and risk of head and neck cancer. Cancer Causes Control 2016, 27, 1105–1115. [Google Scholar] [CrossRef]
- Wulaningsih, W.; Holmberg, L.; Garmo, H.; Karagiannis, S.N.; Ahlstedt, S.; Malmstrom, H.; Lambe, M.; Hammar, N.; Walldius, G.; Jungner, I.; et al. Investigating the association between allergen-specific immunoglobulin E, cancer risk and survival. Oncoimmunology 2016, 5, e1154250. [Google Scholar] [CrossRef] [Green Version]
- Taghizadeh, N.; Vonk, J.M.; Hospers, J.J.; Postma, D.S.; de Vries, E.G.; Schouten, J.P.; Boezen, H.M. Objective allergy markers and risk of cancer mortality and hospitalization in a large population-based cohort. Cancer Causes Control 2015, 26, 99–109. [Google Scholar] [CrossRef]
- Van Hemelrijck, M.; Karagiannis, S.N.; Rohrmann, S. Atopy and prostate cancer: Is there a link between circulating levels of IgE and PSA in humans? Cancer Immunol. Immunother. 2017, 66, 1557–1562. [Google Scholar] [CrossRef]
- Kural, Y.B.; Su, O.; Onsun, N.; Uras, A.R. Atopy, IgE and eosinophilic cationic protein concentration, specific IgE positivity, eosinophil count in cutaneous T Cell lymphoma. Int. J. Dermatol. 2010, 49, 390–395. [Google Scholar] [CrossRef]
- Kretschmer, A.; Schwanbeck, R.; Valerius, T.; Rösner, T. Antibody Isotypes for Tumor Immunotherapy. Transfus. Med. Hemother. 2017, 44, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Leusen, J.H. IgA as therapeutic antibody. Mol. Immunol. 2015, 68, 35–39. [Google Scholar] [CrossRef]
- Lohse, S.; Meyer, S.; Meulenbroek, L.A.; Jansen, J.H.; Nederend, M.; Kretschmer, A.; Klausz, K.; Möginger, U.; Derer, S.; Rösner, T.; et al. An Anti-EGFR IgA That Displays Improved Pharmacokinetics and Myeloid Effector Cell Engagement In Vivo. Cancer Res. 2016, 76, 403–417. [Google Scholar] [CrossRef]
- Josephs, D.H.; Spicer, J.F.; Karagiannis, P.; Gould, H.J.; Karagiannis, S.N. IgE immunotherapy: A novel concept with promise for the treatment of cancer. mAbs 2014, 6, 54–72. [Google Scholar] [CrossRef]
- Waldmann, T.A.; Iio, A.; Ogawa, M.; McIntyre, O.R.; Strober, W. The metabolism of IgE. Studies in normal individuals and in a patient with IgE myeloma. J. Immunol. 1976, 117, 1139–1144. [Google Scholar]
- Lawrence, M.G.; Woodfolk, J.A.; Schuyler, A.J.; Stillman, L.C.; Chapman, M.D.; Platts-Mills, T.A. Half-life of IgE in serum and skin: Consequences for anti-IgE therapy in patients with allergic disease. J. Allergy Clin. Immunol. 2017, 139, 422–428. [Google Scholar] [CrossRef]
- Verwaerde, C.; Joseph, M.; Capron, M.; Pierce, R.J.; Damonneville, M.; Velge, F.; Auriault, C.; Capron, A. Functional properties of a rat monoclonal IgE antibody specific for Schistosoma mansoni. J. Immunol. 1987, 138, 4441–4446. [Google Scholar]
- Vouldoukis, I.; Riveros-Moreno, V.; Dugas, B.; Ouaaz, F.; Bécherel, P.; Debré, P.; Moncada, S.; Mossalayi, M.D. The killing of Leishmania major by human macrophages is mediated by nitric oxide induced after ligation of the Fc epsilon RII/CD23 surface antigen. Proc. Natl. Acad. Sci. USA 1995, 92, 7804–7808. [Google Scholar] [CrossRef]
- Vouldoukis, I.; Mazier, D.; Moynet, D.; Thiolat, D.; Malvy, D.; Mossalayi, M.D. IgE mediates killing of intracellular Toxoplasma gondii by human macrophages through CD23-dependent, interleukin-10 sensitive pathway. PLoS ONE 2011, 6, e18289. [Google Scholar] [CrossRef]
- Hagan, P.; Blumenthal, U.J.; Dunn, D.; Simpson, A.J.; Wilkins, H.A. Human IgE, IgG4 and resistance to reinfection with Schistosoma haematobium. Nature 1991, 349, 243–245. [Google Scholar] [CrossRef]
- Dunne, D.W.; Butterworth, A.E.; Fulford, A.J.; Ouma, J.H.; Sturrock, R.F. Human IgE responses to Schistosoma mansoni and resistance to reinfection. Mem. Inst. Oswaldo Cruz 1992, 87, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, N.; Bruschi, F.; Korenaga, M. IgE: A question of protective immunity in Trichinella spiralis infection. Trends Parasitol. 2005, 21, 175–178. [Google Scholar] [CrossRef]
- Gounni, A.S.; Lamkhioued, B.; Ochiai, K.; Tanaka, Y.; Delaporte, E.; Capron, A.; Kinet, J.P.; Capron, M. High-affinity IgE receptor on eosinophils is involved in defence against parasites. Nature 1994, 367, 183–186. [Google Scholar] [CrossRef]
- Kamisawa, T.; Zen, Y.; Pillai, S.; Stone, J.H. IgG4-related disease. Lancet 2015, 385, 1460–1471. [Google Scholar] [CrossRef]
- Weindorf, S.C.; Frederiksen, J.K. IgG4-Related Disease: A Reminder for Practicing Pathologists. Arch. Pathol. Lab. Med. 2017, 141, 1476–1483. [Google Scholar] [CrossRef] [Green Version]
- Wallace, Z.S.; Mattoo, H.; Carruthers, M.; Mahajan, V.S.; Della Torre, E.; Lee, H.; Kulikova, M.; Deshpande, V.; Pillai, S.; Stone, J.H. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann. Rheum. Dis. 2015, 74, 190–195. [Google Scholar] [CrossRef]
- Crescioli, S.; Correa, I.; Karagiannis, P.; Davies, A.M.; Sutton, B.J.; Nestle, F.O.; Karagiannis, S.N. IgG4 Characteristics and Functions in Cancer Immunity. Curr. Allergy Asthma Rep. 2016, 16, 7. [Google Scholar] [CrossRef]
- Liu, Q.; Niu, Z.; Li, Y.; Wang, M.; Pan, B.; Lu, Z.; Liao, Q.; Zhao, Y. Immunoglobulin G4 (IgG4)-positive plasma cell infiltration is associated with the clinicopathologic traits and prognosis of pancreatic cancer after curative resection. Cancer Immunol. Immunother. 2016, 65, 931–940. [Google Scholar] [CrossRef]
- Harada, K.; Shimoda, S.; Kimura, Y.; Sato, Y.; Ikeda, H.; Igarashi, S.; Ren, X.; Sato, H.; Nakanuma, Y. Significance of immunoglobulin G4 (IgG4)-positive cells in extrahepatic cholangiocarcinoma: Molecular mechanism of IgG4 reaction in cancer tissue. Hepatology 2012, 56, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, M.; Yoshizawa, A.; Sumiyoshi, S.; Sonobe, M.; Kobayashi, M.; Koyanagi, I.; Aini, W.; Tsuruyama, T.; Date, H.; Haga, H. Stromal plasma cells expressing immunoglobulin G4 subclass in non-small cell lung cancer. Hum. Pathol. 2013, 44, 1569–1576. [Google Scholar] [CrossRef]
- Karagiannis, P.; Gilbert, A.E.; Josephs, D.H.; Ali, N.; Dodev, T.; Saul, L.; Correa, I.; Roberts, L.; Beddowes, E.; Koers, A.; et al. IgG4 subclass antibodies impair antitumor immunity in melanoma. J. Clin. Investig. 2013, 123, 1457–1474. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, P.; Villanova, F.; Josephs, D.H.; Correa, I.; Van Hemelrijck, M.; Hobbs, C.; Saul, L.; Egbuniwe, I.U.; Tosi, I.; Ilieva, K.M.; et al. Elevated IgG4 in patient circulation is associated with the risk of disease progression in melanoma. Oncoimmunology 2015, 4, e1032492. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, P.; Gilbert, A.E.; Nestle, F.O.; Karagiannis, S.N. IgG4 antibodies and cancer-associated inflammation: Insights into a novel mechanism of immune escape. Oncoimmunology 2013, 2, e24889. [Google Scholar] [CrossRef]
- Jensen-Jarolim, E.; Bax, H.J.; Bianchini, R.; Capron, M.; Corrigan, C.; Castells, M.; Dombrowicz, D.; Daniels-Wells, T.R.; Fazekas, J.; Fiebiger, E.; et al. AllergoOncology—The impact of allergy in oncology: EAACI position paper. Allergy 2017, 72, 866–887. [Google Scholar] [CrossRef]
- Platzer, B.; Elpek, K.G.; Cremasco, V.; Baker, K.; Stout, M.M.; Schultz, C.; Dehlink, E.; Shade, K.T.; Anthony, R.M.; Blumberg, R.S.; et al. IgE/FcεRI-Mediated Antigen Cross-Presentation by Dendritic Cells Enhances Anti-Tumor Immune Responses. Cell Rep. 2015, 10, 1487–1495. [Google Scholar] [CrossRef] [Green Version]
- Platzer, B.; Dehlink, E.; Turley, S.J.; Fiebiger, E. How to connect an IgE-driven response with CTL activity? Cancer Immunol. Immunother. 2012, 61, 1521–1525. [Google Scholar] [CrossRef]
- Kamta, J.; Chaar, M.; Ande, A.; Altomare, D.A.; Ait-Oudhia, S. Advancing Cancer Therapy with Present and Emerging Immuno-Oncology Approaches. Front. Oncol. 2017, 7, 64. [Google Scholar] [CrossRef]
- Jensen-Jarolim, E.; Turner, M.C.; Karagiannis, S.N. AllergoOncology: IgE- and IgG4-mediated immune mechanisms linking allergy with cancer and their translational implications. J. Allergy Clin. Immunol. 2017, 140, 982–984. [Google Scholar] [CrossRef]
- Jensen-Jarolim, E.; Bax, H.J.; Bianchini, R.; Crescioli, S.; Daniels-Wells, T.R.; Dombrowicz, D.; Fiebiger, E.; Gould, H.J.; Irshad, S.; Janda, J.; et al. AllergoOncology: Opposite outcomes of immune tolerance in allergy and cancer. Allergy 2018, 73, 328–340. [Google Scholar] [CrossRef]
- Jensen-Jarolim, E.; Achatz, G.; Turner, M.C.; Karagiannis, S.; Legrand, F.; Capron, M.; Penichet, M.L.; Rodríguez, J.A.; Siccardi, A.G.; Vangelista, L.; et al. AllergoOncology: The role of IgE-mediated allergy in cancer. Allergy 2008, 63, 1255–1266. [Google Scholar] [CrossRef]
- Karagiannis, S.N.; Josephs, D.H.; Karagiannis, P.; Gilbert, A.E.; Saul, L.; Rudman, S.M.; Dodev, T.; Koers, A.; Blower, P.J.; Corrigan, C.; et al. Recombinant IgE antibodies for passive immunotherapy of solid tumours: From concept towards clinical application. Cancer Immunol. Immunother. 2012, 61, 1547–1564. [Google Scholar] [CrossRef]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Kunert, R.; Reinhart, D. Advances in recombinant antibody manufacturing. Appl. Microbiol. Biotechnol. 2016, 100, 3451–3461. [Google Scholar] [CrossRef] [Green Version]
- Gould, H.J.; Mackay, G.A.; Karagiannis, S.N.; O’Toole, C.M.; Marsh, P.J.; Daniel, B.E.; Coney, L.R.; Zurawski, V.R., Jr.; Joseph, M.; Capron, M.; et al. Comparison of IgE and IgG antibody-dependent cytotoxicity in vitro and in a SCID mouse xenograft model of ovarian carcinoma. Eur. J. Immunol. 1999, 29, 3527–3537. [Google Scholar] [CrossRef]
- Dodev, T.S.; Karagiannis, P.; Gilbert, A.E.; Josephs, D.H.; Bowen, H.; James, L.K.; Bax, H.J.; Beavil, R.; Pang, M.O.; Gould, H.J.; et al. A tool kit for rapid cloning and expression of recombinant antibodies. Sci. Rep. 2014, 4, 5885. [Google Scholar] [CrossRef]
- Bantleon, F.; Wolf, S.; Seismann, H.; Dam, S.; Lorentzen, A.; Miehe, M.; Jabs, F.; Jakob, T.; Plum, M.; Spillner, E. Human IgE is efficiently produced in glycosylated and biologically active form in lepidopteran cells. Mol. Immunol. 2016, 72, 49–56. [Google Scholar] [CrossRef]
- Montero-Morales, L.; Maresch, D.; Castilho, A.; Turupcu, A.; Ilieva, K.M.; Crescioli, S.; Karagiannis, S.N.; Lupinek, C.; Oostenbrink, C.; Altmann, F.; et al. Recombinant plant-derived human IgE glycoproteomics. J. Proteom. 2017, 161, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Ilieva, K.M.; Fazekas-Singer, J.; Achkova, D.Y.; Dodev, T.S.; Mele, S.; Crescioli, S.; Bax, H.J.; Cheung, A.; Karagiannis, P.; Correa, I.; et al. Functionally Active Fc Mutant Antibodies Recognizing Cancer Antigens Generated Rapidly at High Yields. Front. Immunol. 2017, 8, 1112. [Google Scholar] [CrossRef]
- Fazekas-Singer, J.; Singer, J.; Ilieva, K.M.; Matz, M.; Herrmann, I.; Spillner, E.; Karagiannis, S.N.; Jensen-Jarolim, E. AllergoOncology: Generating a canine anticancer IgE against the epidermal growth factor receptor. J. Allergy Clin. Immunol. 2018, 142, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Crescioli, S.; Chiaruttini, G.; Mele, S.; Ilieva, K.M.; Pellizzari, G.; Spencer, D.I.R.; Gardner, R.A.; Lacy, K.E.; Spicer, J.F.; Tutt, A.N.J.; et al. Engineering and stable production of recombinant IgE for cancer immunotherapy and AllergoOncology. J. Allergy Clin. Immunol. 2018, 141, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Boscolo, S.; Mion, F.; Licciulli, M.; Macor, P.; De Maso, L.; Brce, M.; Antoniou, M.N.; Marzari, R.; Santoro, C.; Sblattero, D. Simple scale-up of recombinant antibody production using an UCOE containing vector. New Biotechnol. 2012, 29, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.S.; Hung, A.F.; Lin, C.J.; Chen, J.B.; Chen, C.; Shiung, Y.Y.; Tsai, C.Y.; Chang, T.W. Generating allergen-specific human IgEs for immunoassays by employing human ε gene knockin mice. Allergy 2015, 70, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Hecker, J.; Diethers, A.; Schulz, D.; Sabri, A.; Plum, M.; Michel, Y.; Mempel, M.; Ollert, M.; Jakob, T.; Blank, S.; et al. An IgE epitope of Bet v 1 and fagales PR10 proteins as defined by a human monoclonal IgE. Allergy 2012, 67, 1530–1537. [Google Scholar] [CrossRef]
- Correa, I.; Ilieva, K.M.; Crescioli, S.; Lombardi, S.; Figini, M.; Cheung, A.; Spicer, J.F.; Tutt, A.N.J.; Nestle, F.O.; Karagiannis, P.; et al. Evaluation of Antigen-Conjugated Fluorescent Beads to Identify Antigen-Specific B Cells. Front. Immunol. 2018, 9, 493. [Google Scholar] [CrossRef]
- Nagy, E.; Berczi, I.; Sehon, A.H. Growth inhibition of murine mammary carcinoma by monoclonal IgE antibodies specific for the mammary tumor virus. Cancer Immunol. Immunother. 1991, 34, 63–69. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Darcy, P.K.; Trapani, J.A.; MacGregor, D.; Smyth, M.J. Tumor-specific IgE-mediated inhibition of human colorectal carcinoma xenograft growth. Oncol. Res. 1998, 10, 133–142. [Google Scholar]
- Daniels, T.R.; Leuchter, R.K.; Quintero, R.; Helguera, G.; Rodríguez, J.A.; Martínez-Maza, O.; Schultes, B.C.; Nicodemus, C.F.; Penichet, M.L. Targeting HER2/neu with a fully human IgE to harness the allergic reaction against cancer cells. Cancer Immunol. Immunother. 2012, 61, 991–1003. [Google Scholar] [CrossRef]
- Teo, P.Z.; Utz, P.J.; Mollick, J.A. Using the allergic immune system to target cancer: Activity of IgE antibodies specific for human CD20 and MUC1. Cancer Immunol. Immunother. 2012, 61, 2295–2309. [Google Scholar] [CrossRef]
- Daniels-Wells, T.R.; Helguera, G.; Leuchter, R.K.; Quintero, R.; Kozman, M.; Rodríguez, J.A.; Ortiz-Sánchez, E.; Martínez-Maza, O.; Schultes, B.C.; Nicodemus, C.F.; et al. A novel IgE antibody targeting the prostate specific antigen as a potential prostate cancer therapy. BMC Cancer 2013, 13, 195. [Google Scholar] [CrossRef]
- Karagiannis, P.; Singer, J.; Hunt, J.; Gan, S.K.; Rudman, S.M.; Mechtcheriakova, D.; Knittelfelder, R.; Daniels, T.R.; Hobson, P.S.; Beavil, A.J.; et al. Characterisation of an engineered trastuzumab IgE antibody and effector cell mechanisms targeting HER2/neu-positive tumour cells. Cancer Immunol. Immunother. 2009, 58, 915–930. [Google Scholar] [CrossRef]
- Spillner, E.; Plum, M.; Blank, S.; Miehe, M.; Singer, J.; Braren, I. Recombinant IgE antibody engineering to target EGFR. Cancer Immunol. Immunother. 2012, 61, 1565–1573. [Google Scholar] [CrossRef]
- Chung, C.H.; Mirakhur, B.; Chan, E.; Le, Q.T.; Berlin, J.; Morse, M.; Murphy, B.A.; Satinover, S.M.; Hosen, J.; Mauro, D.; et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N. Engl. J. Med. 2008, 358, 1109–1117. [Google Scholar] [CrossRef]
- Lammerts van Bueren, J.J.; Rispens, T.; Verploegen, S.; van der Palen-Merkus, T.; Stapel, S.; Workman, L.J.; James, H.; van Berkel, P.H.; van de Winkel, J.G.; Platts-Mills, T.A.; et al. Anti-galactose-α-1,3-galactose IgE from allergic patients does not bind α-galactosylated glycans on intact therapeutic antibody Fc domains. Nat. Biotechnol. 2011, 29, 574–576. [Google Scholar] [CrossRef]
- Galili, U. Anti-Gal: An abundant human natural antibody of multiple pathogeneses and clinical benefits. Immunology 2013, 140, 1–11. [Google Scholar] [CrossRef]
- Miotti, S.; Canevari, S.; Ménard, S.; Mezzanzanica, D.; Porro, G.; Pupa, S.M.; Regazzoni, M.; Tagliabue, E.; Colnaghi, M. Characterization of human ovarian carcinoma-associated antigens defined by novel monoclonal antibodies with tumor-restricted specificity. Int. J. Cancer 1987, 39, 297–303. [Google Scholar] [CrossRef]
- Coney, L.R.; Tomassetti, A.; Carayannopoulos, L.; Frasca, V.; Kamen, B.A.; Colnaghi, M.; Zurawski, V.R., Jr. Cloning of a tumor-associated antigen: MOv18 and MOv19 antibodies recognize a folate-binding protein. Cancer Res. 1991, 51, 6125–6132. [Google Scholar]
- Molthoff, C.F.; Prinssen, H.M.; Kenemans, P.; van Hof, A.C.; den Hollander, W.; Verheijen, R.H. Escalating protein doses of chimeric monoclonal antibody MOv18 immunoglobulin G in ovarian carcinoma patients: A phase I study. Cancer 1997, 80, 2712–2720. [Google Scholar] [CrossRef]
- Buijs, W.C.; Tibben, J.G.; Boerman, O.C.; Molthoff, C.F.; Massuger, L.F.; Koenders, E.B.; Schijf, C.P.; Siegel, J.A.; Corstens, F.H. Dosimetric analysis of chimeric monoclonal antibody cMOv18 IgG in ovarian carcinoma patients after intraperitoneal and intravenous administration. Eur. J. Nucl. Med. 1998, 25, 1552–1561. [Google Scholar] [CrossRef]
- Van Zanten-Przybysz, I.; Molthoff, C.; Gebbinck, J.K.; von Mensdorff-Pouilly, S.; Verstraeten, R.; Kenemans, P.; Verheijen, R. Cellular and humoral responses after multiple injections of unconjugated chimeric monoclonal antibody MOv18 in ovarian cancer patients: A pilot study. J. Cancer Res. Clin. Oncol. 2002, 128, 484–492. [Google Scholar] [CrossRef]
- Van Zanten-Przybysz, I.; Molthoff, C.F.; Roos, J.C.; Verheijen, R.H.; van Hof, A.; Buist, M.R.; Prinssen, H.M.; den Hollander, W.; Kenemans, P. Influence of the route of administration on targeting of ovarian cancer with the chimeric monoclonal antibody MOv18: i.v. vs. i.p. Int. J. Cancer 2001, 92, 106–114. [Google Scholar] [CrossRef]
- Bell-McGuinn, K.M.; Konner, J.; Pandit-Taskar, N.; Gerst, S.; Nicolaides, N.; Sass, P.; Grasso, L.; Weil, S.; Phillips, M.; Aghajanian, C. A phase I study of MORAb-003, a fully humanized monoclonal antibody against folate receptor alpha, in advanced epithelial ovarian cancer. J. Clin. Oncol. 2007, 25, 5553. [Google Scholar] [CrossRef]
- Konner, J.A.; Bell-McGuinn, K.M.; Sabbatini, P.; Hensley, M.L.; Tew, W.P.; Pandit-Taskar, N.; Vander, E.N.; Phillips, M.D.; Schweizer, C.; Weil, S.C.; et al. Farletuzumab, a humanized monoclonal antibody against folate receptor alpha, in epithelial ovarian cancer: A phase I study. Clin. Cancer Res. 2010, 16, 5288–5295. [Google Scholar] [CrossRef]
- Farrell, C.; Schweizer, C.; Wustner, J.; Weil, S.; Namiki, M.; Nakano, T.; Nakai, K.; Phillips, M.D. Population pharmacokinetics of farletuzumab, a humanized monoclonal antibody against folate receptor alpha, in epithelial ovarian cancer. Cancer Chemother. Pharmacol. 2012, 70, 727–734. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.; Opzoomer, J.; Ilieva, K.M.; Gazinska, P.; Hoffmann, R.M.; Mirza, H.; Marlow, R.; Francesch-Domenech, E.; Fittall, M.; Dominguez Rodriguez, D.; et al. Anti-Folate Receptor Alpha-Directed Antibody Therapies Restrict the Growth of Triple-negative Breast Cancer. Clin. Cancer Res. 2018, 24, 5098–5111. [Google Scholar] [CrossRef]
- Tochowicz, A.; Dalziel, S.; Eidam, O.; O’Connell, J.D., 3rd; Griner, S.; Finer-Moore, J.S.; Stroud, R.M. Development and binding mode assessment of N-[4-[2-propyn-1-yl[(6S)-4,6,7,8-tetrahydro-2-(hydroxymethyl)-4-oxo-3H-cyclopenta[g]quinazolin-6-yl] amino]benzoyl]-l-γ-glutamyl-D-glutamic acid (BGC 945), a novel thymidylate synthase inhibitor that targets tumor cells. J. Med. Chem. 2013, 56, 5446–5455. [Google Scholar] [CrossRef]
- Karagiannis, S.N.; Wang, Q.; East, N.; Burke, F.; Riffard, S.; Bracher, M.G.; Thompson, R.G.; Durham, S.R.; Schwartz, L.B.; Balkwill, F.R.; et al. Activity of human monocytes in IgE antibody-dependent surveillance and killing of ovarian tumor cells. Eur. J. Immunol. 2003, 33, 1030–1040. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, S.N.; Bracher, M.G.; Beavil, R.L.; Beavil, A.J.; Hunt, J.; McCloskey, N.; Thompson, R.G.; East, N.; Burke, F.; Sutton, B.J.; et al. Role of IgE receptors in IgE antibody-dependent cytotoxicity and phagocytosis of ovarian tumor cells by human monocytic cells. Cancer Immunol. Immunother. 2008, 57, 247–263. [Google Scholar] [CrossRef]
- Karagiannis, S.N.; Bracher, M.G.; Hunt, J.; McCloskey, N.; Beavil, R.L.; Beavil, A.J.; Fear, D.J.; Thompson, R.G.; East, N.; Burke, F.; et al. IgE-antibody-dependent immunotherapy of solid tumors: Cytotoxic and phagocytic mechanisms of eradication of ovarian cancer cells. J. Immunol. 2007, 179, 2832–2843. [Google Scholar] [CrossRef]
- Rudman, S.M.; Josephs, D.H.; Cambrook, H.; Karagiannis, P.; Gilbert, A.E.; Dodev, T.; Hunt, J.; Koers, A.; Montes, A.; Taams, L.; et al. Harnessing engineered antibodies of the IgE class to combat malignancy: Initial assessment of FcɛRI-mediated basophil activation by a tumour-specific IgE antibody to evaluate the risk of type I hypersensitivity. Clin. Exp. Allergy 2011, 41, 1400–1413. [Google Scholar] [CrossRef]
- Kayaba, H.; Dombrowicz, D.; Woerly, G.; Papin, J.P.; Loiseau, S.; Capron, M. Human eosinophils and human high affinity IgE receptor transgenic mouse eosinophils express low levels of high affinity IgE receptor, but release IL-10 upon receptor activation. J. Immunol. 2001, 167, 995–1003. [Google Scholar] [CrossRef]
- Muraki, M.; Gleich, G.J.; Kita, H. Antigen-specific IgG and IgA, but not IgE, activate the effector functions of eosinophils in the presence of antigen. Int. Arch. Allergy Immunol. 2011, 154, 119–127. [Google Scholar] [CrossRef]
- Marquet, R.L.; Westbroek, D.L.; Jeekel, J. Interferon treatment of a transplantable rat colon adenocarcinoma: Importance of tumor site. Int. J. Cancer 1984, 33, 689–692. [Google Scholar] [CrossRef]
- Josephs, D.H.; Bax, H.J.; Dodev, T.; Georgouli, M.; Nakamura, M.; Pellizzari, G.; Saul, L.; Karagiannis, P.; Cheung, A.; Herraiz, C.; et al. Anti-Folate Receptor-α IgE but not IgG Recruits Macrophages to Attack Tumors via TNFα/MCP-1 Signaling. Cancer Res. 2017, 77, 1127–1141. [Google Scholar] [CrossRef] [Green Version]
- Bracher, M.; Gould, H.J.; Sutton, B.J.; Dombrowicz, D.; Karagiannis, S.N. Three-colour flow cytometric method to measure antibody-dependent tumour cell killing by cytotoxicity and phagocytosis. J. Immunol. Methods 2007, 323, 160–171. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Josephs, D.H.; Nakamura, M.; Bax, H.J.; Dodev, T.S.; Muirhead, G.; Saul, L.; Karagiannis, P.; Ilieva, K.M.; Crescioli, S.; Gazinska, P.; et al. An immunologically relevant rodent model demonstrates safety of therapy using a tumour-specific IgE. Allergy 2018, 73, 2328–2341. [Google Scholar] [CrossRef]
- Kraft, S.; Novak, N.; Katoh, N.; Bieber, T.; Rupec, R.A. Aggregation of the high-affinity IgE receptor FcεRI on human monocytes and dendritic cells induces NF-κB activation. J. Investig. Dermatol. 2002, 118, 830–837. [Google Scholar] [CrossRef]
- Karagiannis, S.N.; Josephs, D.H.; Bax, H.J.; Spicer, J.F. Therapeutic IgE Antibodies: Harnessing a Macrophage-Mediated Immune Surveillance Mechanism against Cancer. Cancer Res. 2017, 77, 2779–2783. [Google Scholar] [CrossRef] [Green Version]
- Ishizaka, T.; Ishizaka, K.; Tomioka, H. Release of histamine and slow reacting substance of anaphylaxis (SRS-A) by IgE-anti-IgE reactions on monkey mast cells. J. Immunol. 1972, 108, 513–520. [Google Scholar]
- Schwartz, L.B. Effector cells of anaphylaxis: Mast cells and basophils. Novartis Found. Symp. 2004, 257, 65–79. [Google Scholar]
- Dombrowicz, D.; Brini, A.T.; Flamand, V.; Hicks, E.; Snouwaert, J.N.; Kinet, J.P.; Koller, B.H. Anaphylaxis mediated through a humanized high affinity IgE receptor. J. Immunol. 1996, 157, 1645–1651. [Google Scholar]
- Collins, A.M.; Basil, M.; Nguyen, K.; Thelian, D. Rat basophil leukaemia (RBL) cells sensitized with low affinity IgE respond to high valency antigen. Clin. Exp. Allergy 1996, 26, 964–970. [Google Scholar] [CrossRef]
- Jensen-Jarolim, E.; Singer, J. Why could passive Immunoglobulin E antibody therapy be safe in clinical oncology? Clin. Exp. Allergy 2011, 41, 1337–1340. [Google Scholar] [CrossRef] [Green Version]
- Basal, E.; Eghbali-Fatourechi, G.Z.; Kalli, K.R.; Hartmann, L.C.; Goodman, K.M.; Goode, E.L.; Kamen, B.A.; Low, P.S.; Knutson, K.L. Functional folate receptor alpha is elevated in the blood of ovarian cancer patients. PLoS ONE 2009, 4, e6292. [Google Scholar] [CrossRef]
- Hoffmann, H.J.; Santos, A.F.; Mayorga, C.; Nopp, A.; Eberlein, B.; Ferrer, M.; Rouzaire, P.; Ebo, D.G.; Sabato, V.; Sanz, M.L.; et al. The clinical utility of basophil activation testing in diagnosis and monitoring of allergic disease. Allergy 2015, 70, 1393–1405. [Google Scholar] [CrossRef]
- Marraccini, P.; Pignatti, P.; D’Alcamo, A.; Salimbeni, R.; Consonni, D. Basophil Activation Test Application in Drug Hypersensitivity Diagnosis: An Empirical Approach. Int. Arch. Allergy Immunol. 2018, 177, 160–166. [Google Scholar] [CrossRef]
- Seremet, T.; Haccuria, A.; Lienard, D.; Del Marmol, V.; Neyns, B. Anaphylaxis-like reaction to anti-BRAF inhibitor dabrafenib confirmed by drug provocation test. Melanoma Res. 2019, 29, 95–98. [Google Scholar] [CrossRef]
- Ornelas, C.; Caiado, J.; Campos Melo, A.; Pereira Barbosa, M.; Castells, M.C.; Pereira Dos Santos, M.C. The Contribution of the Basophil Activation Test to the Diagnosis of Hypersensitivity Reactions to Oxaliplatin. Int. Arch. Allergy Immunol. 2018, 177, 274–280. [Google Scholar] [CrossRef]
- De Week, A.L.; Sanz, M.L.; Gamboa, P.M.; Aberer, W.; Bienvenu, J.; Blanca, M.; Demoly, P.; Ebo, D.G.; Mayorga, L.; Monneret, G.; et al. Diagnostic tests based on human basophils: More potentials and perspectives than pitfalls. II. Technical issues. J. Investig. Allergol. Clin. Immunol. 2008, 18, 143–155. [Google Scholar]
- Saul, L.; Josephs, D.H.; Cutler, K.; Bradwell, A.; Karagiannis, P.; Selkirk, C.; Gould, H.J.; Jones, P.; Spicer, J.F.; Karagiannis, S.N. Comparative reactivity of human IgE to cynomolgus monkey and human effector cells and effects on IgE effector cell potency. mAbs 2014, 6, 509–522. [Google Scholar] [CrossRef]
- Riemer, A.B.; Untersmayr, E.; Knittelfelder, R.; Duschl, A.; Pehamberger, H.; Zielinski, C.C.; Scheiner, O.; Jensen-Jarolim, E. Active induction of tumor-specific IgE antibodies by oral mimotope vaccination. Cancer Res. 2007, 67, 3406–3411. [Google Scholar] [CrossRef]
- Herrmann, I.; Gotovina, J.; Fazekas-Singer, J.; Fischer, M.B.; Hufnagl, K.; Bianchini, R.; Jensen-Jarolim, E. Canine macrophages can like human macrophages be in vitro activated toward the M2a subtype relevant in allergy. Dev. Comp. Immunol. 2018, 82, 118–127. [Google Scholar] [CrossRef]
- Singer, J.; Jensen-Jarolim, E. IgE-based Immunotherapy of Cancer -A Comparative Oncology Approach. J. Carcinog. Mutagen. 2014, 5, 1000176. [Google Scholar] [CrossRef]
- Carvalho, M.I.; Silva-Carvalho, R.; Pires, I.; Prada, J.; Bianchini, R.; Jensen-Jarolim, E.; Queiroga, F.L. A Comparative Approach of Tumor-Associated Inflammation in Mammary Cancer between Humans and Dogs. BioMed Res. Int. 2016, 2016, 4917387. [Google Scholar] [CrossRef]
- Santos, R.B.; Galvão, V.R. Monoclonal Antibodies Hypersensitivity: Prevalence and Management. Immunol. Allergy Clin. N. Am. 2017, 37, 695–711. [Google Scholar] [CrossRef]
- Cheifetz, A.; Smedley, M.; Martin, S.; Reiter, M.; Leone, G.; Mayer, L.; Plevy, S. The incidence and management of infusion reactions to infliximab: A large center experience. Am. J. Gastroenterol. 2003, 98, 1315–1324. [Google Scholar] [CrossRef]
- Chen, X.; Churchill, M.J.; Nagar, K.K.; Tailor, Y.H.; Chu, T.; Rush, B.S.; Jiang, Z.; Wang, E.B.; Renz, B.W.; Wang, H.; et al. IL-17 producing mast cells promote the expansion of myeloid-derived suppressor cells in a mouse allergy model of colorectal cancer. Oncotarget 2015, 6, 32966–32979. [Google Scholar] [CrossRef] [Green Version]
- Welsh, T.J.; Green, R.H.; Richardson, D.; Waller, D.A.; O’Byrne, K.J.; Bradding, P. Macrophage and mast-cell invasion of tumor cell islets confers a marked survival advantage in non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 8959–8967. [Google Scholar] [CrossRef]
- Nakae, S.; Suto, H.; Iikura, M.; Kakurai, M.; Sedgwick, J.D.; Tsai, M.; Galli, S.J. Mast cells enhance T cell activation: Importance of mast cell costimulatory molecules and secreted TNF. J. Immunol. 2006, 176, 2238–2248. [Google Scholar] [CrossRef]
- Brown, C.E.; Vishwanath, R.P.; Aguilar, B.; Starr, R.; Najbauer, J.; Aboody, K.S.; Jensen, M.C. Tumor-derived chemokine MCP-1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J. Immunol. 2007, 179, 3332–3341. [Google Scholar] [CrossRef]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutton, B.J.; Davies, A.M.; Bax, H.J.; Karagiannis, S.N. IgE Antibodies: From Structure to Function and Clinical Translation. Antibodies 2019, 8, 19. https://doi.org/10.3390/antib8010019
Sutton BJ, Davies AM, Bax HJ, Karagiannis SN. IgE Antibodies: From Structure to Function and Clinical Translation. Antibodies. 2019; 8(1):19. https://doi.org/10.3390/antib8010019
Chicago/Turabian StyleSutton, Brian J., Anna M. Davies, Heather J. Bax, and Sophia N. Karagiannis. 2019. "IgE Antibodies: From Structure to Function and Clinical Translation" Antibodies 8, no. 1: 19. https://doi.org/10.3390/antib8010019
APA StyleSutton, B. J., Davies, A. M., Bax, H. J., & Karagiannis, S. N. (2019). IgE Antibodies: From Structure to Function and Clinical Translation. Antibodies, 8(1), 19. https://doi.org/10.3390/antib8010019