Dynamic Views of the Fc Region of Immunoglobulin G Provided by Experimental and Computational Observations

 , , ,
, , , 
{kind=link}
{kind=link}
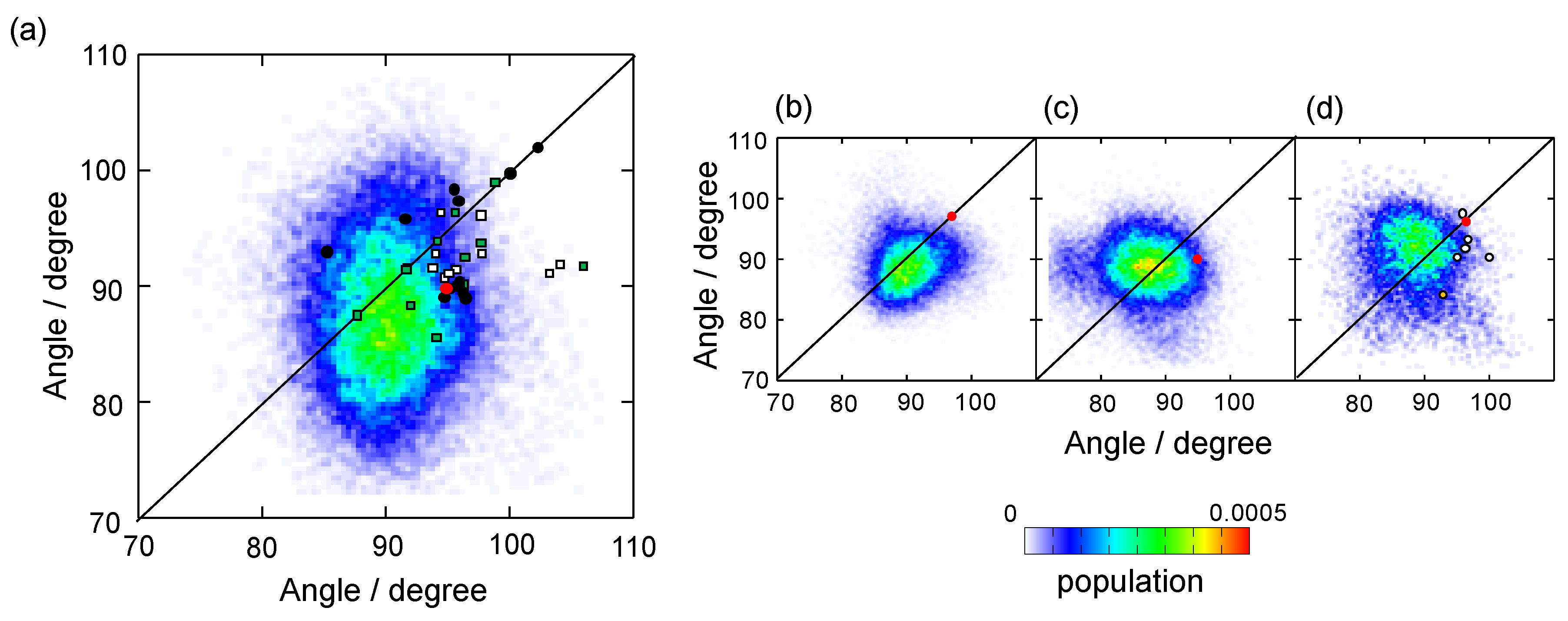{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics Simulation
2.2. Sample Preparation
2.3. Small-Angle X-Ray Scattering
2.4. NMR Measurement
3. Results and Discussion
3.1. Overall Conformation of IgG-Fc
3.2. Intramolecular Interaction Networks of N-Glycans
3.3. Effects of Fc Defucosylation of its Dynamic Conformation and FcγR Interaction
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADCC | Antibody-dependent cell-mediated cytotoxicity |
| FcγR | Fcγ receptor |
| Fuc | Fucose |
| Gal | Galactose |
| GlcNAc | N- Acetylglucosamine |
| HSQC | Heteronuclear single-quantum correlation |
| IgG | Immunoglobulin G |
| Man | Mannose |
| MD | Molecular dynamics |
| NMR | Nuclear magnetic resonance |
| NOE | Nuclear Overhauser effect |
| NOESY | Nuclear Overhauser effect spectroscopy |
| PDB | Protein Data Bank |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| SAXS | Small-angle X-ray scattering |
References
- Dorrington, K.J.; Klein, M.H. Binding sites for Fcγ receptors on immunoglobulin G and factors influencing their expression. Mol. Immunol. 1982, 19, 1215–1221. [Google Scholar] [CrossRef]
- Burton, D.R. Immunoglobulin G: Functional sites. Mol. Immunol. 1985, 22, 161–206. [Google Scholar] [CrossRef]
- Jefferis, R.; Lund, J.; Pound, J.D. IgG-Fc-mediated effector functions: Molecular definition of interaction sites for effector ligands and the role of glycosylation. Immunol. Rev. 1998, 163, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Kroe-Barrett, R.; Singh, S.; Roberts, C.J.; Laue, T.M. IgG cooperativity—Is there allostery? Implications for antibody functions and therapeutic antibody development. mAbs 2017, 9, 1231–1252. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.C.K.; Lian, L.Y.; Barsukov, I.L.; Derrick, J.P.; Kato, K.; Arata, Y. Interactions of bacterial cell-surface proteins with antibodies: A versatile set of protein-protein interactions. Tech. Prot. Chem. 1995, 6, 409–416. [Google Scholar]
- Nezlin, R. Internal movements in immunoglobulin molecules. Adv. Immunol. 1990, 48, 1–40. [Google Scholar] [PubMed]
- Jay, J.W.; Bray, B.; Qi, Y.; Igbinigie, E.; Wu, H.; Li, J.; Ren, G. IgG antibody 3D structures and dynamics. Antibodies 2018, 7, 18. [Google Scholar] [CrossRef]
- Arata, Y.; Kato, K.; Takahashi, H.; Shimada, I. Nuclear-magnetic-resonance study of antibodies—A multinuclear approach. Methods Enzymol. 1994, 239, 440–464. [Google Scholar]
- Nakasako, M.; Oka, T.; Mashumo, M.; Takahashi, H.; Shimada, I.; Yamaguchi, Y.; Kato, K.; Arata, Y. Conformational dynamics of complementarity-determining region H3 of an anti-dansyl Fv fragment in the presence of its hapten. J. Mol. Biol. 2005, 351, 627–640. [Google Scholar] [CrossRef]
- Fernández-Quintero, M.L.; Loeffler, J.R.; Kraml, J.; Kahler, U.; Kamenik, A.S.; Liedl, K.R. Characterizing the diversity of the CDR-H3 loop conformational ensembles in relationship to antibody binding properties. Front. Immunol. 2018, 9, 3065. [Google Scholar] [CrossRef]
- Foote, J.; Milstein, C. Conformational isomerism and the diversity of antibodies. Proc. Natl. Acad. Sci. USA 1994, 91, 10370–10374. [Google Scholar] [CrossRef] [PubMed]
- Sela-Culang, I.; Kunik, V.; Ofran, Y. The structural basis of antibody-antigen recognition. Front. Immunol. 2013, 4, 302. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, S.; Yagi, H.; Yogo, R.; Yagi-Utsumi, M.; Kato, K. Stable isotope labeling approaches for NMR characterization of glycoproteins using eukaryotic expression systems. J. Biomol. NMR 2018, 71, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Yamaguchi, Y.; Arata, Y. Stable-isotope-assisted NMR approaches to glycoproteins using immunoglobulin G as a model system. Prog. Nucl. Magn. Reson. Spectrosc. 2010, 56, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.J.; Zhang, H.L.; Krueger, S.; Lee, H.J.; Ketchem, R.R.; Kerwin, B.; Kanapuram, S.R.; Treuheit, M.J.; McAuley, A.; Curtis, J.E.; et al. Small-angle neutron scattering study of a monoclonal antibody using free-energy constraints. J. Phys. Chem. B 2013, 117, 14029–14038. [Google Scholar] [CrossRef] [PubMed]
- Eryilmaz, E.; Janda, A.; Kim, J.; Cordero, R.J.; Cowburn, D.; Casadevall, A. Global structures of IgG isotypes expressing identical variable regions. Mol. Immunol. 2013, 56, 588–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inouye, H.; Houde, D.; Temel, D.B.; Makowski, L. Utility of solution X-ray scattering for the development of antibody biopharmaceuticals. J. Pharm. Sci. 2016, 105, 3278–3289. [Google Scholar] [CrossRef]
- Tian, X.; Vestergaard, B.; Thorolfsson, M.; Yang, Z.; Rasmussen, H.B.; Langkilde, A.E. In-depth analysis of subclass-specific conformational preferences of IgG antibodies. IUCrJ 2015, 2, 9–18. [Google Scholar] [CrossRef]
- Castellanos, M.M.; Howell, S.C.; Gallagher, D.T.; Curtis, J.E. Characterization of the NISTmAb reference material using small-angle scattering and molecular simulation. Anal. Bioanal. Chem. 2018, 410, 2141–2159. [Google Scholar] [CrossRef]
- Ugurlar, D.; Howes, S.C.; de Kreuk, B.J.; Koning, R.I.; de Jong, R.N.; Beurskens, F.J.; Schuurman, J.; Koster, A.J.; Sharp, T.H.; Parren, P.W.H.I.; et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science 2018, 359, 794–797. [Google Scholar] [CrossRef] [Green Version]
- Preiner, J.; Kodera, N.; Tang, J.L.; Ebner, A.; Brameshuber, M.; Blaas, D.; Gelbmann, N.; Gruber, H.J.; Ando, T.; Hinterdorfer, P.; et al. IgGs are made for walking on bacterial and viral surfaces. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.P.; Patapoff, T.W.; Aragon, S.R. Construction, md simulation, and hydrodynamic validation of an all-atom model of a monoclonal IgG antibody. Biophys. J. 2010, 99, 905–913. [Google Scholar] [CrossRef]
- Krapp, S.; Mimura, Y.; Jefferis, R.; Huber, R.; Sondermann, P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J. Mol. Biol. 2003, 325, 979–989. [Google Scholar] [CrossRef]
- Frank, M.; Walker, R.C.; Lanzilotta, W.N.; Prestegard, J.H.; Barb, A.W. Immunoglobulin G1 Fc domain motions: Implications for Fc engineering. J. Mol. Biol. 2014, 426, 1799–1811. [Google Scholar] [CrossRef] [PubMed]
- Caaveiro, J.M.; Kiyoshi, M.; Tsumoto, K. Structural analysis of Fc/FcγR complexes: A blueprint for antibody design. Immunol. Rev. 2015, 268, 201–221. [Google Scholar] [CrossRef]
- Jefferis, R. Glycoforms of human IgG in health and disease. Trends Glycosci. Glycotechnol. 2009, 21, 105–117. [Google Scholar]
- Barb, A.W.; Prestegard, J.H. NMR analysis demonstrates immunoglobulin G N-glycans are accessible and dynamic. Nat. Chem. Biol. 2011, 7, 147–153. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Takahashi, N.; Kato, K. Antibody Structures; Elsevier: Oxford, UK, 2007; Volume 3. [Google Scholar]
- Dekkers, G.; Treffers, L.; Plomp, R.; Bentlage, A.E.H.; de Boer, M.; Koeleman, C.A.M.; Lissenberg-Thunnissen, S.N.; Visser, R.; Brouwer, M.; Mok, J.Y.; et al. Decoding the human immunoglobulin G-Glycan repertoire reveals a spectrum of Fc-receptor- and complement-mediated-effector activities. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Niwa, R.; Shoji-Hosaka, E.; Sakurada, M.; Shinkawa, T.; Uchida, K.; Nakamura, K.; Matsushima, K.; Ueda, R.; Hanai, N.; Shitara, K.; et al. Defucosylated chimeric anti-CC chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Res. 2004, 64, 2127–2133. [Google Scholar] [CrossRef]
- Yamane-Ohnuki, N.; Satoh, M. Production of therapeutic antibodies with controlled fucosylation. mAbs 2009, 1, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, C.; Stuart, F.; Sondermann, P.; Brunker, P.; Umana, P. The carbohydrate at FcγRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J. Biol. Chem. 2006, 281, 5032–5036. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Babin, V.; Berryman, J.T. Amber14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14sb: Improving the accuracy of protein side chain and backbone parameters from ff99sb. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeirino, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. Glycam06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Kato, K.; Yanaka, S.; Yagi, H. Technical Basis for Nuclear Magnetic Resonance Approach for Glycoproteins; Springer: Singapore, 2018; pp. 415–438. [Google Scholar]
- Deisenhofer, J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8- resolution. Biochemistry 1981, 20, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Matsumiya, S.; Yamaguchi, Y.; Saito, J.; Nagano, M.; Sasakawa, H.; Otaki, S.; Satoh, M.; Shitara, K.; Kato, K. Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J. Mol. Biol. 2007, 368, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Borrok, M.J.; Jung, S.T.; Kang, T.H.; Monzingo, A.F.; Georgiou, G. Revisiting the role of glycosylation in the structure of human IgG Fc. ACS Chem. Biol. 2012, 7, 1596–1602. [Google Scholar] [CrossRef] [PubMed]
- Remesh, S.G.; Armstrong, A.A.; Mahan, A.D.; Luo, J.; Hammel, M. Conformational plasticity of the immunoglobulin Fc domain in solution. Structure 2018, 26, 1007–1014.e2. [Google Scholar] [CrossRef]
- Yageta, S.; Imamura, H.; Shibuya, R.; Honda, S. CH2 domain orientation of human immunoglobulin G in solution: Structural comparison of glycosylated and aglycosylated Fc regions using small-angle X-ray scattering. mAbs 2019, 11, 453–462. [Google Scholar] [CrossRef]
- Idusogie, E.E.; Presta, L.G.; Gazzano-Santoro, H.; Totpal, K.; Wong, P.Y.; Ultsch, M.; Meng, Y.G.; Mulkerrin, M.G. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J. Immunol. 2000, 164, 4178–4184. [Google Scholar] [CrossRef] [PubMed]
- Ramsland, P.A.; Farrugia, W.; Bradford, T.M.; Sardjono, C.T.; Esparon, S.; Trist, H.M.; Powell, M.S.; Tan, P.S.; Cendron, A.C.; Wines, B.D.; et al. Structural basis for FcγRIIa recognition of human IgG and formation of inflammatory signaling complexes. J. Immunol. 2011, 187, 3208–3217. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Nishimura, M.; Nagano, M.; Yagi, H.; Sasakawa, H.; Uchida, K.; Shitara, K.; Kato, K. Glycoform-dependent conformational alteration of the Fc region of human immunoglobulin G1 as revealed by NMR spectroscopy. Biochim. Biophys. Acta 2006, 1760, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Subedi, G.P.; Barb, A.W. The structural role of antibody N-glycosylation in receptor interactions. Structure 2015, 23, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Wormald, M.R.; Petrescu, A.J.; Pao, Y.L.; Glithero, A.; Elliott, T.; Dwek, R.A. Conformational studies of oligosaccharides and glycopeptides: Complementarity of NMR, X-ray crystallography, and molecular modelling. Chem. Rev. 2002, 102, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, Y.; Satoh, T.; Kato, K. Recent advances in glycoprotein production for structural biology: Toward tailored design of glycoforms. Curr. Opin. Struct. Biol. 2014, 26, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Sakae, Y.; Satoh, T.; Yagi, H.; Yanaka, S.; Yamaguchi, T.; Isoda, Y.; Iida, S.; Okamoto, Y.; Kato, K. Conformational effects of N-Glycan core fucosylation of immunoglobulin G Fc region on its interaction with Fcγ receptor IIIa. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Im, W. Effects of N-Glycan composition on structure and dynamics of IgG1 Fc and their implications for antibody engineering. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Isoda, Y.; Yagi, H.; Satoh, T.; Shibata-Koyama, M.; Masuda, K.; Satoh, M.; Kato, K.; Iida, S. Importance of the side chain at position 296 of antibody Fc in interactions with FcγRIIIa and other Fcγ receptors. PLoS ONE 2015, 10, e0140120. [Google Scholar] [CrossRef]
- Mizushima, T.; Yagi, H.; Takemoto, E.; Shibata-Koyama, M.; Isoda, Y.; Iida, S.; Masuda, K.; Satoh, M.; Kato, K. Structural basis for improved efficacy of therapeutic antibodies on defucosylation of their Fc glycans. Genes Cells Devoted Mol. Cell. Mech. 2011, 16, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, A.; Shoji-Hosaka, E.; Nakamura, K.; Wakitani, M.; Uchida, K.; Kakita, S.; Tsumoto, K.; Kumagai, I.; Shitara, K. Fucose depletion from human IgG1 oligosaccharide enhances binding enthalpy and association rate between IgG1 and FcγRIIIa. J. Mol. Biol. 2004, 336, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Mimoto, F.; Igawa, T.; Kuramochi, T.; Katada, H.; Kadono, S.; Kamikawa, T.; Shida-Kawazoe, M.; Hattori, K. Novel asymmetrically engineered antibody Fc variant with superior FcγR binding affinity and specificity compared with afucosylated Fc variant. mAbs 2013, 5, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.O.; Karki, S.; Lazar, G.A.; Chen, H.; Dang, W.; Desjarlais, J.R. Optimization of antibody binding to FcγRIIa enhances macrophage phagocytosis of tumor cells. Mol. Cancer Ther. 2008, 7, 2517–2527. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanaka, S.; Yogo, R.; Inoue, R.; Sugiyama, M.; Itoh, S.G.; Okumura, H.; Miyanoiri, Y.; Yagi, H.; Satoh, T.; Yamaguchi, T.; et al. Dynamic Views of the Fc Region of Immunoglobulin G Provided by Experimental and Computational Observations. Antibodies 2019, 8, 39. https://doi.org/10.3390/antib8030039
Yanaka S, Yogo R, Inoue R, Sugiyama M, Itoh SG, Okumura H, Miyanoiri Y, Yagi H, Satoh T, Yamaguchi T, et al. Dynamic Views of the Fc Region of Immunoglobulin G Provided by Experimental and Computational Observations. Antibodies. 2019; 8(3):39. https://doi.org/10.3390/antib8030039
Chicago/Turabian StyleYanaka, Saeko, Rina Yogo, Rintaro Inoue, Masaaki Sugiyama, Satoru G. Itoh, Hisashi Okumura, Yohei Miyanoiri, Hirokazu Yagi, Tadashi Satoh, Takumi Yamaguchi, and et al. 2019. "Dynamic Views of the Fc Region of Immunoglobulin G Provided by Experimental and Computational Observations" Antibodies 8, no. 3: 39. https://doi.org/10.3390/antib8030039
APA StyleYanaka, S., Yogo, R., Inoue, R., Sugiyama, M., Itoh, S. G., Okumura, H., Miyanoiri, Y., Yagi, H., Satoh, T., Yamaguchi, T., & Kato, K. (2019). Dynamic Views of the Fc Region of Immunoglobulin G Provided by Experimental and Computational Observations. Antibodies, 8(3), 39. https://doi.org/10.3390/antib8030039