Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells


Abstract
:1. Introduction and History
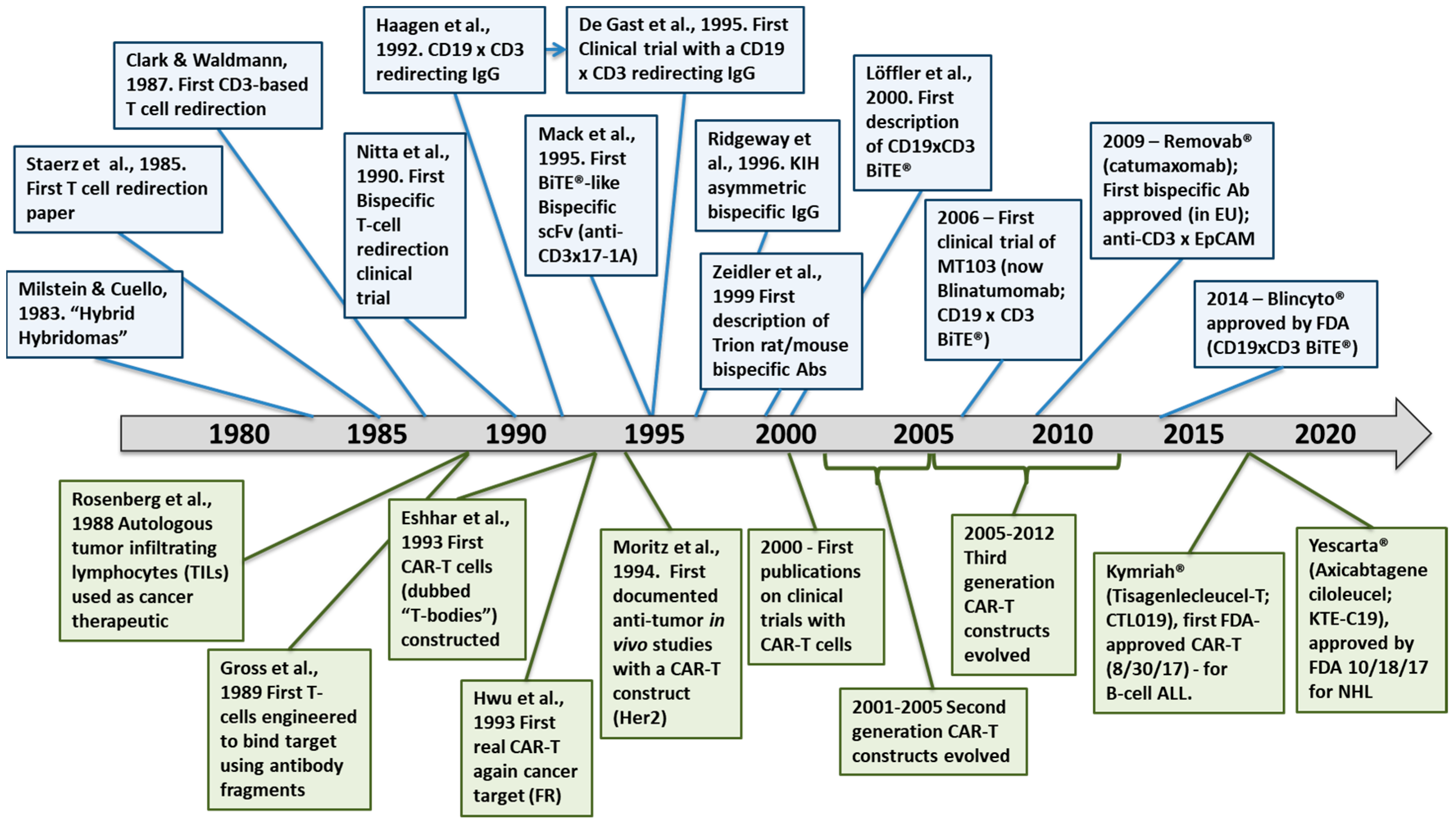
1.1. Historical Context for Immunotherapy
1.2. Brief History of T-Cell Redirecting Bispecific Antibodies
1.3. Brief History of CAR-T Cells
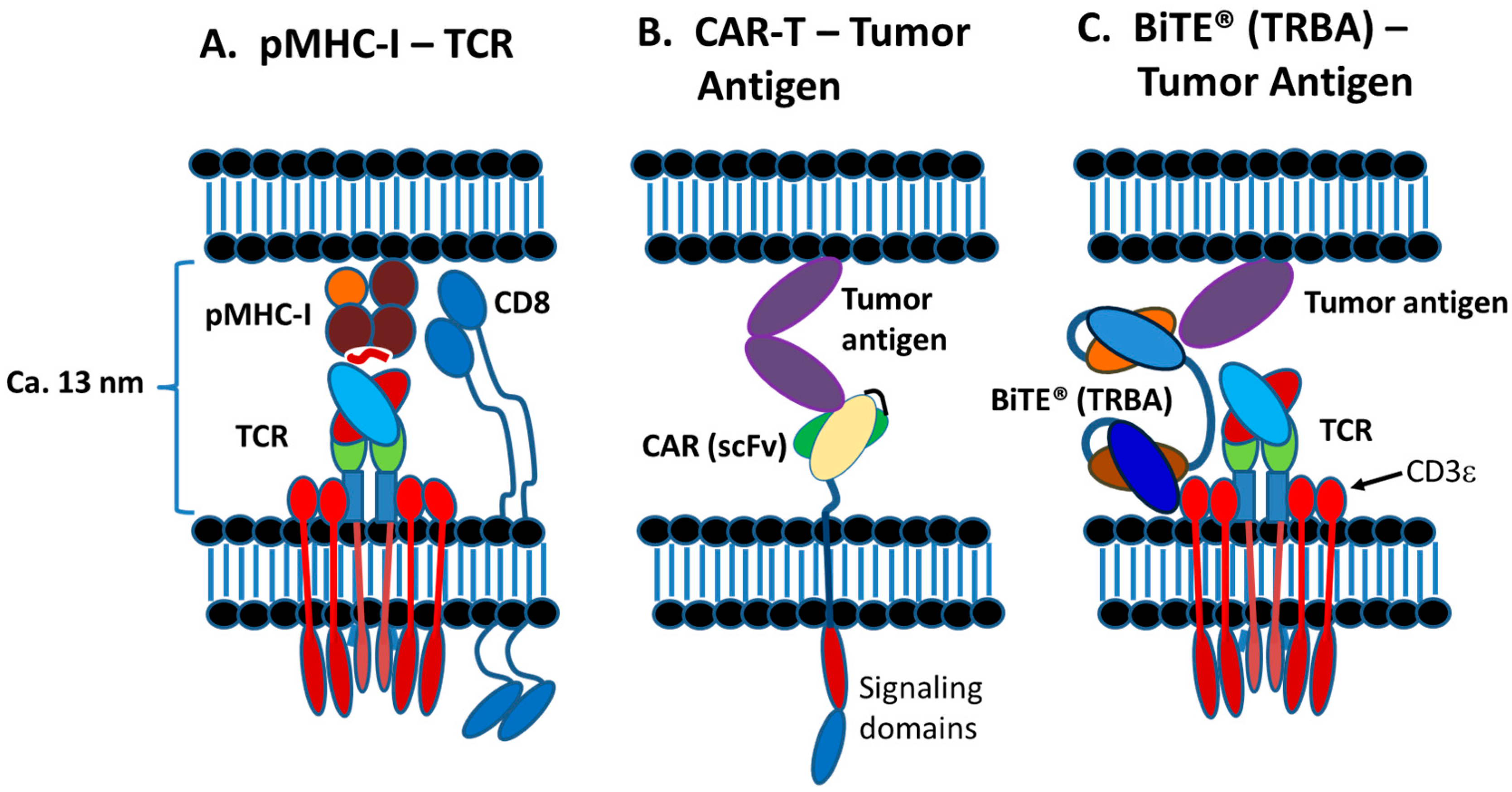
2. T-Cell Synapse and Killing Target Cells
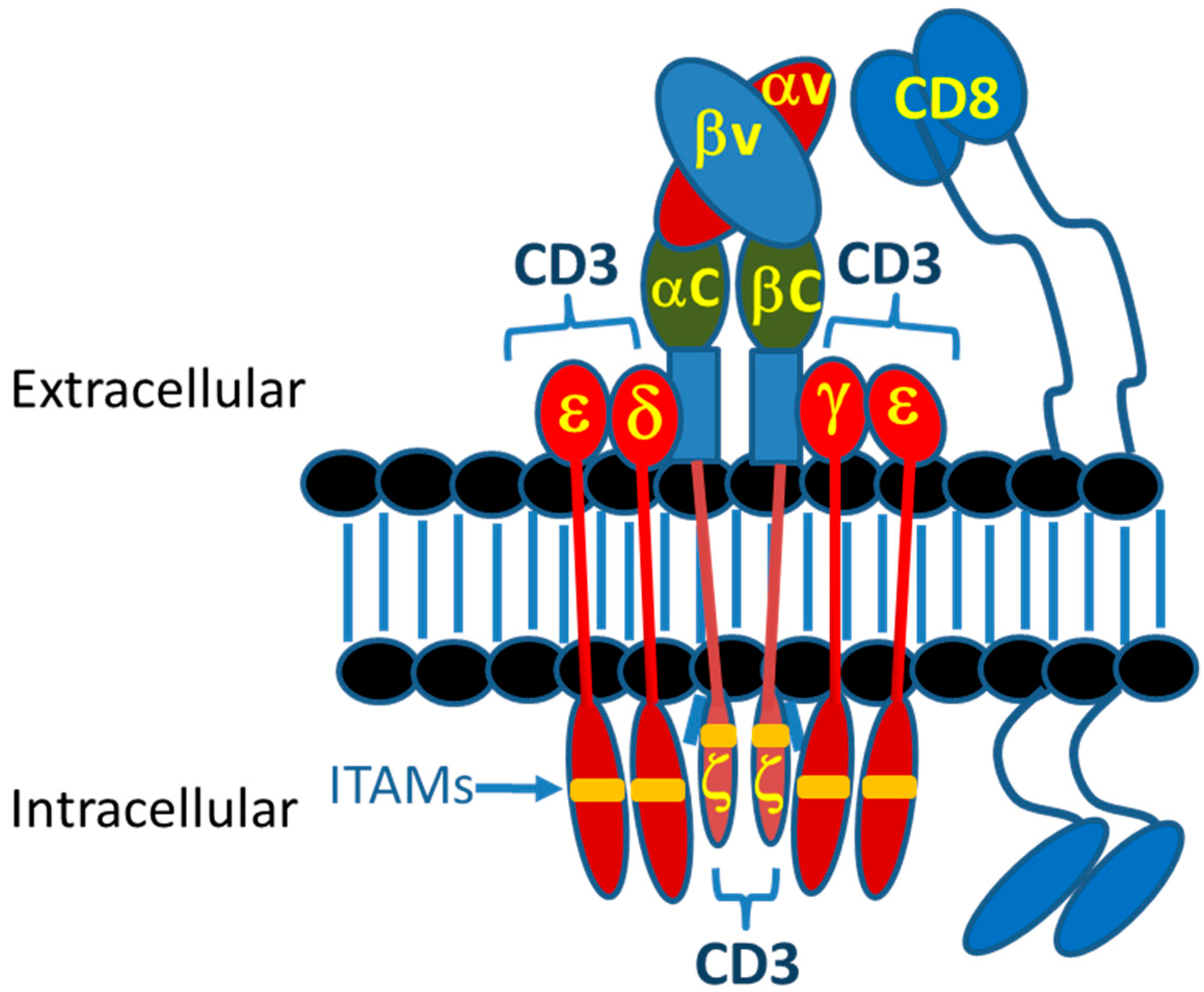
2.1. Introduction to Immunological Synapse

2.2. Normal TCR-pMHC Synapses vs. CAR-T and TRBA-Induced Synapses
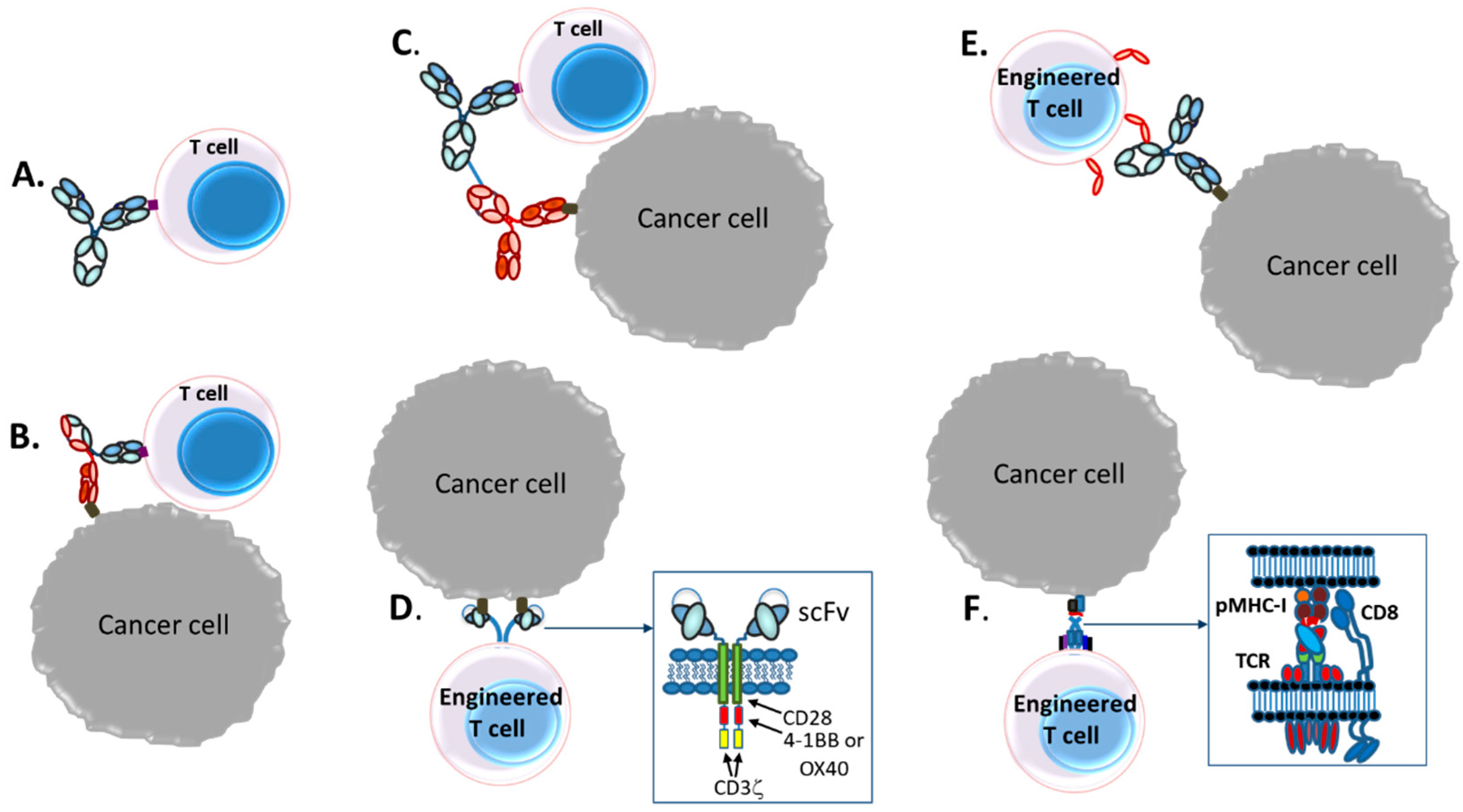
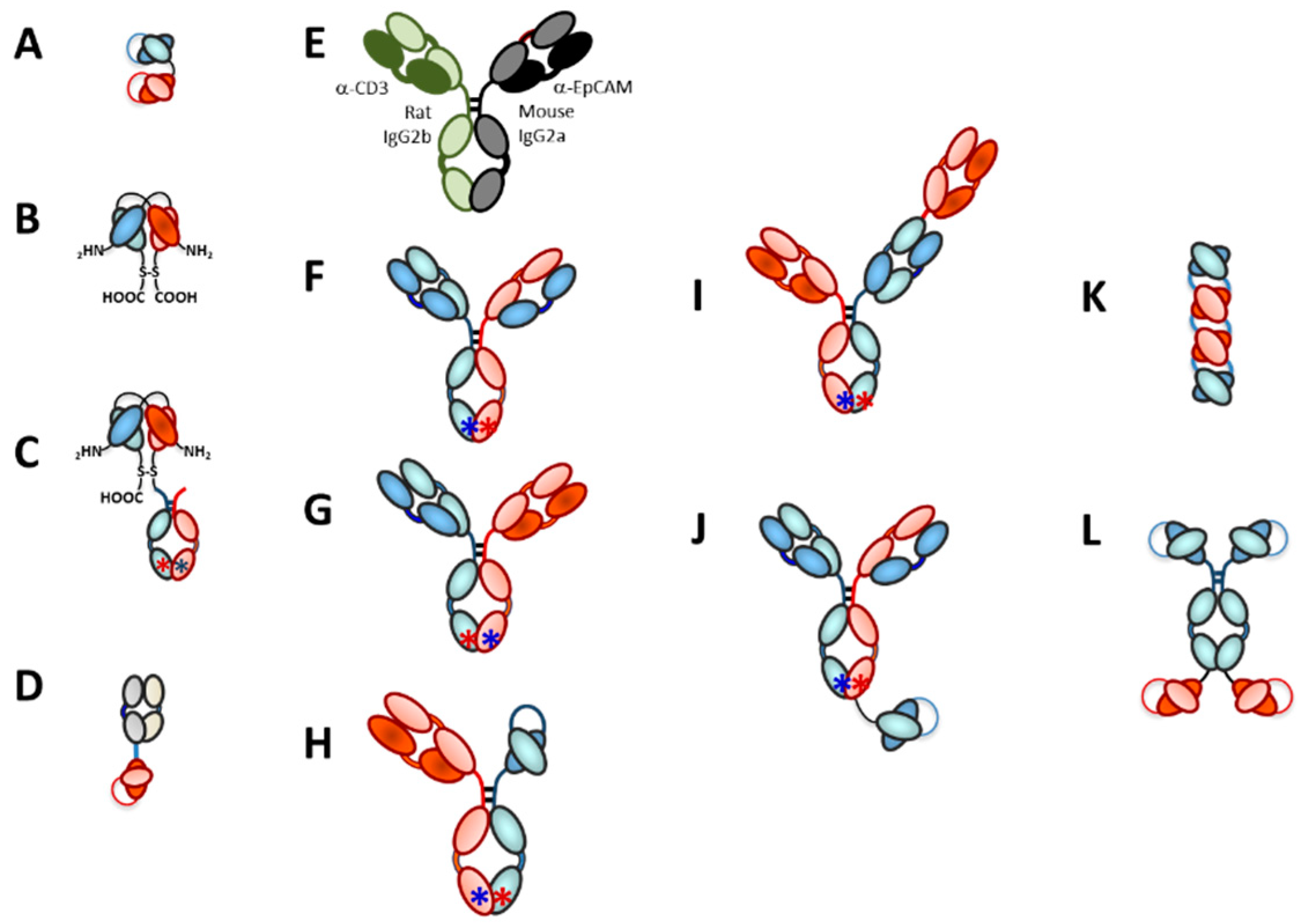
3. T-Cell Redirecting Bispecific Antibodies (TRBAs)
3.1. Introduction
3.2. Bispecific Bivalent Antibody Fragments Used to Make TRBAs
3.3. Bispecific Bivalent Asymmetric IgG-Like Antibodies Used to Make TRBAs
3.3.1. Asymmetric Pairing of HCs
3.3.2. LC Issue for Asymmetric Heterobispecific IgG-Like Antibodies
3.3.3. Trivalent, Bispecific Antibody Platforms
3.3.4. Tetravalent Bispecific Antibody Platforms
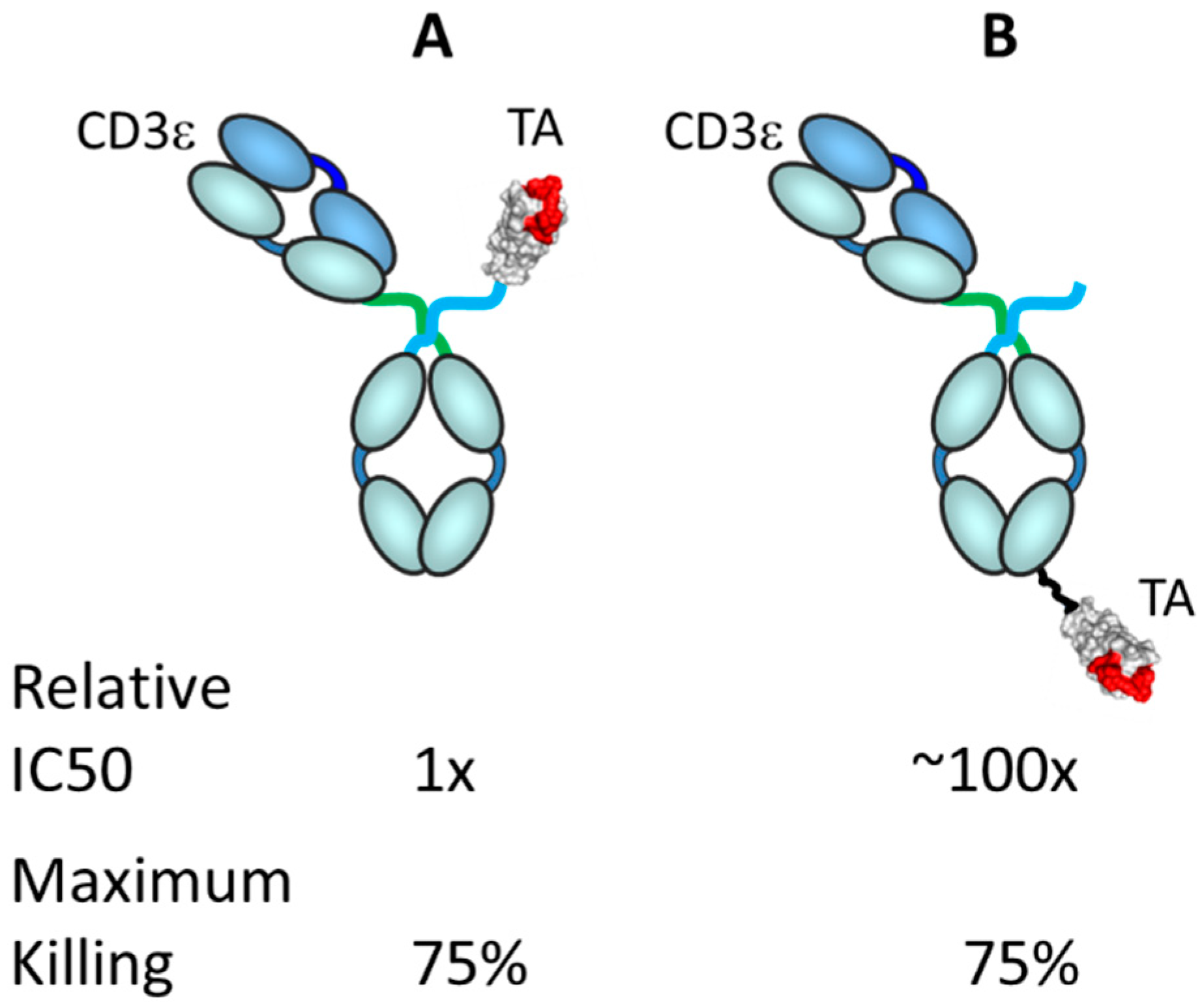
3.4. Factors Affecting TRBA Potency
4. Ex Vivo T-Cell–Bispecific Antibody Approaches
4.1. T Cells Armed Ex Vivo with Bispecific Antibody Conjugates
4.2. Cytokine-Induced Killer Cells
5. Other Examples of Immune Cell Redirection
5.1. Early Immune Cell Redirection Efforts
5.2. NK Cell Redirection, BiKEs and TriKEs
5.3. Combining Engineered Cells with mAb Therapy
5.4. Engineered T or NK Cells with Recombinant Target-Specific TCRs
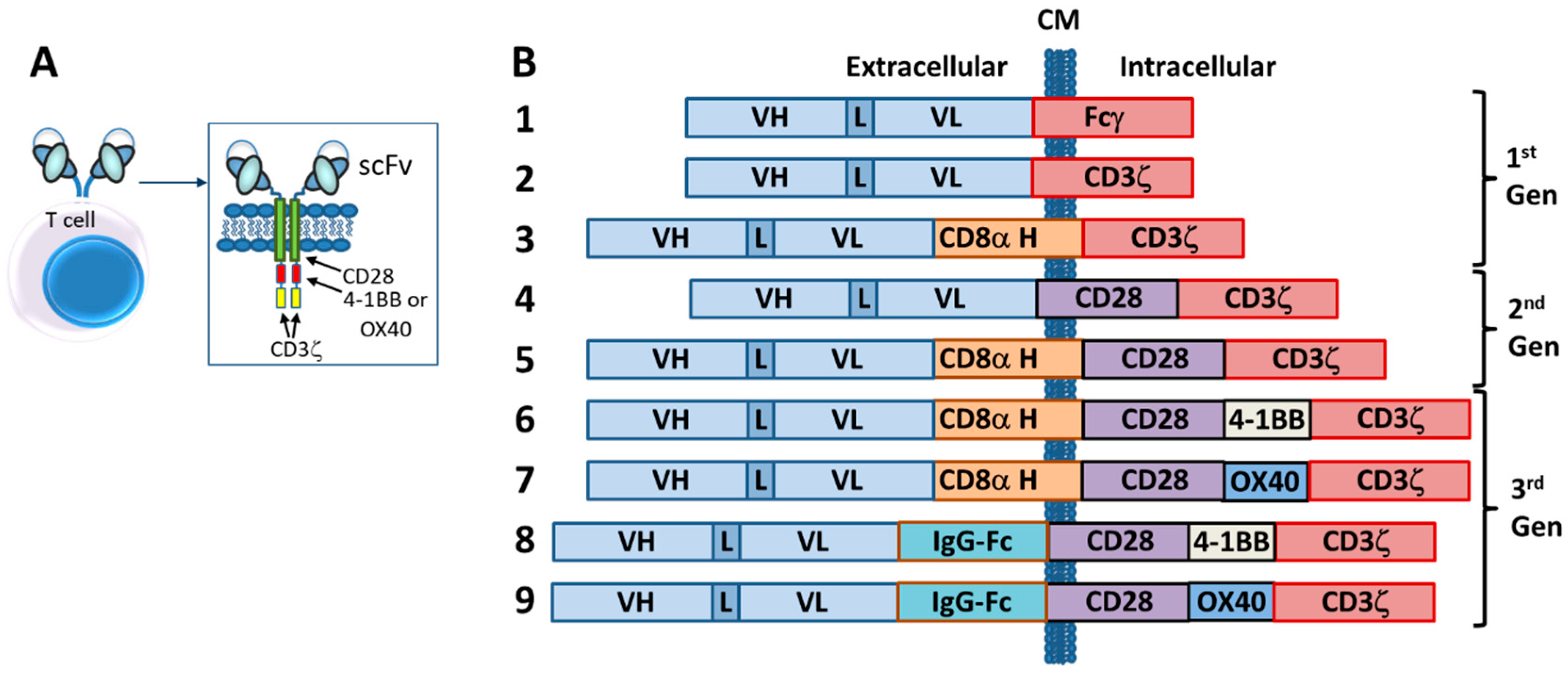
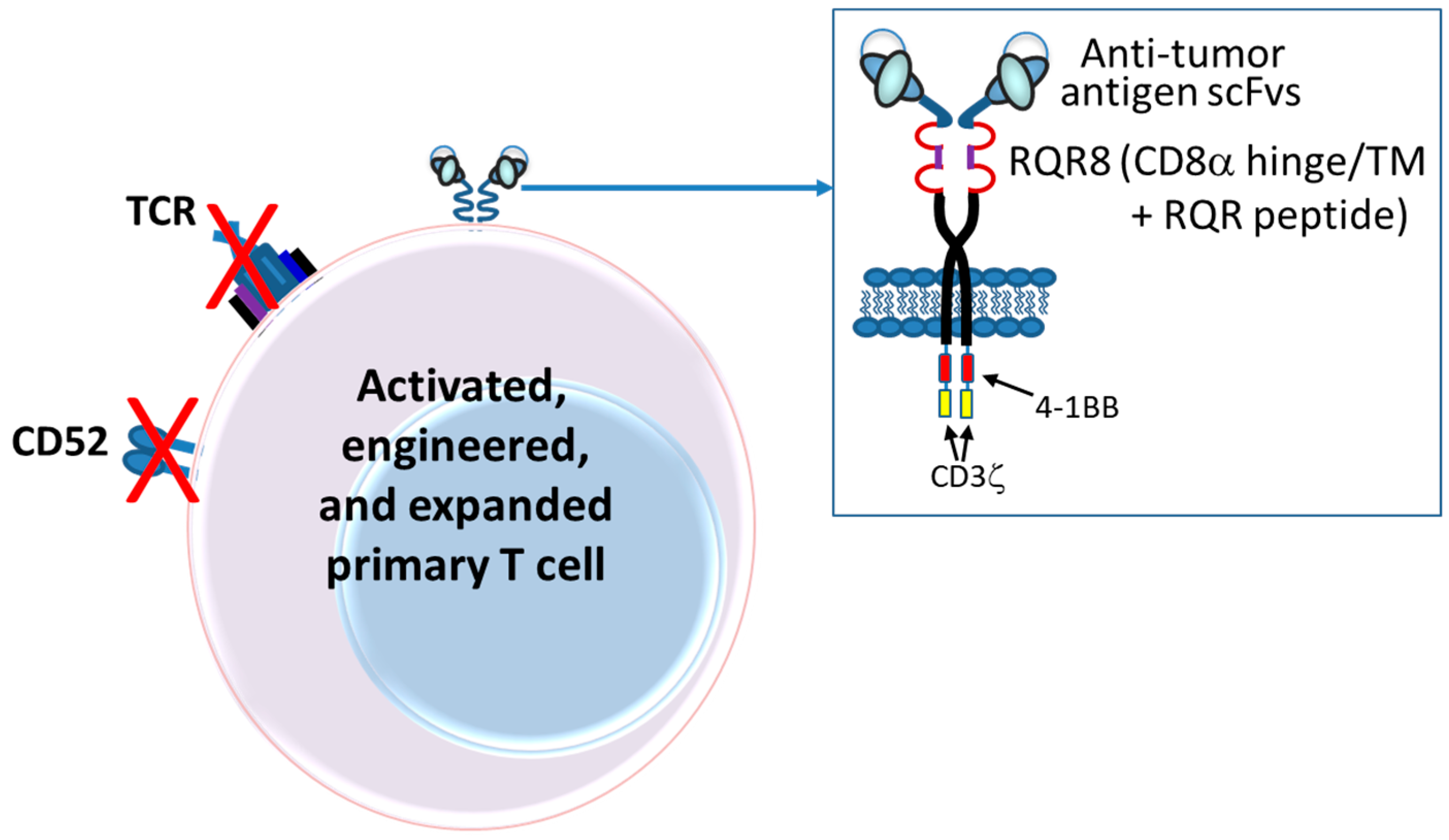
6. Chimeric Antigen Receptor (CAR)-T and NK Cells
6.1. Introduction
6.2. Autologous CARs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | CAR-T Constructs | |||
|---|---|---|---|---|
| Kymriah® (Tisagenlecleucel-T; CTL019) | Yescarta® (Axicabtagene Ciloleucel; KTE-C19) | Lisocaptagene Maraleucel (Liso-cel, JCAR-017) | bb2121 | |
| Sponsor | Novartis | Gilead (Kite) | Celgene | Celgene/bluebird |
| KEGG Number # | D11386 | D11144 | Na | Na |
| Clinical stage | Approved by USFDA | Approved by USFDA | Phase III (NCT03575351) | Phase III (NCT03651128) |
| Base cost (US) | $475,000 for B-ALL; $373,000 for R/R DLBCL | $373,000 | Na | Na |
| Indication | B-ALL, R/R DLBCL | R/R DLBCL; PMBCL | R/R DLBCL; CLL | MM |
| T-cell source | Patient PBMCs; autologous; unspecified | Patient PBMCs; autologous; unspecified | Patient CD4 and CD8 T cells 1:1 ratio; autologous | Patient PBMCs; autologous |
| Vector | Lentivirus | Retrovirus | Lentivirus | Lentivirus |
| Antibody | Anti-CD19 mouse scFv FMC63 | Anti-CD19 mouse scFv FMC63 | Anti-CD19 mouse scFv FMC63 | Anti-BCMA |
| Costimulatory domain | 4-1BB | CD28 | 4-1BB | 4-1BB |
| Signaling domain | CD3ε | CD3ε | CD3ε | CD3ε |
| Hinge and transmembrane | CD8α | IgG1 Fc | IgG4 Fc spacer; CD28tm | CD8α |
| Other Markers | Nk | nk | EGFRt | nk |
| Ex vivo activation | CD3, CD28 | CD3, IL-2 | nk | CD3, CD28 |
| Lymphodepletion | Yes | yes | yes | yes |
| Time from leukapheresis to infusion | 21–28 days | 17 days | nk | 10 days |
| Dose | 0.2 to 5 × 106 CAR-positive viable T cells/kg | 0.4–2 × 106 anti-CD19 CAR-positive viable T cells/kg | 5 × 107 CD8+ and 5 × 107 CD4+ CAR-positive (not weight based) | 50–800 × 106 CAR-positive T cells (not weight based) |
6.3. Allogeneic CARs
6.4. Alternative Cell Types for CAR Expression
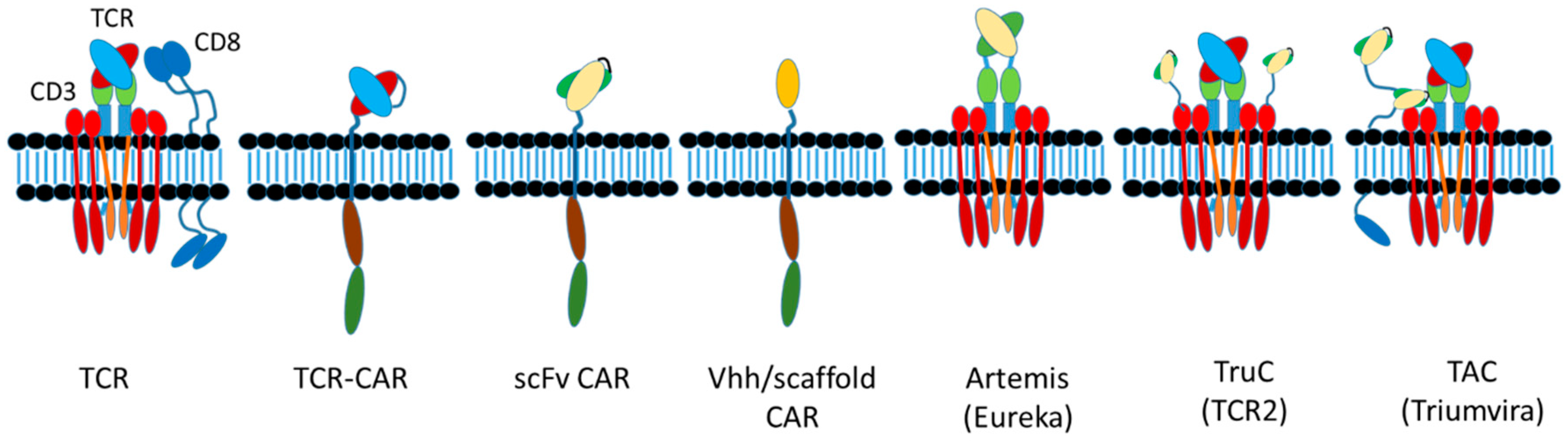
6.5. CAR Designs
6.5.1. scFvs
6.5.2. Domain Antibodies and Alternative Scaffolds
6.5.3. Multiple CAR Designs
6.6. Additional Enhancements for Tuning CAR-T Cells
6.6.1. Safety Switches
6.6.2. Adapters
6.6.3. Homing Receptors
6.6.4. Counteracting PD-1/PD-L1-Based Immunosuppression
6.6.5. Cytokine-Expressing CAR-Ts
7. Targets for Clinical Stage TRBAs and CAR-T Cells
8. Cytokine Release Syndrome (CRS) and Its Effect on Treatment
9. Comparison of TRBAs and CAR-Ts Therapeutic Approaches
9.1. General Comparison of TRBAs with CAR-T Cells
| Properties | Therapeutic Approach | ||
|---|---|---|---|
| T cell Redirection with TRBAs | Autologous CAR-T or NK Cells | Allogeneic CAR-T or NK Cells (Projected) | |
| Currently approved and marketed (as of 20 June 2019) | 1; Blincyto® (anti-CD19 × CD3 BiTE®) | 2; Kymriah® and Yescarta®, both CD19-targeting autologous CAR-Ts | None |
| Current indications covered | R/R B-ALL | DLBCL, R/R NHL, B-ALL | None |
| Structure | Bispecific antibodies that bind both a tumor antigen and CD3ε on T cells | T cells engineered with synthetic gene construct encoding scFv fused to linker and activation domains | T cells engineered with synthetic gene construct encoding scFv fused to linker and activation domains |
| Source and homogeneity of T cell component | Endogenous T cells; No homogeneity (i.e., all CD3+ T cells may be engaged) | Expanded and activated endogenous T cells; homogeneity depends on process used | Could be homogeneous CD8+ T-cells, depending on cell type and approach |
| Antibody | Short half-life vs long half-life formats | Currently, mostly scFvs; possible unfolding, aggregation, tonic signaling; need for better binding constructs | Currently mostly scFvs—possible unfolding, aggregation, tonic signaling; need for better binding constructs |
| T-cell signaling domain(s) | CD3ζ | CD3ζ + 4-1BB (or OX40) and/or CD28 | CD3ζ + 4-1BB (or OX40) and/or CD28 |
| PD-1 inhibition of CD28 activity | Likely significant issue; may need to co-dose with PD-1 inhibitor | Use of 4-1-BB signaling domain should alleviate | Use of 4-1-BB signaling domain should alleviate |
| Drug-like properties | “Off-the-shelf” drug | Must be engineered from patient’s T cells (2–4 week process) | Depends on cell type and construct |
| Dosing | Multiple dosing; short half-life formats may require continuous dosing via pumps | Single dose | Single dose; multiple dose potentially available if engineered to eliminate HLA |
| Route of administration | IV; possible subcutaneous for future candidates | IV only | IV only |
| Long-term persistence and memory | Short half-life – only as long as continuously infused; long half-life – typically measured in weeks | Yes, but variable; longer persistence correlated with activity | Unknown but likely to be similar to autologous T-cells |
| Immune synapse | Normal and concentric; normal detachment | Abnormal and multifocal; fast detachment | Expected to be similar to autologous CAR-T cells |
| T cell signals at synapse | Signals 1, 3 | Signals 1, 2 (sometimes), 3 | Expected to be similar to autologous CAR-T cells |
| Killing mechanisms | Perforin and granzyme; Secondary: cytokine modulation of TME [123] | Perforin and granzyme; Fas/FasL axis; Secondary: cytokine modulation of TME [123] | Expected to be similar to autologous CAR-T cells |
| Serial killing | Yes, similar to CTLs | Yes, faster than TRBAs an CTLs | Expected to be the same as autologous T cells |
| None; related to dosing and half-life | Yes, in responders | Unknown but expected | |
| Bystander killing of antigen-negative cells | Demonstrated, as long as antigen-negative cells were in direct contact with antigen-positive cells [381] | Demonstrated, as long as antigen-negative cells were in direct contact with antigen-positive cells [380] | Unknown but expected based on CAR-T results |
| Toxicity | CRS, neurotoxicity | Higher CRS and neurotoxicity than TRBAs | Unknown but expected |
| Ability to attack solid tumors | To be determined; early data are mixed but not encouraging | To be determined; early data are mixed but not encouraging | Potential based on TIL correlation data |
| Trafficking | Passive | Active but limited; can be engineered to match tumor needs | Active; possible to engineered to match tumor needs |
| Trafficking into CNS | Not demonstrated; Unlikely if BBB is intact [395] | Demonstrated trafficking into CNS [399] | Unknown but expected based on CAR-T results |
| Need for lymphodepletion prior to treatment | No | Yes | Yes |
| Technical risk | Moderate; many platforms are working well | High but may be manageable | Currently very high |
| Need for “kill switch” or turn-off methodology | No but nice to have, especially for long half-life formats | Moderate; nice to have | Very high; must have for safety |
| Accessibility | High–off-the-shelf biologic drug | Only available at specific medical centers thus far; 2–4 week process time before therapy | Projected to eventually have availability similar to biologic drugs |
| Cost of goods | Relatively low; Antibody-like or slightly higher depending on type of TRBA platform | Very high (more than a $75,000 process) | Projected to be low to medium once cell manufacturing process is established |
| Cost to patient/payers | Medium ($89,000/course; $178,000 for predicted two course therapy) * | Very high ($373,000 for treatment of DLBCL; $475,000 for Kymriah® treatment of B-cell ALL) ** | Projected as medium to high, depending on cell type and construct |
9.2. Clinical Comparison of TRBAs vs. CAR-T Cells
9.3. Future Improvements
9.3.1. TRBAs
9.3.2. CARs
10. Summary and Future State
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AML | adult acute myeloid leukemia |
| APC | antigen presenting cell |
| ART-Ig | asymmetric re-engineering technology—immunoglobulin |
| ATTACK | asymmetric tandem trimerbody for T cell activation and cancer killing |
| B-ALL | B cell acute lymphoblastic leukemia |
| BBB | blood-brain barrier |
| BEAT | bispecific engagement by antibodies based on the T cell receptor |
| BiKE | bispecific killer engager |
| BiTE | bispecific T-cell engager |
| CAR | chimeric antigen receptor |
| CD | cluster of differentiation |
| CIKs | cytokine-induced killers |
| CLL | chronic lymphocytic leukemia |
| CNS | central nervous system |
| CR | complete response |
| CRS | cytokine released syndrome |
| CTL | cytotoxic T lymphocytes |
| DART | dual affinity retargeting (antibody) |
| DLBCL | diffuse large B cell lymphoma |
| DVD-Ig | dual variable domain immunoglobulins |
| EGFRt | truncated version of epidermal growth factor receptor |
| EMA | European Medicines Agency |
| EpCAM | epithelial cell adhesion molecule |
| Fc | fragment, crystallizable |
| FIT-Ig | Fabs-in-tandem immunoglobulins |
| GVH, GVHD | graft-versus-host (disease) |
| HC | heavy chain |
| HLA | human leukocyte antigen |
| HSV-TK | herpes simplex virus thymidine kinase |
| iCasp9 | inducible caspase-9 |
| Ig | immunoglobulin |
| ImmTAC | immune-mobilizing monoclonal TCR against cancer |
| iNKT | invariant NKT (cells) |
| ITAM | immunoreceptor tyrosine activation motif |
| KIH | knobs-into-holes |
| LC | light chain |
| MATH | mutant-allele tumor heterogeneity |
| MHC | major histocompatibility complex |
| MM | multiple myeloma |
| NK | natural killer (cell) |
| NKG2D | natural killer group 2D |
| NKT | natural killer T (cell) |
| NSCLC | non-small cell lung cancer |
| OR | objective response |
| PBMCs | peripheral blood mononuclear cells |
| pMHC | peptide-MHC complex |
| PR | partial response |
| R/R | relapsed/refractory |
| sCAR | switchable chimeric antigen receptor |
| scFv | single chain, fragment variable |
| SEED | strand exchange engineered domain |
| SMAC | supramolecular activation cluster |
| TandAb | tandem diabody |
| TBE | target cell-biologic-effector cell (complex) |
| TCB | T-cell bispecifics |
| TCR | T-cell receptor |
| TILs | tumor infiltrating lymphocytes |
| TME | tumor microenvironment |
| TRBA | T-cell redirecting bispecific antibody |
| TriKE | trispecific killer engager |
| TITAC | trispecific T cell activating construct |
| US-FDA | United States Food and Drug Administration |
References
- Coley, W.B. Contribution to the knowledge of sarcoma. Ann. Surg. 1891, 14, 199–220. [Google Scholar] [CrossRef] [PubMed]
- Hoption Cann, S.A.; Van Netten, J.J.; Van Netten, C. Dr William Coley and tumor regression: A place in history or in the future. Postgrad. Med. 2003, 79, 672–680. [Google Scholar]
- Vernon, L.F. William Bradley Coley, MD and the phenomenon of spontaneous regression. Immunotargets Ther. 2018, 7, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.-Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Reisser, D.; Pance, A.; Jeannin, J.F. Mechanisms of the antitumoral effect of lipid A. Bioessays 2002, 24, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Aptsiauri, N.; Ruiz-Cabello, F.; Garrido, F. The transition from HLA-I positive to HLA-I negative primary tumors: The road to escape from T-cell responses. Curr. Opin. Immunol. 2018, 51, 123–132. [Google Scholar] [CrossRef]
- Isaaz, S.; Baetz, K.; Olsen, K.; Podack, E.; Griffiths, G.M. Serial killing by cytotoxic T lymphocytes: T cell receptor triggers degranulation, re-filling of the lytic granules and secretion of lytic proteins via a non-granule pathway. Eur. J. Immunol. 1995, 25, 1071–1079. [Google Scholar] [CrossRef]
- De La Roche, M.; Asano, Y.; Griffiths, G.M. Origins of the cytolytic synapse. Nat. Rev. Immunol. 2016, 16, 421–432. [Google Scholar] [CrossRef]
- Han, X.; Vesely, M.D. Stimulating T cells against cancer with agonist immunostimulatory monoclonal antibodies. Int. Rev. Cell Mol. Biol. 2019, 342, 1–25. [Google Scholar]
- Kim, M.T.; Harty, J.T. Impact of inflammatory cytokines on effector and memory CD8+ T cells. Front. Immunol. 2014, 5, 295. [Google Scholar] [CrossRef]
- Kammertoens, T.; Blankenstein, T. It’s the peptide-MHC affinity, stupid. Cancer Cell 2013, 23, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Bubenik, J. Tumour MHC class I downregulation and immunotherapy (Review). Oncol. Rep. 2003, 10, 2005–2008. [Google Scholar] [CrossRef] [PubMed]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Stopeck, A.T.; Gessner, A.; Miller, T.P.; Hersh, E.M.; Johnson, C.S.; Cui, H.; Frutiger, Y.; Grogan, T.M. Loss of B7.2 (CD86) and intracellular adhesion molecule 1 (CD54) expression is associated with decreased tumor-infiltrating T lymphocytes in diffuse B-cell large-cell lymphoma. Clin. Cancer Res. 2000, 6, 3904–3909. [Google Scholar] [PubMed]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Theate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van Den Eynde, B.J. Evidence for atumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Ellerman, D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 2019, 154, 102–117. [Google Scholar] [CrossRef]
- Chen, D.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Lum, L.G.; Thakur, A. Targeting T cells with bispecific antibodies for cancer therapy. BioDrugs 2011, 25, 365–379. [Google Scholar] [CrossRef]
- Clynes, R.A.; Desjarlais, J.R. Redirected T cell cytotoxicity in cancer therapy. Annu. Rev. Med. 2018, 70, 437–450. [Google Scholar] [CrossRef]
- Lum, L.G.; Thakur, A.; Al-Kadhimi, Z.; Colvin, G.A.; Cummings, F.J.; Legare, R.D.; Dizon, D.S.; Kouttab, N.; Maizei, A.; Colaiace, W.; et al. Targeted T-cell therapy in stage IV breast cancer: A phase I clinical trial. Clin. Cancer Res. 2015, 21, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Brentjens, R.J. Adoptive immunotherapy for B-cell malignancies with autologous chimeric antigen receptor modified tumor targeted T cells. Discov. Med. 2010, 9, 277–288. [Google Scholar] [PubMed]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Antigen-specific T-cell activation independently of the MHC: Chimeric antigen receptor-redirected T cells. Front. Immunol. 2013, 4, 371. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Fujii, R.; Morillon, Y.M., 2nd; Greiner, J.W.; Padget, M.R.; Tritsch, S.R.; Tsang, K.Y.; Campbell, K.S.; et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016, 7, 86359–86373. [Google Scholar] [CrossRef] [PubMed]
- Satta, A.; Mezzanzanica, D.; Turatti, F.; Canevari, S.; Figini, M. Redirection of T-cell effector functions for cancer therapy: Bispecific antibodies and chimeric antigen receptors. Future Oncol. 2013, 9, 527–539. [Google Scholar] [CrossRef]
- Zhukovsky, E.A.; Morse, R.J.; Maus, M.V. Bispecific antibodies and CARs: Generalized immunotherapeutics harnessing T cell redirection. Curr. Opin. Immunol. 2016, 40, 24–35. [Google Scholar] [CrossRef]
- Sahu, G.K.; Sango, K.; Selliah, N.; Ma, Q.; Skowron, G.; Junghans, R.P. Anti-HIV designer T cells progressively eradicate a latently infected cell line by sequentially inducing HIV reactivation then killing the newly gp120-positive cells. Virology 2013, 446, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zou, F.; Lu, L.; Chen, C.; He, D.; Zhang, X.; Tang, X.; Liu, C.; Li, L.; Zhang, H. Chimeric antigen receptor T cells guided by the single-chain Fv of a broadly neutralizing antibody specifically and effectively eradicate virus reactivated from latency in CD4+ T lymphocytes isolated from HIV-1-infected individuals receiving suppressive combined antiretroviral therapy. J. Virol. 2016, 90, 9712–9724. [Google Scholar]
- Hale, M.; Mesojednik, T.; Romano Ibarra, G.S.; Sahni, J.; Bernard, A.; Sommer, K.; Scharenberg, A.M.; Rawlings, D.J.; Wagner, T.A. Engineering HIV-resistant, anti-HIV chimeric antigen receptor T cells. Mol. Ther. 2017, 25, 570–579. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Martz, E. Multiple target cell killing by the cytolytic T lymphocyte and the mechanism of cytotoxicity. Transplantation 1976, 21, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, T.L.; Mage, M.; Jones, G.; McHugh, L.L. Cytotoxic T lymphocyte sequential killing of immobilized allogeneic tumor target cells measured by time-lapse microcinematography. J. Immunol. 1978, 121, 1652–1656. [Google Scholar] [PubMed]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse: A molecular machine controlling T cell activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Gross, G. Chimeric T cell receptor which incorporates the anti-tumour specificity of a monoclonal antibody with the cytolytic activity of T cells: A model system for immunotherapeutical approach. Br. J. Cancer Suppl. 1990, 10, 27–29. [Google Scholar] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Milstein, C.; Cuello, A.C. Hybrid hybridomas and their use in immunohistochemistry. Nature 1983, 305, 537–540. [Google Scholar] [CrossRef]
- Jantscheff, P.; Winkler, L.; Karawajew, L.; Kaiser, G.; Böttger, V.; Micheel, B. Hybrid hybridomas producing bispecific antibodies to CEA and peroxidase isolated by a combination of HAT medium selection and fluorescence activated cell sorting. J. Immunol. Methods 1993, 163, 91–97. [Google Scholar] [CrossRef]
- Brennan, M.; Davison, P.F.; Paulus, H. Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin G1 fragments. Science 1985, 229, 81–83. [Google Scholar] [CrossRef]
- Staerz, U.D.; Kanagaw, O.; Bevan, M.J. Hybrid antibodies can target sites for attack by T cells. Nature 1985, 314, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Perez, P.; Hoffman, R.W.; Shaw, S.; Bluestone, J.A.; Segal, D.M. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature 1985, 316, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Staerz, U.D.; Bevan, M.J. Hybrid hybridoma producing a bispecific monoclonal antibody that can focus effector T-cell activity. Proc. Natl. Acad. Sci. USA 1986, 83, 1453–1457. [Google Scholar] [CrossRef] [PubMed]
- Holliger, P.; Prospero, T.; Winter, G. “Diabodies”: Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.; Riethmüller, G.; Kufer, P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 7021–7025. [Google Scholar] [CrossRef] [PubMed]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-chain antigen-binding proteins. Science 1988, 242, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotný, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R.; et al. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar] [CrossRef]
- Clark, M.; Waldmann, H. T-cell killing of target cells induced by hybrid antibodies: Comparison of two bispecific monoclonal antibodies. J. Natl. Cancer Inst. 1987, 79, 1393–1401. [Google Scholar]
- Nitta, T.; Sato, K.; Yagita, H.; Okumura, K.; Ishii, S. Preliminary trial of specific targeting therapy against malignant glioma. Lancet 1990, 335, 368–371. [Google Scholar] [CrossRef]
- Haagen, I.A.; Van De Griend, R.; Clark, M.; Geerars, A.; Bast, B.; De Gast, B. Killing of human leukaemia/lymphoma B cells by activated cytotoxic T lymphocytes in the presence of a bispecific monoclonal antibody (alpha CD3/alpha CD19). Clin. Exp. Immunol. 1992, 90, 368–375. [Google Scholar] [CrossRef]
- De Gast, G.C.; Van Houten, A.A.; Haagen, I.A.; Klein, S.; De Weger, R.A.; Van Dijk, A.; Phillips, J.; Clark, M.; Bast, B.J. Clinical experience with CD3 × CD19 bispecific antibodies in patients with B cell malignancies. J. Hematother. 1995, 4, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Ridgeway, J.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef]
- Zeidler, R.; Reisbach, G.; Wollenberg, B.; Lang, S.; Chaubel, S.; Schmitt, B.; Lindhofer, H. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J. Immunol. 1999, 163, 1246–1252. [Google Scholar] [PubMed]
- Löffler, A.; Kufer, P.; Lutterbüse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmüller, G.; Dörken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 × CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. Preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Hwu, P.; Shafer, G.E.; Treisman, J.; Schindler, G.; Gross, G.; Cowherd, R.; Rosenberg, S.A.; Eshhar, Z. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J. Exp. Med. 1993, 178, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Moritz, D.; Wels, W.; Mattern, J.; Groner, B. Cytotoxic T lymphocytes with a grafted recognition specificity for ERBB2-expressing tumor cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4318–4322. [Google Scholar] [CrossRef]
- Atwell, S.; Ridgway, J.B.; Wells, J.A.; Carter, P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J. Mol. Biol. 1997, 270, 26–35. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Ha, J.H.; Kim, J.E.; Kim, Y.S. Immunoglobulin Fc heterodimer platform technology: From design to applications in therapeutic antibodies and proteins. Front. Immunol. 2016, 7, 394. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Tesar, M. Recombinant antibodies to arm cytotoxic lymphocytes in cancer immunotherapy. Transfus. Med. Hemother. 2017, 44, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, M.; Kiewe, P.; Schuette, W.; Brust, D.; Peschel, C.; Schneller, F.; Rühle, K.H.; Nilius, G.; Ewert, R.; Lodziewski, S.; et al. Treatment of malignant pleural effusion with the trifunctional antibody catumaxomab (Removab) (anti-EpCAM × Anti-CD3): Results of a phase 1/2 study. J. Immunother. 2009, 32, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Burges, A.; Wimberger, P.; Kümper, C.; Gorbounova, V.; Sommer, H.; Schmalfeldt, B.; Pfisterer, J.; Lichinitser, M.; Makhson, A.; Moiseyenko, V.; et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM × anti-CD3 antibody: A phase I/II study. Clin. Cancer Res. 2007, 13, 3899–3905. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, M. Review of catumaxomab in the treatment of malignant ascites. Cancer Manag. Res. 2010, 2, 283–286. [Google Scholar] [CrossRef]
- Mølhøj, M.; Crommer, S.; Brischwein, K.; Rau, D.; Sriskandarajah, M.; Hoffmann, P.; Kufer, P.; Hofmeister, R.; Baeuerle, P.A. CD19-/CD3-bispecific antibody of the BiTE class is far superior to tandem diabody with respect to redirected tumor cell lysis. Mol. Immunol. 2007, 44, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Przepiorka, D.; Ko, C.W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA approval blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef]
- Kiewe, P.; Thiel, E. Ertumaxomab: A trifunctional antibody for breast cancer treatment. Expert Opin. Investig. Drugs 2008, 17, 1553–1558. [Google Scholar] [CrossRef]
- Topalian, S.L.; Solomon, D.; Avis, F.P.; Chang, A.E.; Freerksen, D.L.; Linehan, W.M.; Lotze, M.T.; Robertson, C.N.; Seipp, C.A.; Simon, P.; et al. Immunotherapy of patients with advanced cancer using tumor-infiltrating lymphocytes and recombinant interleukin-2: A pilot study. J. Clin. Oncol. 1988, 6, 839–853. [Google Scholar] [CrossRef]
- Eshhar, Z.; Waks, T.; Gross, G. The emergence of T-bodies/CAR T cells. Cancer J. 2014, 20, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.A.; June, C.H. The principles of engineering immune cells to treat cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef] [PubMed]
- Hwu, P.; Yang, J.C.; Cowherd, R.; Treisman, J.; Shafer, G.E.; Eshhar, Z.; Rosenberg, S.A. In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell receptor genes. Cancer Res. 1995, 55, 3369–3373. [Google Scholar] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyasu, R.T.; Anton, P.A.; Deeks, S.G.; Scadden, D.T.; Connick, E.; Downs, M.T.; Bakker, A.; Roberts, M.R.; June, C.H.; Jalali, S.; et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood 2000, 96, 785–793. [Google Scholar] [PubMed]
- Junghans, R.P.; Safar, M.; Huberman, M.S. Preclinical and phase I data of anti-CEA ‘‘designer T cell’’ therapy for cancer: A new immunotherapeutic modality. Proc. Am. Assoc. Cancer Res. 2000, 41, 543. [Google Scholar]
- Eshhar, Z. The T-body approach: Redirecting T cells with antibody specificity. Handb. Exp. Pharmacol. 2008, 181, 329–342. [Google Scholar]
- Brentjens, R.J.; Latouche, J.B.; Santos, E.; Marti, F.; Gong, M.C.; Lyddane, C.; King, P.D.; Larson, S.; Weiss, M.; Riviere, I.; et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. 2003, 9, 279–286. [Google Scholar] [CrossRef]
- Cooper, L.J.; Topp, M.S.; Serrano, L.M.; Gonzalez, S.; Chang, W.C.; Naranjo, A.; Wright, C.; Popplewell, L.; Raubitschek, A.; Forman, S.J.; et al. T-cell clones can be rendered specific for CD19: Toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood 2003, 101, 1637–1644. [Google Scholar] [CrossRef]
- Pulè, M.A.; Straathof, K.C.; Dotti, G.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 2005, 12, 933–941. [Google Scholar] [CrossRef]
- Gimmi, C.D.; Freeman, G.J.; Gribben, J.G.; Gray, G.; Nadler, L.M. Human T-cell clonal anergy is induced by antigen presentation in the absence of B7 co-stimulation. Proc. Natl. Acad. Sci. USA 1993, 90, 6586–6590. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.K.; Chen, C.A.; Jung, G.; Mueller, D.L.; Schwartz, R.H. Inhibition of antigen-specific proliferation of type 1 murine T cell clones after stimulation with immobilized anti-CD3 monoclonal antibody. J. Immunol. 1990, 144, 16–22. [Google Scholar]
- Hombach, A.; Sent, D.; Schneider, C.; Heuser, C.; Koch, D.; Pohl, C.; Seliger, B.; Abken, H. T-cell activation by recombinant receptors: CD28 co-stimulation is required for interleukin 2 secretion and receptor-mediated T-cell proliferation but does not affect receptor-mediated target cell lysis. Cancer Res. 2001, 61, 1976–1982. [Google Scholar] [PubMed]
- Finney, H.M.; Akbar, A.N.; Lawson, A.D. Activation of resting human primary T cells with chimeric receptors: Co-stimulation from CD28, inducible costimulator, CD134 and CD137 in series with signals from the TCR zeta chain. J. Immunol. 2004, 172, 104–113. [Google Scholar] [CrossRef]
- Kowolik, C.M.; Topp, M.S.; Gonzalez, S.; Pfeiffer, T.; Olivares, S.; Gonzalez, N.; Smith, D.D.; Forman, S.J.; Jensen, M.C.; Cooper, L.J. CD28 co-stimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006, 66, 10995–11004. [Google Scholar] [CrossRef]
- Hombach, A.A.; Abken, H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int. J. Cancer 2011, 129, 2935–2944. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Moritz, D.; Groner, B. A spacer region between the single chain antibody and the CD3 zeta-chain domain of chimeric T cell receptor components is required for efficient ligand binding and signaling activity. Gene Ther. 1995, 2, 539–546. [Google Scholar]
- Guest, R.D.; Hawkins, R.E.; Kirillova, N.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O’Neill, A.; Irlam, J.; Chester, K.A.; Kemshead, J.T.; et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: Evaluation of four different scFvs and antigens. J. Immunother. 2005, 28, 203–211. [Google Scholar] [CrossRef]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef]
- Qin, L.; Lai, Y.; Zhao, R.; Wei, X.; Weng, J.; Lai, P.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; et al. Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells. J. Hematol. Oncol. 2017, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Condomines, M.; Van Der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural design of engineered co-stimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Prinzing, B.; Cao, F.; Gottschalk, S.; Krenciute, G. Optimizing EphA2-CAR T cells for the adoptive immunotherapy of glioma. Mol. Ther. Methods Clin. Dev. 2018, 9, 70–80. [Google Scholar]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M. Enhancing CAR T cell persistence through ICOS and 4-1BB co-stimulation. JCI Insight 2018, 3, 96976. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Hou, X.; Yang, C.; Liu, Y.; Jiang, G. Advances on chimeric antigen receptor-modified T-cell therapy for oncotherapy. Mol. Cancer 2018, 17, 91. [Google Scholar] [CrossRef] [PubMed]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef]
- Ramello, M.C.; Benzaïd, I.; Kuenzi, B.M.; Lienlaf-Moreno, M.; Kandell, W.M.; Santiago, D.N.; Pabón-Saldaña, M.; Darville, L.; Fang, B.; Rix, U. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal. 2019, 12, eaap9777. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. CAR T cells transform to trucks: Chimeric antigen receptor–redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol. Immunother. 2012, 61, 1269–1277. [Google Scholar] [CrossRef]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powel, D.J., Jr.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef]
- Rossig, C.; Bollard, C.M.; Nuchtern, J.G.; Rooney, C.M.; Brenner, M.K. Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: Potential for improved immunotherapy. Blood 2002, 99, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Pulè, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Rossig, C.; Pulè, M.; Altvater, B.; Saiagh, S.; Wright, G.; Ghorashian, S.; Clifton-Hadley, L.; Champion, K.; Sattar, Z.; Popova, B.; et al. Vaccination to improve the persistence of CD19CAR gene-modified T cells in relapsed pediatric acute lymphoblastic leukemia. Leukemia 2017, 31, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.B. First CAR to pass the road test. Tisagenlecleucel’s drive to FDA approval. Clin. Cancer Res. 2019, 25, 1133–1135. [Google Scholar] [CrossRef] [PubMed]
- Bouchkouj, N.; Kasamon, Y.L.; De Claro, R.A.; George, B.; Lin, X.; Lee, S.; Blumenthal, G.M.; Bryan, W.; McKee, A.E.; Pazdur, R. FDA approval summary: Axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin. Cancer Res. 2019, 25, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, J.C.; Majorovits, E.; Bossi, G.; Fuller, S.; Griffiths, G.M. Centrosome polarization delivers secretory granules to the immunological synapse. Nature 2006, 443, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Kabanova, A.; Zurli, V.; Baldari, C.T. Signals controlling lytic granule polarization at the cytotoxic immune synapse. Front. Immunol. 2018, 9, 307. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Tian, S.; Zhang, K.; Xiong, W.; Lubaki, N.M.; Chen, Z.; Han, W. Chimeric antigen receptor (CAR)-modified natural killer cell-based immunotherapy and immunological synapse formation in cancer and HIV. Protein Cell 2017, 8, 861–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stinchcombe, J.C.; Bossi, G.; Booth, S.; Griffiths, G.M. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 2001, 15, 751–761. [Google Scholar] [CrossRef]
- Huppa, J.B.; Davis, M.M. T-cell-antigen recognition and the immunological synapse. Nat. Rev. 2003, 3, 973–983. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D., Jr.; June, C.H. Expanding the therapeutics window for CAR-T cell therapy in solid tumors: The knowns and unknowns of CAR-T cell biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, A.J.; Jenkins, M.R. Programming a serial killer: CAR T cells form non-classical immune synapses. Oncoscience 2018, 5, 69–70. [Google Scholar] [PubMed]
- Wucherpfennig, K.W.; Gagnon, E.; Call, M.J.; Huseby, E.S.; Call, M.E. Structural biology of the T-cell receptor: Insights into receptor assembly, ligand recognition and initiation of signaling. Cold Spring Harb. Perspect. Biol. 2009, 2, a005140. [Google Scholar] [CrossRef] [PubMed]
- Artyomov, M.N.; Lis, M.; Devadas, S.; Davis, M.M.; Chakraborty, A.K. CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. Proc. Natl. Acad. Sci. USA 2010, 107, 16916–16921. [Google Scholar] [CrossRef] [Green Version]
- Engels, B.; Engelhard, V.H.; Sidney, J.; Sette, A.; Binder, D.C.; Liu, R.B.; Kranz, D.M.; Meredith, S.C.; Rowley, D.A.; Schreiber, H. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell 2013, 23, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Furlan, G.; Minowa, T.; Hanagata, N.; Kataoka-Hamai, C.; Kaizuka, Y. Phosphatase CD45 both positively and negatively regulates T cell receptor phosphorylation in reconstituted membrane protein clusters. J. Biol. Chem. 2014, 289, 28514–28525. [Google Scholar] [CrossRef]
- Penninger, J.M.; Irie-Sasaki, J.; Sasaki, T.; Oliveira-dos-Santos, A.J. CD45: New jobs for an old acquaintance. Nat. Immunol. 2001, 2, 389–396. [Google Scholar] [CrossRef]
- Choudhuri, K.; Wiseman, D.; Brown, M.H.; Gould, K.; Van Der Merwe, P.A. T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature 2005, 436, 578–582. [Google Scholar] [CrossRef]
- Sykulev, Y.; Joo, M.; Vturina, I.; Tsomides, T.J.; Eisen, H.N. Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity 1996, 4, 565–571. [Google Scholar] [CrossRef]
- Purbhoo, M.A.; Irvine, D.J.; Huppa, J.B.; Davis, M.M. T cell killing does not require the formation of a stable mature immunological synapse. Nat. Immunol. 2004, 5, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Gwalani, L.A.; Orange, J.S. Single degranulations in NK cells can mediate target cell killing. J. Immunol. 2018, 200, 3231–3243. [Google Scholar] [CrossRef] [PubMed]
- Benmebarek, M.R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef] [PubMed]
- Cazaux, M.; Grandjean, C.L.; Lemaître, F.; Garcia, Z.; Beck, R.J.; Milo, I.; Postat, J.; Beltman, J.B.; Cheadle, E.J.; Bousso, J. Single-cell imaging of CAR T cell activity in vivo reveals extensive functional and anatomical heterogeneity. J. Exp. Med. 2019, 216, 1038. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Chen, Y.; Kang, X.; Chen, Z.; Zheng, P.; Hsu, Y.H.; Jang, J.H.; Qin, L.; Liu, H.; Dotti, G.; et al. Immunological synapse predicts effectiveness of chimeric antigen receptor cells. Mol. Ther. 2018, 26, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Purbhoo, M.A.; Sutton, D.H.; Brewer, J.E.; Mullings, R.E.; Hill, M.E.; Mahon, T.M.; Karbach, J.; Jäger, E.; Cameron, B.J.; Lissin, N.; et al. Quantifying and imaging NY-ESO-1/LAGE-1-derived epitopes on tumzr cells using high affinity T cell receptors. J. Immunol. 2006, 176, 7308–7316. [Google Scholar] [CrossRef]
- Stone, J.D.; Aggen, D.H.; Schietinger, A.; Schreiber, H.; Kranz, D.M. A sensitivity scale for targeting T cells with chimeric antigen receptors (CARs) and bispecific T-cell Engagers (BiTEs). Oncoimmunology 2012, 1, 863–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Terakura, S.; Martens, A.C.; Van Meerten, T.; Uchiyama, S.; Imai, M.; Sakemura, R.; Goto, T.; Hanajiri, R.; Imahashi, N.; et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J. Immunol. 2015, 194, 911–920. [Google Scholar] [CrossRef]
- Au-Yeung, B.B.; Zikherman, J.; Mueller, J.L.; Ashouri, J.F.; Matloubian, M.; Cheng, D.A.; Chen, Y.; Shokat, K.M.; Weiss, A. A sharp T-cell antigen receptor signaling threshold for T-cell proliferation. Proc. Natl. Acad. Sci. USA 2014, 111, E3679–E3688. [Google Scholar] [CrossRef] [Green Version]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; Van Der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef]
- Wolf, E.; Hofmeister, R.; Kufer, P.; Schlereth, B.; Baeuerle, P.A. BiTEs: Bispecific antibody constructs with unique anti-tumor activity. Drug Discov. Today 2005, 10, 1237–1244. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, M.P.; Szoor, A.; Vaidya, A.; Thakkar, A.; Nguyen, P.; Wu, M.-F.; Liu, H.; Gottschalk, S. CD28 and 41BB co-stimulation enhances the effector function of CD19-specific engager T cells. Cancer Immunol. Res. 2017, 5, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Quintarelli, C.; Orlando, D.; Boffa, I.; Guercio, M.; Polito, V.A.; Petretto, A.; Lavarello, C.; Sinibaldi, M.; Weber, G.; Del Bufalo, F.; et al. Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology 2018, 7, e1433518. [Google Scholar] [CrossRef] [PubMed]
- Feucht, J.; Kayser, S.; Gorodezki, D.; Hamieh, M.; Döring, M.; Blaeschke, F.; Schlegel, P.; Bösmüller, H.; Quintanilla-Fend, L.; Ebinger, M.; et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget 2016, 7, 76902–76919. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Krupka, C.; Deiser, K.; Brauchle, B.; Marcinek, A.; Ogrinc Wagner, A.; Rataj, F.; Mocikat, R.; Metzeler, K.H.; Spiekermann, K.; et al. Bifunctional PD-1 × αCD3 × αCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood 2018, 132, 2484–2494. [Google Scholar] [CrossRef]
- Kobold, S.; Pantelyushin, S.; Rataj, F.; Berg, J.V. Rationale for combining bispecific T cell activating antibodies with checkpoint blockade for cancer therapy. Front. Oncol. 2018, 8, 285. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 8, 3130–3144. [Google Scholar] [CrossRef]
- Blinatumomab Relapsed/Refractory Acute Leukemia or Lymphoma—Clinical Trial: NCT03605589. Available online: https://clinicaltrials.gov/ct2/ show/NCT03605589?term=NCT03605589&rank=1 (accessed on 4 April 2019).
- Safety and Efficacy of Blinatumomab—KEYNOTE-348 Clinical Trial: NCT03340766. Available online: https://clinicaltrials.gov/ct2/show/NCT03340766?term=NCT03340766&rank=1 (accessed on 4 April 2019).
- Blinatumomab in CD19+ Precursor B-Lymphoblastic Leukemia. Clinical Trial: NCT02879695. Available online: https://clinicaltrials.gov/ct2/show/NCT02879695?term=NCT02879695&rank=1 (accessed on 4 April 2019).
- CART-EGFR-vIII+ Pembrolizumab Clinical Trial: NCT03726515. Available online: https://clinicaltrials.gov/ct2/show/NCT03726515?term=NCT03726515&rank=1 (accessed on 4 April 2019).
- Suarez, E.R.; Chang, D.-K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- CTLA-4 and PD-1 Expressing EGFR-CAR-T Cells Clinical Trial: NCT03182816. Available online: https://clinicaltrials.gov/ct2/show/NCT03182816?term=NCT03182816&rank=1 (accessed on 4 April 2019).
- PD-1 Gene-Knocked Out Mesothelin-Directed CAR-T Cells. Clinical Trial: NCT03747965. Available online: https://clinicaltrials.gov/ct2/show/NCT03747965?term=NCT03747965&rank=1 (accessed on 4 April 2019).
- CD16/IL-15/CD33 Tri-Specific Killer Engagers (TriKEs) Clinical Trial: NCT03214666. Available online: https://clinicaltrials.gov/ct2/show/NCT03214666?term=NCT03214666&rank=1 (accessed on 10 April 2019).
- AFM13 in Relapsed/Refractory Cutaneous Lymphomas Clinical Trial: NCT03192202. Available online: https://clinicaltrials.gov/ct2/show/NCT03192202?term=NCT03192202&rank=1 (accessed on 10 April 2019).
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and biomanufacturing of bispecific antibodies. Antibodies 2019. under review. [Google Scholar]
- Husain, B.; Ellerman, D. Expanding the boundaries of biotherapeutics with bispecific antibodies. BioDrugs 2018, 32, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, P.; Humphreys, D.F. Relative contribution of framework and CDR regions in antibody variable domains to multimerisation of Fv- and scFv-containing bispecific antibodies. Antibodies 2018, 7, 35. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Disc. 2019, 1. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Burke, S.; Huang, L.; Gorlatov, S.; Li, H.; Wang, W.; Zhang, W.; Tuaillon, N.; Rainey, J.; Barat, B.; et al. Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J. Mol. Biol. 2010, 399, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lam, C.K.; Long, V.; Widjaja, L.; Yang, Y.; Li, H.; Jin, L.; Burke, S.; Gorlatov, S.; Brown, J.; et al. MGD011, a CD19 × CD3 dual-affinity retargeting bi-specific molecule incorporating extended circulating half-life for the treatment of B-cell malignancies. Clin. Cancer Res. 2017, 23, 1506–1518. [Google Scholar] [CrossRef]
- Bossi, G.; Buisson, S.; Oates, J.; Jakobsen, B.K.; Hassan, N.J. ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol. Immunother. 2014, 63, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Meesters, J.I.; De Goeij, B.E.; Van Den Bremer, E.T.; Neijssen, J.; Van Kampen, M.D.; Strumane, K.; Verploegen, S.; Kundu, A.; Gramer, M.J.; et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc. Natl. Acad. Sci. USA 2013, 110, 5145–5250. [Google Scholar] [CrossRef]
- Skegro, D.; Stutz, C.; Ollier, R.; Svensson, E.; Wassmann, P.; Bourquin, F.; Monney, T.; Gn, S.; Blein, S. Immunoglobulin domain interface exchange as a platform technology for the generation of Fc heterodimers and bispecific antibodies. J. Biol. Chem. 2017, 292, 9745–9759. [Google Scholar] [CrossRef] [Green Version]
- Moore, G.L.; Bautista, C.; Pong, E.; Nguyen, D.H.; Jacinto, J.; Eivazi, A.; Muchhal, U.S.; Karki, S.; Chu, S.Y.; Lazar, G.A. A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. MAbs 2011, 3, 546–557. [Google Scholar] [CrossRef]
- Bacac, M.; Klein, C.; Umaña, P. CEA TCB: A novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunol. 2016, 5, e1203498. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Umaña, P.; Herter, S.; Colombetti, S.; Sam, J.; Le Clech, M.; Freimoser-Grundschober, A.; Richard, M.; Nicolini, V.; Gerdes, C.; et al. CD20 Tcb (RG6026), a novel “2:1” T cell bispecific antibody for the treatment of B cell malignancies. Blood 2016, 128, 1836. [Google Scholar]
- Sampei, Z.; Igawa, T.; Soeda, T.; Okuyama-Nishida, Y.; Moriyama, C.; Wakabayashi, T.; Tanaka, E.; Muto, A.; Kojima, T.; Kitazawa, T.; et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS ONE 2013, 8, e57479. [Google Scholar] [CrossRef] [PubMed]
- Shiraiwa, H.; Narita, A.; Kamata-Sakurai, M.; Ishiguro, T.; Sano, Y.; Hironiwa, N.; Tsushima, T.; Segawa, H.; Tsunenari, T.; Ikeda, Y.; et al. Engineering a bispecific antibody with a common light chain: Identification and optimization of an anti-CD3 epsilon and anti-GPC3 bispecific antibody, ERY974. Methods 2019, 154, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Kipriyanov, S.M.; Moldenhauer, G.; Schuhmacher, J.; Cochlovius, B.; Von Der Lieth, C.-W.; Matys, E.R.; Little, M. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J. Mol. Biol. 1999, 293, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Hoyos, G.; Sewell, T.; Bader, R.; Bannink, J.; Chenault, R.A.; Daugherty, M.; Dasovich, M.; Fang, H.; Gottschalk, R.; Kumer, J.; et al. MOR209/ES414, a novel bispecific antibody targeting PSMA for the treatment of metastatic castration-resistant prostate cancer. Mol. Cancer Ther. 2016, 15, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Madrenas, J.; Chau, L.A.; Teft, W.A.; Wu, P.W.; Jussif, J.; Kasaian, M.; Carreno, B.M.; Ling, V. Conversion of CTLA-4 from inhibitor to activator of T cells with a bispecific tandem single-chain Fv ligand. J. Immunol. 2004, 172, 5948–5956. [Google Scholar] [CrossRef] [PubMed]
- Stieglmaier, J.; Benjamin, J.; Nagorsen, D. Utilizing the BiTE (bispecific T-cell engager) platform for immunotherapy of cancer. Expert Opin. Biol. Ther. 2015, 15, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Conrath, K.E.; Lauwereys, M.; Wyns, L.; Muyldermans, S. Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J. Biol. Chem. 2001, 276, 7346–7350. [Google Scholar] [CrossRef]
- Müller, D.; Karle, A.; Meissburger, B.; Höfig, I.; Stork, R.; Kontermann, R.E. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J. Biol. Chem. 2007, 282, 12650–12660. [Google Scholar] [CrossRef]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.S.; Carol, H.; Biro, M. TriKEs and BiKEs join CARs on the cancer immunotherapy highway. Hum. Vaccines Immunother. 2016, 12, 2790–2796. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Lenvik, T.R.; Davis, Z.B.; Miller, J.S.; Vallera, D.A. Generation of BiKEs and TriKEs to improve NK cell-mediated targeting of tumor cells. Meth. Mol. Biol. 2016, 1441, 333–346. [Google Scholar]
- Schmohl, J.U.; Felices, M.; Taras, E.; Miller, J.S.; Vallera, D.A. Enhanced ADCC and NK cell activation of an anticarcinoma bispecific antibody by genetic insertion of a modified IL-15 cross-linker. Mol. Ther. 2016, 24, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Schoffelen, R.; Boerman, O.C.; Goldenberg, D.M.; Sharkey, R.M.; Van Herpen, C.M.; Franssen, G.M.; McBride, W.J.; Chang, C.H.; Rossi, E.A.; Van Der Graaf, W.T.; et al. Development of an imaging-guided CEA-pretargeted radionuclide treatment of advanced colorectal cancer: First clinical results. Br. J. Cancer 2013, 109, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Bates, A.; Power, C.A. David vs. Goliath: The structure, function, and clinical prospects of antibody fragments. Antibodies 2019, 8, 28. [Google Scholar] [CrossRef]
- Austin, R.; Aaron, W.; Baeuerle, P.; Barath, M.; Jones, A.; Jones, S.D.; Law, C.-L.; Kwant, K.; Lemon, B.; Muchnik, A.; et al. HPN536, a T cell-engaging, mesothelin/CD3-specific TriTAC for the treatment of solid tumors (abstract). Cancer Res. 2018, 78, 1781. [Google Scholar]
- Yang, F.; Wen, W.; Qin, W. Bispecific antibodies as a development platform for new concepts and treatment strategies. Int. J. Mol. Sci. 2017, 18, 48. [Google Scholar] [CrossRef]
- Ellerson, J.R.; Yasmeen, D.; Painter, R.H.; Dorrington, K.J. Structure and function of immunoglobulin domains. III. Isolation and characterization of a fragment corresponding to the Cgamma2 homology region of human immunoglobin G1. J. Immunol. 1976, 116, 510–517. [Google Scholar]
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Ng, S.B.; Born, T.; Retter, M.; Manchulenko, K.; et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 2010, 285, 19637–19646. [Google Scholar] [CrossRef]
- Von Kreudenstein, T.S.; Escobar-Carbrera, E.; Lario, P.I.; D’Angelo, I.; Brault, K.; Kelly, J.; Durocher, Y.; Baardsnes, J.; Woods, R.J.; Xie, M.H.; et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: Quality by molecular design. MAbs 2013, 5, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Ho, W.H.; Boustany, L.M.; Abdiche, Y.N.; Lindquist, K.C.; Farias, S.E.; Rickert, M.; Appah, C.T.; Pascua, E.; Radcliffe, T.; et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J. Mol. Biol. 2012, 420, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Sustmann, C.; Thomas, M.; Stubenrauch, K.; Croasdale, R.; Schanzer, J.; Brinkmann, U.; Kettenberger, H.; Regula, J.T.; Schaefer, W. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. MAbs 2012, 4, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navicixizumabum, Proposed INN: List 114. WHO Drug Information 2015. Volume 29, pp. 550–551. Available online: https://www.who.int/medicines/publications/druginformation/issues/PL_114.pdf?ua=1 (accessed on 19 April 2019).
- Moore, G.L.; Bernett, M.J.; Rashid, R.; Pong, E.W.; Nguyen, D.T.; Jacinto, J.; Eivazi, A.; Nisthal, A.; Diaz, J.E.; Chu, S.Y.; et al. A robust heterodimeric Fc platform engineered for efficient development of bispecific antibodies of multiple formats. Methods 2019, 154, 38–50. [Google Scholar] [CrossRef] [PubMed]
- De Nardis, C.; Hendriks, L.J.A.; Poirier, E.; Arvinte, T.; Gros, P.; Bakker, A.B.H.; De Kruif, J. A new approach for generating bispecific antibodies based on a common light chain format and the stable architecture of human immunoglobulin G1. J. Biol. Chem. 2017, 292, 14706–14717. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.J.; Olson, K.; Haber, L.J.; Varghese, B.; Duramad, P.; Tustian, A.D.; Oyejide, A.; Kirshner, J.R.; Canova, L.; Menon, J.; et al. A novel, native-format bispecific antibody triggering T-cell killing of B-cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci. Rep. 2015, 5, 17943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tustian, A.D.; Endicott, C.; Adams, B.; Mattila, J.; Bak, H. Development of purification processes for fully human bispecific antibodies based upon modification of protein A binding avidity. MAbs 2016, 8, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H.; Aperlo, C.; Li, Y.; Kurosawa, E.; Lan, Y.; Lo, K.-M.; Huston, J.S. SEEDbodies: Fusion proteins based on strand-exchange engineered domain (SEED) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng. Des. Sel. 2010, 23, 195–202. [Google Scholar] [CrossRef]
- Muda, M.; Gross, A.W.; Dawson, J.P.; He, C.; Kurosawa, E.; Schweickhardt, R.; Dugas, M.; Soloviev, M.; Bernhardt, A.; Fischer, D.; et al. Therapeutic assessment of SEED: A new engineered antibody platform designed to generate mono- and bispecific antibodies. Protein Eng. Des. Sel. 2011, 24, 447–454. [Google Scholar] [CrossRef]
- Fischer, N.; Elson, G.; Magistrelli, G.; Dheilly, E.; Fouque, N.; Laurendon, A.; Gueneau, F.; Ravn, U.; Depoisier, J.F.; Moine, V.; et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat. Commun. 2015, 6, 6113. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Kim, Y.J.; Choi, D.K.; Kim, Y.S. Engineering of immunoglobulin Fc heterodimers using yeast surface-displayed combinatorial Fc library screening. PLoS ONE 2015, 10, e0145349. [Google Scholar] [CrossRef] [PubMed]
- Leaver-Fay, A.; Froning, K.J.; Atwell, S.; Aldaz, H.; Pustilnik, A.; Lu, F.; Huang, F.; Yuan, R.; Hassanali, S.; Chamberlain, A.K.; et al. Computationally designed bispecific antibodies using negative state repertoires. Structure 2016, 24, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Suresh, M.R.; Cuello, A.C.; Milstein, C. Bispecific monoclonal antibodies from hybrid hybridomas. Meth. Enzymol. 1986, 121, 210–228. [Google Scholar] [PubMed]
- Van Blarcom, T.; Lindquist, K.; Melton, Z.; Cheung, W.L.; Wagstrom, C.; McDonough, D.; Valle Oseguera, C.; Ding, S.; Rossi, A.; Potluri, S.; et al. Productive common light chain libraries yield diverse panels of high affinity bispecific antibodies. MAbs 2018, 10, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Krah, S.; Sellmann, C.; Rhiel, L.; Schröter, C.; Dickgiesser, S.; Beck, J.; Zielonka, S.; Toleikis, L.; Hock, B.; Kolmar, H.; et al. Engineering bispecific antibodies with defined chain pairing. Nat. Biotechnol. 2017, 39, 167–173. [Google Scholar] [CrossRef]
- Schaefer, W.; Regula, J.T.; Bähner, M.; Schanzer, J.; Croasdale, R.; Dürr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenn, S.; Schiller, C.B.; Griese, J.J.; Duerr, H.; Imhof-Jung, S.; Gassner, C.; Moelleken, J.; Regula, J.T.; Schaefer, W.; Thomas, M.; et al. Crystal structure of an anti-Ang2 CrossFab demonstrates complete structural and functional integrity of the variable domain. PLoS ONE 2013, 8, e61953. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T. The use of CrossMab technology for the generation of bi- and multispecific antibodies. MAbs 2016, 8, 1010–1020. [Google Scholar] [CrossRef]
- Lewis, S.M.; Wu, X.; Pustilnik, A.; Sereno, A.; Huang, F.; Rick, H.L.; Guntas, G.; Leaver-Fay, A.; Smith, E.M.; Ho, C.; et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat. Biotechnol. 2014, 32, 191–198. [Google Scholar] [CrossRef]
- Golay, J.; Choblet, S.; Iwaszkiewicz, J.; Cérutti, P.; Ozil, A.; Loisel, S.; Pugnière, M.; Ubiali, G.; Zoete, V.; Michielin, O.; et al. Design and validation of a novel generic platform for the production of tetravalent IgG1-like bispecific antibodies. J. Immunol. 2016, 196, 3199–3211. [Google Scholar] [CrossRef]
- Dillon, M.; Yin, Y.; Zhou, J.; McCarty, L.; Ellerman, D.; Slaga, D.; Junttila, T.T.; Han, G.; Sandoval, W.; Ovacik, M.A.; et al. Efficient production of bispecific IgG of different isotypes and species of origin in single mammalian cells. MAbs 2017, 9, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Corper, A.L.; Urosev, D.; Tom-Yew, S.A.L.; Bleile, D.W.B.; Von Kreudenstein, T.S.; Dixit, S.; Lario, P.I. Engineered Immunoglobulin Heavy Chain-Light Chain Pairs and Uses Thereof. U.S. Patent 2014/0200331, 17 July 2014. [Google Scholar]
- Spiess, C.; Merchant, M.; Huang, A.; Zheng, Z.; Yang, N.Y.; Peng, J.; Ellerman, D.; Shatz, W.; Reilly, D.; Yansura, D.G.; et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat. Biotechnol. 2013, 31, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Bardwell, P.D.; Staron, M.M.; Liu, J.; Tao, Q.; Scesney, S.; Bukofzer, G.; Rodriguez, L.E.; Choi, C.H.; Wang, J.; Chang, Q.; et al. Potent and conditional redirected T cell killing of tumor cells using half DVD-Ig. Protein Cell 2018, 9, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Safety, Pharmacokinetics and Therapeutic Activity of RO6958688. Clinical Trial NCT02650713. Available online: https://clinicaltrials.gov/ct2/show/NCT02650713?term=NCT02650713&rank=1 (accessed on 16 April 2019).
- Dose Escalation Study with RO7082859. Clinical Trial NCT03075696. Available online: https://clinicaltrials.gov/ct2/show/NCT03075696?term=NCT03075696&rank=1 (accessed on 16 April 2019).
- Vu, M.D.; Moser, S.; Delon, C.; Latzko, M.; Gianotti, R.; Lüoend, R.; Friang, C.; Murr, R.; Duerner, L.; Weinzierl, T.; et al. A new class of T-cell bispecific antibodies for the treatment of multiple myeloma, binding to B cell maturation antigen and CD3 and showing potent, specific antitumor activity in myeloma cells and long duration of action in cynomolgus monkeys. Blood 2015, 126, 2998. [Google Scholar]
- Rius Ruiz, I.; Vicario, R.; Morancho, B.; Morales, C.B.; Arenas, E.J.; Herter, S.; Freimoser-Grundschober, A.; Somandin, J.; Sam, J.; Ast, O.; et al. p95HER2–T cell bispecific antibody for breast cancer treatment. Sci. Transl. Med. 2018, 10, eaat1445. [Google Scholar] [CrossRef] [PubMed]
- Study of ERY974 in Patients with Advanced Solid Tumors. Clinical Trial NCT02748837. Available online: https://clinicaltrials.gov/ct2/show/NCT02748837?term=NCT02748837&rank=1 (accessed on 16 April 2019).
- Harwood, S.L.; Alvarez-Cienfuegos, A.; Nunez-Prado, N.; Compte, M.; Hernandez-Perez, S.; Merino, N.; Bonet, J.; Navarro, R.; Van Bergen En Henegouwen, P.M.P.; Lykkemark, S.; et al. ATTACK, a novel bispecific T cell-recruiting antibody with trivalent EGFR binding and monovalent CD3 binding for cancer immunotherapy. Oncoimmunology 2017, 7, e1377874. [Google Scholar] [CrossRef] [PubMed]
- Dickopf, S.; Lauer, M.E.; Ringler, P.; Spick, C.; Kern, P.; Brinkmann, U. Highly flexible, IgG-shaped, trivalent antibodies effectively target tumor cells and induce T cell-mediated killing. Biol. Chem. 2019, 400, 343–350. [Google Scholar] [CrossRef]
- Shiheido, H.; Chen, C.; Hikida, M.; Watanabe, T.; Shimizu, J. Modulation of the human T cell response by a novel non-mitogenic anti-CD3 antibody. PLoS ONE 2014, 9, e94324. [Google Scholar] [CrossRef]
- Phase 1 Study of AMV564 in Patients with Myelodysplastic Syndromes. Clinical Trial NCT03516591. Available online: https://clinicaltrials.gov/ct2/show/NCT03516591?term=NCT03516591&rank=1 (accessed on 18 April 2019).
- Study of ES414 in Metastatic Castration-Resistant Prostate Cancer. Clinical Trial NCT02262910. Available online: https://clinicaltrials.gov/ct2/show/NCT02262910?term=NCT02262910&rank=1 (accessed on 18 April 2019).
- Coloma, M.J.; Morrison, S.L. Design and production of novel tetravalent bispecific antibodies. Nat. Biotechnol. 1997, 15, 159–163. [Google Scholar] [CrossRef]
- Wu, C.; Ting, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007, 25, 1290–1297. [Google Scholar] [CrossRef]
- Wu, C.; Ying, H.; Bose, S.; Miller, R.; Medina, L.; Santora, L.; Ghayur, T. Molecular construction and optimization of anti-human IL-1alpha/beta dual variable domain immunoglobulin (DVD-Ig) molecules. MAbs 2009, 1, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Ren, F.; Wu, D.; Wu, X.; Wu, C. Fabs-in-tandem immunoglobulin is a novel and versatile bispecific design for engaging multiple therapeutic targets. MAbs 2017, 9, 1118–1128. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Zhu, Z. Construction and production of an IgG-like tetravalent bispecific antibody, IgG-single-chain Fv fusion. Meth. Mol. Biol. 2014, 1060, 185–213. [Google Scholar]
- Dong, J.; Sereno, A.; Snyder, W.B.; Miller, B.R.; Tamraz, S.; Doern, A.; Favis, M.; Wu, X.; Tran, H.; Langley, E.; et al. Stable IgG-like bispecific antibodies directed toward the type I insulin-like growth factor receptor demonstrate enhanced ligand blockade and anti-tumor activity. J. Biol. Chem. 2011, 286, 4703–4717. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; Demarest, S.J.; Lugovskoy, A.; Huang, F.; Wu, X.; Snyder, W.B.; Croner, L.J.; Wang, N.; Amatucci, A.; Michaelson, J.S.; et al. Stability engineering of scFvs for the development of bispecific and multivalent antibodies. Protein Eng. Des. Sel. 2010, 23, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Bluemel, C.; Hausmann, S.; Fluhr, P.; Sriskandarajah, M.; Stallcup, W.B.; Baeuerle, P.A.; Kufer, P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol. Immunother. 2010, 59, 1197–1209. [Google Scholar] [CrossRef]
- Jiang, X.; Chen, X.; Carpenter, T.J.; Wang, J.; Zhou, R.; Davis, H.M.; Heald, D.L.; Wang, W. Development of a target cell-biologics-effector cell (TBE) complex-based cell killing model to characterize target cell depletion by T cell redirecting bispecific agents. MAbs 2018, 10, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Slaney, C.Y.; Wang, P.; Darcy, P.K.; Kershaw, M.H. CARs versus BiTEs: A comparison between T cell-redirection strategies for cancer treatment. Cancer Discov. 2018, 8, 924–934. [Google Scholar] [CrossRef]
- James, S.E.; Greenberg, P.D.; Jensen, M.C.; Lin, Y.; Wang, J.; Till, B.G.; Raubitschek, A.A.; Forman, S.J.; Press, O.W. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J. Immunol. 2008, 180, 7028–7038. [Google Scholar] [CrossRef]
- Root, A.; Cao, W.; Li, B.; LaPan, P.; Meade, C.; Sanford, J.; Jin, M.; O’Sullivan, C.; Cummins, E.; Lambert, M.; et al. PF-06671008, a highly potent anti-P-cadherin/anti-CD3 bispecific DART molecule with extended half-life for the treatment of cancer. Antibodies 2016, 5, 6. [Google Scholar] [CrossRef]
- Qi, J.; Li, X.; Peng, H.; Cook, E.M.; Dadashian, E.L.; Wiestner, A.; Park, H.; Rader, C. Potent and selective antitumor activity of a T cell-engaging bispecific antibody targeting a membrane-proximal epitope of ROR1. Proc. Natl. Acad. Sci. USA 2018, 115, E5467–E5476. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Colombetti, S.; Herter, S.; Sam, J.; Perro, M.; Chen, S.; Bianchi, R.; Richard, M.; Schoenle, A.; Nicolini, V.; et al. CD20-TCB with obinutuzumab pretreatment as next-generation treatment of hematologic malignancies. Clin. Cancer Res. 2018, 24, 4785–4797. [Google Scholar] [CrossRef] [PubMed]
- IGM Biosciences Anti-CD20 × CD3 IgM. Available online: http://igmbio.com/pipeline/cd20-x-dc3/ (accessed on 18 April 2019).
- Chelius, D.; Ruf, P.; Gruber, P.; Plöscher, M.; Liedtke, R.; Gansberger, E.; Hess, J.; Wasiliu, M.; Lindhofer, H. Structural and functional characterization of the trifunctional antibody catumaxomab. MAbs 2010, 2, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical development and future directions. MAbs 2010, 2, 129–136. [Google Scholar] [CrossRef]
- Lee, K.J.; Chow, V.; Weissman, A.; Tulpule, S.; Aldoss, I.; Akhtari, M. Clinical use of blinatumomab for B-cell acute lymphoblastic leukemia in adults. Ther. Clin. Risk Manag. 2016, 12, 1301–1310. [Google Scholar]
- Nisonoff, A.; Rivers, M.M. Recombination of a mixture of univalent antibody fragments of different specificity. Arch. Biochem. Biophys. 1961, 93, 460–462. [Google Scholar] [CrossRef]
- Karpovsky, B.; Titus, J.A.; Stephany, D.A.; Segal, D.M. Production of target-specific effector cells using hetero-cross-linked aggregates containing anti-target cell and anti-Fc gamma receptor antibodies. J. Exp. Med. 1984, 160, 1686–1701. [Google Scholar] [CrossRef]
- Glennie, M.J.; McBride, H.M.; Worth, A.T.; Stevenson, G.T. Preparation and performance of bispecific F(ab’ gamma)2 antibody containing thioether-linked Fab’ gamma fragments. J. Immunol. 1987, 139, 2367–2375. [Google Scholar]
- Sen, M.; Wankowski, D.M.; Garlie, N.K.; Siebenlist, R.E.; Van Epps, D.; LeFever, A.V.; Lum, L.G. Use of anti-CD3 × anti-HER2/neu bispecific antibody for redirecting cytotoxicity of activated T cells toward HER2/neu+ tumors. J. Hematother. Stem Cell Res. 2001, 10, 247–260. [Google Scholar] [CrossRef]
- Reusch, U.; Sundaram, M.; Davol, P.A.; Olson, S.D.; Davis, J.B.; Demel, K.; Nissim, J.; Rathore, R.; Liu, P.Y.; Lum, L.G. Anti-CD3 × anti-epidermal growth factor receptor (EGFR) bispecific antibody redirects T-cell cytolytic activity to EGFR-positive cancers in vitro and in an animal model. Clin. Cancer Res. 2006, 12, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Yankelevich, M.; Kondadasula, S.V.; Thakur, A.; Buck, S.; Cheung, N.K.; Lum, L.G. Anti-CD3 × anti-GD2 bispecific antibody redirects T-cell cytolytic activity to neuroblastoma targets. Pediatr. Blood Cancer 2012, 59, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Han, H.; Liu, D.; Li, W.; Feng, H.; Xue, X.; Wu, X.; Niu, G.; Zhang, G.; Zhao, Y.; et al. HER2 as a promising target for cytotoxicity T cells in human melanoma therapy. PLoS ONE 2013, 8, e73261. [Google Scholar]
- HER2Bi-Armed Activated T Cells for Castration Resistant Prostate Cancer. Clinical Trial NCT03406858. Available online: https://clinicaltrials.gov/ct2/show/NCT03406858?term=NCT03406858&rank=1 (accessed on 20 June 2019).
- Bispecific Antibody Armed Activated T-Cells. Clinical Trial NCT02620865. Available online: https://clinicaltrials.gov/ct2/show/NCT02620865?term=NCT02620865&rank=1 (accessed on 20 April 2019).
- Activated T Cells Armed with GD2 Bispecific Antibody. Clinical Trial NCT02173093. Available online: https://clinicaltrials.gov/ct2/show/NCT02173093?term=NCT02173093&rank=1 (accessed on 20 April 2019).
- Thakur, A.; Sorenson, C.; Norkina, O.; Schalk, D.; Ratanatharathorn, V.; Lum, L.G. Activated T cells from umbilical cord blood armed with anti-CD3 × anti-CD20 bispecific antibody mediate specific cytotoxicity against CD20+ targets with minimal allogeneic reactivity: A strategy for providing antitumor effects after cord blood transplants. Transfusion 2012, 52, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Kung, P.; Goldstein, G.; Reinherz, E.L.; Schlossman, S.F. Monoclonal antibodies defining distinctive human T cell surface antigens. Science 1979, 206, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Kwakkenbos, M.J.; Claassen, Y.B.; Maijoor, K.; Böhne, M.; Van Der Sluijs, K.F.; Witte, M.D.; Van Zoelen, D.J.; Cornelissen, L.A.; Beaumont, T.; et al. Bispecific antibody generated with sortase and click chemistry has broad antiinfluenza virus activity. Proc. Natl. Acad. Sci. USA 2014, 111, 16820–16825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Mi, Y.; Guo, N.; Xu, H.; Xu, L.; Gou, X.; Jin, W. Cytokine-induced killer cells as pharmacological tools for cancer immunotherapy. Front. Immunol. 2017, 8, 774. [Google Scholar] [CrossRef]
- Study of Activated Cytokine-Induced Killer. Clinical Trial NCT03554395. Available online: https://clinicaltrials.gov/ct2/show/NCT03554395?term=NCT03554395&rank=1 (accessed on 20 April 2019).
- James, N.D.; Atherton, P.J.; Jones, J.; Howie, A.J.; Tchekmedyian, S.; Curnow, R.T. A phase II study of the bispecific antibody MDX-H210 (anti-HER2 × CD64) with GM-CSF in HER2+ advanced prostate cancer. Br. J. Cancer 2001, 85, 152–156. [Google Scholar] [CrossRef]
- Repp, R.; Van Ojik, H.H.; Valerius, T.; Groenewegen, G.; Wieland, G.; Oetzel, C.; Stockmeyer, B.; Becker, W.; Eisenhut, M.; Steininger, H.; et al. Phase I clinical trial of the bispecific antibody MDX-H210 (anti-FcgammaRI × anti-HER-2/neu) in combination with Filgrastim (G-CSF) for treatment of advanced breast cancer. Br. J. Cancer 2003, 89, 2234–2243. [Google Scholar] [CrossRef]
- Balaian, L.; Ball, E.D. Inhibition of acute myeloid leukemia cell growth by mono-specific and bi-specific anti-CD33 × anti-CD64 antibodies. Leuk. Res. 2004, 28, 821–829. [Google Scholar] [CrossRef]
- Stockmeyer, B.; Dechant, M.; Van Egmond, M.; Tutt, A.L.; Sundarapandiyan, K.; Graziano, R.F.; Repp, R.; Kalden, J.R.; Gramatzki, M.; Glennie, M.J.; et al. Triggering Fc alpha-receptor I (CD89) recruits neutrophils as effector cells for CD20-directed antibody therapy. J. Immunol. 2000, 165, 5954–5961. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; Hartshorn, K.L.; White, M.R.; Van Kooten, C.; Van De Winkel, J.G.; Reid, K.B.; Batenburg, J.J. Effective targeting of pathogens to neutrophils via chimeric surfactant protein D/anti-CD89 protein. J. Immunol. 2004, 172, 4934–4940. [Google Scholar] [CrossRef] [PubMed]
- Guettinger, Y.; Barbin, K.; Peipp, M.; Bruenke, J.; Dechant, M.; Horner, H.; Thierschmidt, D.; Valerius, T.; Repp, R.; Fey, G.H.; et al. A recombinant bispecific single-chain fragment variable specific for HLA class II and Fc alpha RI (CD89) recruits polymorphonuclear neutrophils for efficient lysis of malignant B lymphoid cells. J. Immunol. 2010, 184, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Boross, P.; Lohse, S.; Nederend, M.; Jansen, J.H.; Van Tetering, G.; Dechant, M.; Peipp, M.; Royle, L.; Liew, L.P.; Boon, L.; et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol. Med. 2013, 5, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Duval, M.; Gawron, M.; Posner, M.R.; Cavacini, L.A. Overcoming the constraints of anti-HIV/CD89 bispecific antibodies that limit viral inhibition. J. Immunol. Res. 2016, 2016, 1–5. [Google Scholar] [CrossRef]
- Germain, C.; Campigna, E.; Salhi, I.; Morisseau, S.; Navarro-Teulon, I.; Mach, J.P.; Pèlegrin, A.; Robert, B. Redirecting NK cells mediated tumor cell lysis by a new recombinant bifunctional protein. Protein Eng. Des. Sel. 2008, 21, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silla, L.M.; Chen, J.; Zhong, R.K.; Whiteside, T.L.; Ball, E.D. Potentiation of lysis of leukaemia cells by a bispecific antibody to CD33 and CD16 (Fc gamma RIII) expressed by human natural killer (NK) cells. Br. J. Haematol. 1995, 89, 712–718. [Google Scholar] [CrossRef]
- Hartmann, F.; Renner, C.; Jung, W.; Da Costa, L.; Tembrink, S.; Held, G.; Sek, A.; König, J.; Bauer, S.; Kloft, M.; et al. Anti-CD16/CD30 bispecific antibody treatment for Hodgkin’s disease: Role of infusion schedule and costimulation with cytokines. Clin. Cancer Res. 2001, 7, 1873–1881. [Google Scholar]
- Lo Nigro, C.; Macagno, M.; Sangiolo, D.; Bertolaccini, L.; Aglietta, M.; Merlano, M.C. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 2019, 7, 105. [Google Scholar] [CrossRef]
- Chiang, S.C.; Theorell, J.; Entesarian, M.; Meeths, M.; Mastafa, M.; Al-Herz, W.; Frisk, P.; Gilmour, K.C.; Ifversen, M.; Langenskiöld, C.; et al. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood 2013, 121, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Yang, H.; Dimitrov, D.S. Identification of high-affinity anti-CD16A allotype-independent human antibody domains. Exp. Mol. Pathol. 2016, 101, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Fu, J.; Zhang, M.; Liu, D. AFM13: A first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. J. Hematol. Oncol. 2015, 8, 96. [Google Scholar] [CrossRef] [PubMed]
- GHSG-AFM13 an Open-Label, Multicenter Phase II Trial. Clinical Trial NCT02321592. Available online: https://clinicaltrials.gov/ct2/show/NCT02321592?term=NCT02321592&rank=1 (accessed on 21 April 2019).
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion and enhanced function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Tsang, K.Y.; Vandeveer, A.J.; Gulley, J.L.; Schlom, J. ADCC employing an NK cell line (haNK) expressing the high affinity CD16 allele with avelumab, an anti-PD-L1 antibody. Int. J. Cancer 2017, 141, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Quilt-3.028: Study of HANK™. Clinical Trial NCT03027128. Available online: https://clinicaltrials.gov/ct2/show/NCT03027128?term=NCT03027128&rank=1 (accessed on 21 April 2019).
- Evaluate Efficacy of Avelumab, HaNK and N-803. Clinical Trial NCT03853317. Available online: https://clinicaltrials.gov/ct2/show/NCT03853317?term=NCT03853317&rank=1 (accessed on 21 April 2019).
- Motz, G.; Whiteman, K.; Shin, J.; Pai, T.; Judge, C.; Barnitz, A.; Hemphill, J.; Kim, J.; Ranger, A.; Huet, H.; et al. ACTR707: A novel T-cell therapy for the treatment of relapsed or refractory CD20+ B cell lymphoma in combination with rituximab. Mol. Cancer Ther. 2018, 17, B105. [Google Scholar] [CrossRef]
- Study of ACTR707 for Relapsed or Refractory B Cell Lymphoma. Clinical Trial NCT03189836. Available online: https://clinicaltrials.gov/ct2/show/NCT03189836?term=NCT03189836&rank=1 (accessed on 21 April 2019).
- Akard, L.P.; Jaglowski, S.; Devine, S.M.; McKinney, M.S.; Vasconcelles, M.; Huet, H.; Ettenberg, S.; Ranger, A.; Abramson, J.S. ACTR087, autologous T lymphocytes expressing antibody coupled T-cell receptors (ACTR), induces complete responses in patients with relapsed or refractory CD20-positive B-cell lymphoma, in combination with rituximab. Blood 2017, 130, 580. [Google Scholar]
- Study of ACTR087 for Relapsed or Refractory B Cell Lymphoma. Clinical Trial NCT02776813. Available online: https://clinicaltrials.gov/ct2/show/NCT02776813?term=NCT02776813&rank=1 (accessed on 21 April 2019).
- Study of ACTR087 for Relapsed or Refractory Multiple Myeloma. Clinical Trial NCT03266692. Available online: https://clinicaltrials.gov/ct2/show/NCT03266692?term=NCT03266692&rank=1 (accessed on 21 April 2019).
- Van Epps, H.; Anderson, M.; Yu, C.; Klussman, K.; Westendorf, L.; Carosino, C.; Manlove, L.; Cochran, J.; Neale, J.; Benjamin, D.; et al. SEA-BCMA: A highly active enhanced antibody for multiple myeloma. Cancer Res. 2018, 78, 3833. [Google Scholar]
- Fujio, K.; Okamura, T.; Sumitomo, S.; Yamamoto, K. Regulatory T cell-mediated control of autoantibody-induced inflammation. Front. Immunol. 2012, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Getts, D.; Hofmeister, R.; Quintás-Cardama, A. Synthetic T cell receptor-based lymphocytes for cancer therapy. Adv. Drug Deliv. Rev. 2019. [Google Scholar] [CrossRef]
- Mensali, N.; Dillard, P.; Hebeisen, M.; Lorenz, S.; Theodossiou, T.; Myhre, M.R.; Fåne, A.; Gaudernack, G.; Kvalheim, G.; Myklebust, J.H.; et al. NK cells specifically TCR-dressed to kill cancer cells. EBioMedicine 2019, 40, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Walseng, E.; Köksal, H.; Sektioglu, I.M.; Fåne, A.; Skorstad, G.; Kvalheim, G.; Gaudernack, G.; Inderberg, E.M.; Wälchli, S. A TCR-based chimeric antigen receptor. Sci. Rep. 2017, 7, 10713. [Google Scholar] [CrossRef] [PubMed]
- Efficacy and Safety Study of Bb2121. Clinical Trial NCT03651128. Available online: https://clinicaltrials.gov/ct2/show/NCT03651128?term=NCT03651128&rank=1 (accessed on 21 April 2019).
- A Study to Compare Efficacy and Safety of JCAR017 to Standard of Care. Clinical Trial NCT03575351. Available online: https://clinicaltrials.gov/ct2/show/NCT03575351?term=NCT03575351&rank=1 (accessed on 21 April 2019).
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells—Challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- Salmikangas, P.; Kinsella, N.; Chamberlain, P. Chimeric antigen receptor T-cells (CAR T-cells) for cancer immunotherapy—Moving target for industry? Pharm. Res. 2018, 35, 152. [Google Scholar] [CrossRef] [PubMed]
- Makita, S.; Imaizumi, K.; Kurosawa, S.; Tobinai, K. Chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma: Opportunities and challenges. Drugs Context 2019, 8, 212567. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; June, C.H. Driving gene-engineered T cell immunotherapy of cancer. Cell Research 2017, 27, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Murad, J.M.; Baumeister, S.H.; Werner, L.; Daley, H.; Trébéden-Negre, H.; Reder, J.; Sentman, C.L.; Gilham, D.; Lehmann, F.; Snykers, S.; et al. Manufacturing development and clinical production of NKG2D chimeric antigen receptor-expressing T cells for autologous adoptive cell therapy. Cytotherapy 2018, 20, 952–963. [Google Scholar] [CrossRef]
- He, J.; Zhang, Z.; Lv, S.; Liu, X.; Cui, L.; Jiang, D.; Zhang, Q.; Li, L.; Qin, W.; Jin, H. Engineered CAR T cells targeting mesothelin by piggyBac transposon system for the treatment of pancreatic cancer. Cell. Immunol. 2018, 329, 31–40. [Google Scholar] [CrossRef]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar]
- Chow, V.A.; Shadman, M.; Gopal, A.K. Translating anti-CD19 CAR T-cell therapy into clinical practice for relapsed/refractory diffuse large B-Cell lymphoma. Blood 2018, 132, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Kymriah European Public Assessment Reports (EPAR) Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/kymriah-epar-product-information_en.pdf (accessed on 21 April 2019).
- Yescarta European Public Assessment Reports (EPAR) Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/yescarta-epar-product-information_en.pdf (accessed on 21 April 2019).
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective targeting of multiple B-cell maturation antigen-expressing hematological malignances by anti-B-cell maturation antigen chimeric antigen receptor T cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Bb2121 Website. Available online: https://www.researchoncology.com/translational-research/bb2121-car-t/ (accessed on 21 April 2019).
- Ramsborg, C.G.; Guptill, P.; Weber, C.; Christin, B.; Larson, R.P.; Lewis, K.; Mallaney, M.; Bowen, M.; Higham, E.; Albertson, T. JCAR017 is a defined composition CAR T cell product with product and process controls that deliver precise doses of CD4 and CD8 CAR T cell to patients with NHL. Blood 2017, 130, 4471. [Google Scholar]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Torikai, H.; Reik, A.; Soldner, F.; Warren, E.H.; Yuen, C.; Zhou, Y.; Crossland, D.L.; Huls, H.; Littman, N.; Zhang, Z.; et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013, 122, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, B.; Ono, M.; Yoshida, Y.; Kaneko, S.; Hotta, A. Targeted disruption of HLA genes via CRISPR-Cas9 generates iPSCs with enhanced immune compatibility. Cell Stem Cell 2019, 24, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Torikai, H.; Reik, A.; Yuen, C.; Zhou, Y.; Kellar, S.; Huls, H.; Warren, E.E., III; Tykodi, S.S.; Gregory, P.D.; Holmes, M.C.; et al. HLA and TCR knockout by zinc finger nucleases: Toward “off-the-shelf” allogeneic T-cell therapy for CD19+ malignancies. Blood 2010, 116, 3766. [Google Scholar]
- Mehta, R.S.; Rezvani, K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front. Immunol. 2018, 9, 283. [Google Scholar] [CrossRef]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immmunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef]
- Sommer, C.; Boldajipour, B.; Kuo, T.C.; Bentley, T.; Sutton, J.; Chen, A.; Geng, T.; Dong, H.; Galetto, R.; Valton, J.; et al. Preclinical evaluation of allogeneic CAR T cells targeting BCMA for the treatment of multiple myeloma. Mol. Ther. 2019, 27, 1126–1138. [Google Scholar] [CrossRef]
- Philip, B.; Kokalaki, E.; Mekkaoui, L.; Thomas, S.; Straathof, K.; Flutter, B.; Marin, V.; Marafioti, T.; Chakraverty, R.; Linch, D.; et al. A highly compact epitope based marker/suicide gene for easier and safer T-cell therapy. Blood 2014, 124, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Valton, J.; Guyot, V.; Boldajipour, B.; Sommer, C.; Pertel, T.; Juillerat, A.; Duclert, A.; Sasu, B.J.; Duchateau, P.; Poirot, L. A versatile safeguard for chimeric antigen receptor T-cell immunotherapies. Sci. Rep. 2018, 8, 8972. [Google Scholar] [CrossRef] [PubMed]
- Gautron, A.S.; Juillerat, A.; Guyot, V.; Filhol, J.-M.; Dessez, E.; Duclert, A.; Duchateau, P.; Poirot, L. Fine and predictable tuning of TALEN gene editing targeting for improved T cell adoptive immunotherapy. Mol. Ther. Nucleic Acids 2017, 9, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Harrer, D.C.; Dörrie, J.; Schaft, N. Chimeric antigen receptors in different cell types: New vehicles join the race. Hum. Gene Ther. 2018, 29, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Narni-Mancinelli, E.; Vivier, E.; Kerdiles, Y.M. The ‘T-cell-ness’ of NK cells: Unexpected similarities between NK cells and T cells. Internat. Immunol. 2011, 23, 427–431. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural killer cells: Development, maturation and clinical utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef]
- Wolf, B.J.; Choi, J.E.; Exley, M.A. Novel approaches to exploiting invariant NKT cells in cancer immunotherapy. Front. Immunol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Fisher, J.; Anderson, J. Engineering approaches in human gamma delta T cells for cancer immunotherapy. Front. Immunol. 2018, 9, 1409. [Google Scholar] [CrossRef]
- Gamma Delta T Cells in AML. Clinical Trial NCT03885076. Available online: https://clinicaltrials.gov/ct2/show/NCT03885076?term=NCT03885076&rank=1 (accessed on 21 April 2019).
- Capsomidis, A.; Benthall, G.; Van Acker, H.H.; Fisher, J.; Kramer, A.M.; Abeln, Z.; Majani, Y.; Gileadi, T.; Wallace, R.; Gustafsson, K.; et al. Chimeric antigen receptor-engineered human gamma delta T cells: Enhanced cytotoxicity with retention of cross presentation. Mol. Ther. 2018, 26, 354–365. [Google Scholar] [CrossRef]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7, e36688. [Google Scholar] [CrossRef]
- Allen, E.S.; Stroncek, D.F.; Ren, J.; Eder, A.F.; West, K.A.; Fry, T.J.; Lee, D.W.; Mackall, C.L.; Conry-Cantilena, C. Autologous lymphapheresis for the production of chimeric antigen receptor (CAR) T Cells. Transfusion 2017, 57, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Q.J.; Yang, S.; Kochenderfer, J.N.; Zheng, Z.; Zhong, X.; Sadelain, M.; Eshhar, Z.; Rosenberg, S.A.; Morgan, R.A. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J. Immunol. 2009, 183, 5563–5574. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunol. 2016, 5, e1253656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajina, A.; Maher, J. Strategies to address chimeric antigen receptor tonic signaling. Mol. Cancer Ther. 2018, 17, 1795–1815. [Google Scholar] [CrossRef] [PubMed]
- Kulemzin, S.V.; Kuznetsova, V.V.; Mamonkin, M.; Taranin, A.V.; Gorchakov, A.A. Engineering chimeric antigen receptors. Acta Naturae 2017, 9, 6–14. [Google Scholar] [CrossRef]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and nanobody-based human heavy chain antibodies as antitumor therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef] [PubMed]
- Rahbarizadeh, F.; Ahmadvand, D.; Moghimi, S.M. CAR T-cell bioengineering: Single variable domain of heavy chain antibody targeted CARs. Adv. Drug Deliv. Rev. 2019. [Google Scholar] [CrossRef]
- Chin, C.-N.; Lee, J.; McCabe, T.; Mooney, J.; Naso, M.; Strohl, W.R. Chimeric Antigen Receptors Comprising BCMA-Specific Fibronectin Type III Domains and Uses Thereof. Patent WO/2018/052828, 22 March 2018. [Google Scholar]
- Han, X.; Cinay, G.E.; Zhao, Y.; Guo, Y.; Zhang, X.; Wang, P. Adnectin-based design of chimeric antigen receptor for T cell engineering. Mol. Ther. 2017, 25, 2466–2476. [Google Scholar] [CrossRef]
- Hammill, J.A.; VanSeggelen, H.; Helsen, C.W.; Denisova, G.F.; Evelegh, C.; Tantalo, D.G.; Bassett, J.D.; Bramson, J.L. Designed ankyrin repeat proteins are effective targeting elements for chimeric antigen receptors. J. Immunother. Cancer 2015, 3, 55. [Google Scholar] [CrossRef] [Green Version]
- Hermanson, D.L.; Barnett, B.E.; Rengarajan, S.; Codde, R.; Wang, X.; Tan, Y.; Martin, C.E.; Smith, J.B.; He, J.; Mathur, R.; et al. A novel BCMA-specific, centyrin-based CAR-T product for the treatment of multiple myeloma. Blood 2016, 128, 2127. [Google Scholar]
- Martyniszyn, A.; Krahl, A.-C.; André, M.C.; Hombach, A.A.; Abken, H. CD20-CD19 bispecific CAR T cells for the treatment of B-cell malignancies. Hum. Gene Ther. 2017, 28, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- De Munter, S.; Ingels, J.; Goetgeluk, G.; Bonte, S.; Pille, M.; Weening, K.; Kerre, T.; Abken, H.; Vandekerckhove, B. Nanobody based dual specific CARs. Int. J. Mol. Sci. 2018, 19, 403. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.H.; Liu, J.; Wang, B.Y.; Chen, Y.X.; Cao, X.M.; Yang, Y.; Zhang, Y.L.; Wang, F.X.; Zhang, P.Y.; Lei, B.; et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- A Study of LCAR-B38M CAR-T Cells. Clinical Trial NCT03758417. Available online: https://clinicaltrials.gov/ct2/show/NCT03758417?term=bcma+car+janssen&rank=1 (accessed on 10 May 2019).
- Wilkie, S.; Van Schalkwyk, M.C.; Hobbs, S.; Davies, D.M.; Van Der Stegen, S.J.; Pereira, A.C.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J., Jr. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell 2016, 167, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Ebert, L.M.; Yu, W.; Gargett, T.; Brown, M.P. Logic-gated approaches to extend the utility of chimeric antigen receptor T-cell technology. Biochem. Soc. Trans. 2018, 46, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Patel, E.; Ding, J.; Thorausch, N.; Krishnamurthy, J.; Choudhary, R.; Weiler, S.; Le, B.; Tavares, P.; Zieba, A.; Quinn, J.; et al. Abstract 3589: Preclinical evaluation of mesothelin-specific T cell receptor (TCR) fusion constructs (TRuC™s) utilizing the signaling power of the complete TCR complex: A new opportunity for solid tumor therapy. Cancer Res. 2018, 78, 3589. [Google Scholar]
- Helsen, C.W.; Hammill, J.A.; Lau, V.W.C.; Mwawasi, K.A.; Afsahi, A.; Bezverbnaya, K.; Newhook, L.; Hayes, D.L.; Aarts, C.; Bojovic, B.; et al. The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nat. Commun. 2018, 9, 3049. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, Z.; Horan, L.H.; Zhang, P.; Liu, L.; Zimdahl, B.; Green, S.; Lu, J.; Morales, J.F.; Barrett, D.M.; et al. A novel antibody-TCR (AbTCR) platform combines Fab-based antigen recognition with gamma/delta-TCR signaling to facilitate T-cell cytotoxicity with low cytokine release. Cell Discov. 2018, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Gu, J.; Xu, H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol. Cancer 2018, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in solid tumors: Blueprints for building effective therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T cells for solid tumors: New strategies for finding, infiltrating and surviving in the tumor microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Bonini, C.; Stanghellini, M.T.; Bondanza, A.; Traversari, C.; Salomoni, M.; Turchetto, L.; Colombi, S.; Bernardi, M.; Peccatori, J.; et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): A non-randomised phase I-II study. Lancet Oncol. 2009, 10, 489–500. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef]
- Straathof, K.C.; Pulè, M.A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [Green Version]
- Duong, M.T.; Collinson-Pautz, M.R.; Morschl, E.; Lu, A.; Szymanski, S.P.; Zhang, M.; Brandt, M.E.; Chang, W.C.; Sharp, K.L.; Toler, S.M.; et al. Two-dimensional regulation of CAR-T cell therapy with orthogonal switches. Mol. Ther. Oncolyt. 2018, 12, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Sakemura, R.; Terakura, S.; Watanabe, K.; Julamanee, J.; Takagi, E.; Miyao, K.; Koyama, D.; Goto, T.; Hanajiri, R.; Nishida, T.; et al. A tet-on inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol. Res. 2016, 4, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; He, D.; Li, C.; Wang, H.; Yang, G. Development of inducible CD19-CAR T cells with a tet-on system for controlled activity and enhanced clinical safety. Int. J. Mol. Sci. 2018, 19, 3455. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chang, W.C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Paszkiewicz, P.J.; Fräßle, S.P.; Srivastava, S.; Sommermeyer, D.; Hudecek, M.; Drexler, I.; Sadelain, M.; Liu, L.; Jensen, M.C.; Riddell, S.R.; et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Investig. 2016, 126, 4262–4272. [Google Scholar] [CrossRef] [PubMed]
- Kao, R.L.; Truscott, L.C.; Chiou, T.T.; Tsai, W.; Wu, A.M.; De Oliveira, S.N. A cetuximab-mediated suicide system in chimeric antigen receptor-modified hematopoietic stem cells for cancer therapy. Hum. Gene Ther. 2019, 30, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Darowski, D.; Kobold, S.; Jost, C.; Klein, C. Combining the best of two worlds: Highly flexible chimeric antigen receptor adaptor molecules (CAR-adaptors) for the recruitment of chimeric antigen receptor T cells. MAbs 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Minutolo, N.G.; Hollander, E.E.; Powell, D.J., Jr. The emergence of universal immune receptor T cell therapy for cancer. Front. Oncol. 2019, 9, 176. [Google Scholar] [CrossRef]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468. [Google Scholar] [CrossRef] [Green Version]
- Scarfò, I.; Maus, M.V. Current approaches to increase CAR T cell potency in solid tumors: Targeting the tumor microenvironment. J. Immunother. Cancer 2017, 5, 28. [Google Scholar] [CrossRef]
- Sackstein, R.; Schatton, T.; Barthel, S.R. T-lymphocyte homing: An underappreciated yet critical hurdle for successful cancer immunotherapy. Lab. Investig. 2017, 97, 669–697. [Google Scholar] [CrossRef] [PubMed]
- Chheda, Z.S.; Sharma, R.K.; Jala, V.R.; Luster, A.D.; Haribabu, B. Chemoattractant receptors BLT1 and CXCR3 regulate antitumor immunity by facilitating CD8+ T cell migration into tumors. J. Immunol. 2016, 197, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Kremer, V.; Ligtenberg, M.A.; Zendehdel, R.; Seitz, C.; Duivenvoorden, A.; Wennerberg, E.; Colón, E.; Scherman-Plogell, A.H.; Lundqvist, A. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J. Immunother. Cancer 2017, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.; Erreni, M.; Van Brakel, M.; Debets, R.; Allavena, P. Enhanced recruitment of genetically modified CX3CR1-positive human T cells into Fractalkine/CX3CL1 expressing tumors: Importance of the chemokine gradient. J. Immunother. Cancer 2016, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Rapp, M.; Grassmann, S.; Chaloupka, M.; Layritz, P.; Kruger, S.; Ormanns, S.; Rataj, F.; Janssen, K.P.; Endres, S.; Anz, D.; et al. C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration and therapeutic efficacy of adoptive T cell transfer. Oncoimmunology 2015, 5, e1105428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 enhanced the anti-tumor activity of chimeric antigen receptor T cells against hepatocellular carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O.; et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA 2019, 116, 7624–7631. [Google Scholar] [CrossRef] [Green Version]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Di, S.; Shi, B.; Jiang, H.; Shi, Z.; Liu, Y.; Wang, Y.; Luo, H.; Yu, M.; Wu, X.; et al. Increased antitumor activities of glypican-3-specific chimeric antigen receptor-modified T cells by coexpression of a soluble PD1-CH3 fusion protein. Cancer Immunol. Immunother. 2018, 67, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- CAR-T Cell Immunotherapy. Clinical Trial NCT03330834. Available online: https://clinicaltrials.gov/ct2/show/NCT03330834?term=NCT03330834&rank=1 (accessed on 22 April 2019).
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [PubMed]
- Autologous T Cells Genetically Engineered to Secrete IL-12. Clinical Trial NCT02498912. Available online: https://clinicaltrials.gov/ct2/show/NCT02498912?term=NCT02498912&rank=1 (accessed on 22 April 2019).
- Safety, Tolerability, Pharmacokinetics and Efficacy of AMG 562. Clinical Trial NCT03571828. Available online: https://clinicaltrials.gov/ct2/show/NCT03571828?term=amg562&rank=1 (accessed on 20 June 2019).
- Townsend, M.H.; Shrestha, G.; Robison, R.A.; O’Neill, K.L. The expansion of targetable biomarkers for CAR T cell therapy. J. Exp. Clin. Cancer Res. 2018, 37, 163. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Chen, N.; Li, X.; Chintala, N.K.; Tano, Z.E.; Adusumilli, P.S. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr. Opin. Immunol. 2018, 51, 103–110. [Google Scholar] [CrossRef]
- Hardiman, K.M.; Ulintz, P.J.; Kuick, R.D.; Hovelson, D.H.; Gates, C.M.; Bhasi, A.; Rodrigues Grant, A.; Liu, J.; Cani, A.K.; Greenson, J.K.; et al. Intra-tumor genetic heterogeneity in rectal cancer. Lab. Investig. 2016, 96, 4–15. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Mroz, E.A.; Tward, A.M.; Hammon, R.J.; Ren, Y.; Rocco, J.W. Intra-tumor genetic heterogeneity and mortality in head and neck cancer: Analysis of data from the Cancer Genome Atlas. PLoS Med. 2015, 12, e1001786. [Google Scholar] [CrossRef] [PubMed]
- Mroz, E.A.; Rocco, J.W. MATH, a novel measure of intratumor genetic heterogeneity, is high in poor-outcome classes of head and neck squamous cell carcinoma. Oral Oncol. 2013, 49, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanitis, E.; Poussin, M.; Hagemann, I.S.; Coukos, G.; Sandaltzopoulos, R.; Scholler, N.; Powell, D.J., Jr. Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol. Ther. 2012, 20, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.L.; Sherman, M.; McElroy, P.L.; Lofgren, J.A.; Moody, G.; Baeuerle, P.A.; Coxon, A.; Arvedson, T. Bispecific T cell engager (BiTE®) antibody constructs can mediate bystander tumor cell killing. PLoS ONE 2017, 12, e0183390. [Google Scholar] [CrossRef] [PubMed]
- Shadrin, N.; Shapira, M.G.; Khalfin, B.; Uppalapati, L.; Parola, A.H.; Nathan, I. Serine protease inhibitors interact with IFN-γ through up-regulation of FasR; a novel therapeutic strategy against cancer. Exp. Cell Res. 2015, 330, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; Von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Blincyto Package Insert. 2019. Available online: https://www.pi.amgen.com/~/media/amgen/repositorysites/pi-amgen-com/blincyto/blincyto_pi_hcp_english.pdf (accessed on 26 April 2019).
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef]
- Giavridis, T.; Van Der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, M.; Duchateau, P.; Depil, S.; Poirot, L.; Valton, J. Granulocyte-macrophage colony-stimulating factor inactivation in CAR T-cells prevents monocyte-dependent release of key cytokine release syndrome mediators. J. Biol. Chem. 2019, 294, 5430–5437. [Google Scholar] [CrossRef] [PubMed]
- Yuraszeck, T.; Kasichayanula, S.; Benjamin, J.E. Translation and clinical development of bispecific T-cell engaging antibodies for cancer treatment. Clin. Pharmacol. Ther. 2017, 101, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Anti-BCMA and/or Anti-CD19 CART Cells Treatment. Clinical Trial NCT03767725. Available online: https://clinicaltrials.gov/ct2/show/NCT03767725?term=NCT03767725&rank=1 (accessed on 27 April 2019).
- CD22 Redirected Autologous T Cells for ALL. Clinical Trial NCT02650414. Available online: https://clinicaltrials.gov/ct2/show/NCT02650414?term=NCT02650414&rank=1 (accessed on 27 April 2019).
- Goebeler, M.E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-cell engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non-Hodgkin lymphoma: Final results from a phase I study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Curran, K.J.; Cheung, N.K. Chimeric antigen receptors and bispecific antibodies to retarget T cells in pediatric oncology. Pediatr. Blood Cancer 2015, 62, 1326–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldoss, I.; Bargou, R.C.; Nagorsen, D.; Friberg, G.R.; Baeuerle, P.A.; Forman, S.J. Redirecting T cells to eradicate B-cell acute lymphoblastic leukemia: Bispecific T-cell engagers and chimeric antigen receptors. Leukemia 2017, 31, 777–787. [Google Scholar] [CrossRef]
- Yescarta Sales 2018. Available online: https://www.gilead.com/news-and-press/press-room/press-releases/2019/2/gilead-sciences-announces-fourth-quarter-and-full-year-2018-financial-results (accessed on 4 May 2019).
- Kymriah Sales 2018. Available online: https://www.novartis.com/sites/www.novartis.com/files/q4-2018-media-release-en.pdf (accessed on 4 May 2019).
- Blincyto Sales 2018. Available online: https://www.amgen.com/media/news-releases/2019/01/amgen-reports-fourth-quarter-and-full-year-2018-financial-results/ (accessed on 4 May 2019).
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- New Leukemia Drug Tops the Charts With a $178,000 Price Tag. Available online: https://www.medscape.com/viewarticle/836879 (accessed on 4 May 2019).
- CAR-T: How Will These $400k Therapies be Adapted for Europe? Available online: https://www.pharmaceutical-technology.com/comment/car-t-therapies-europe/ (accessed on 4 May 2019).
- Choi, B.D.; Gedeon, P.C.; Sanchez-Perez, L.; Bigner, D.D.; Sampson, J.H. Regulatory T cells are redirected to kill glioblastoma by an EGFRvIII-targeted bispecific antibody. Oncoimmunology 2013, 2, e26757. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Drent, E.; Poels, R.; Ruiter, R.; Van De Donk, N.W.C.J.; Zweegman, S.; Yuan, H.; De Bruijn, J.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.J.; et al. Combined CD28 and 4-1BB costimulation potentiates affinity-tuned chimeric antigen receptor-engineered T cells. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Pulte, E.D.; Vallejo, J.; Przepiorka, D.; Nie, L.; Farrell, A.T.; Goldberg, K.B.; McKee, A.E.; Pazdur, R. FDA supplemental approval: Blinatumomab for treatment of relapsed and refractory precursor B-cell acute lymphoblastic leukemia. Oncologist 2018, 23, 1366–1371. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Gordon, L.I.; Palomba, M.L.; Lunning, M.; Arnason, J.; Forero-Torres, A.; Wang, M.; Maloney, D.; Sehgal, A.; Andreadis, C.; et al. Updated Safety and Long-Term Clinical Outcomes in TRANSCEND NHL 001, Pivotal Trial of Lisocabtagene Maraleucel (JCAR017) in R/R Aggressive NHL. Available online: https://www.primeoncology.org/app/uploads/hematology-updates-stockholm-2018-dlbcl-s800-abramson.pdf (accessed on 5 May 2019).
- Safety Study of Bispecific T-Cell Engager Blinatumomab. Clinical Trial NCT00274742. Available online: https://clinicaltrials.gov/ct2/show/NCT00274742?term=NCT00274742&rank=1 (accessed on 7 May 2019).
- Study of bb2121 in Multiple Myeloma. Clinical Trial NCT02658929. Available online: https://clinicaltrials.gov/ct2/show/NCT02658929?term=NCT02658929&rank=1 (accessed on 7 May 2019).
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Kroenke, J.; Facon, T.; Einsele, H.; Munzert, G. Treatment with AMG 420, an anti-B-cell maturation antigen (BCMA) bispecific T-cell engager (BiTE®) antibody construct, induces minimal residual disease (MRD) negative complete responses in relapsed and/or refractory (R/R) multiple myeloma (MM) patients: Results of a first-in-human (FIH) phase I dose escalation study. Blood 2018, 132, 1010. [Google Scholar]
- Phase I Dose Escalation of I.V. BI 836909. Clinical Trial NCT02514239. Available online: https://clinicaltrials.gov/ct2/show/NCT02514239?term=NCT02514239&rank=1 (accessed on 7 May 2019).
- Jaspers, J.E.; Brentjens, R.J. Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacol. Ther. 2017, 178, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majzner, R.G.; Mackall, C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Disc. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Mandikian, D.; Takahashi, N.; Lo, A.A.; Li, J.; Eastham-Anderson, J.; Slaga, D.; Ho, J.; Hristopoulos, M.; Clark, R.; Totpal, K.; et al. Relative target affinities of T cell-dependent bispecific antibodies determine biodistribution in a solid tumor mouse model. Mol. Cancer Ther. 2018, 17, 776–785. [Google Scholar] [CrossRef]
- Trinklein, N.D.; Pham, D.; Schellenberger, U.; Buelow, B.; Boudreau, A.; Choudhry, P.; Clarke, S.C.; Dang, K.; Harris, K.E.; Iyer, S.; et al. Efficient tumor killing and minimal cytokine release with novel T-cell agonist bispecific antibodies. MAbs 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Saber, H.; Del Valle, P.; Ricks, T.K.; Leighton, J.K. An FDA oncology analysis of CD3 bispecific constructs and first-in-human dose selection. Regul. Toxicol. Pharmacol. 2017, 90, 144–152. [Google Scholar] [CrossRef]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef]
- Caruso, H.G.; Hurton, L.V.; Najjar, A.; Rushworth, D.; Ang, S.; Olivares, S.; Mi, T.; Switzer, K.; Singh, H.; Huls, H.; et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015, 75, 3505–3518. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. The immunological synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.R.; Rudd-Schmidt, J.A.; Lopez, J.A.; Ramsbottom, K.M.; Mannering, S.I.; Andrews, D.M.; Voskoboinik, I.; Trapani, J.A. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J. Exp. Med. 2015, 212, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenciute, G.; Krebs, S.; Torres, D.; Wu, M.F.; Liu, H.; Dotti, G.; Li, X.N.; Lesniak, M.S.; Balyasnikova, I.V.; Gottschalk, S. Characterization and functional analysis of scFv-based chimeric antigen receptors to redirect T cells to IL-13Ralpha2-positive glioma. Mol. Ther. 2016, 24, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, J.; Rajasekaran, A.K. Biological impediments to monoclonal antibody-based cancer immunotherapy. Mol. Cancer Ther. 2004, 3, 1493–1501. [Google Scholar] [PubMed]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef]
- Autologous CAR T Cells in Relapsed or Refractory B-Cell Lymphoma. Clinical Trial NCT02842138. Available online: https://clinicaltrials.gov/ct2/show/NCT02842138 (accessed on 10 May 2019).
- Sun, S.; Hao, H.; Yang, G.; Zhang, Y.; Fu, Y. Immunotherapy with CAR-modified T cells: Toxicities and overcoming strategies. J. Immunol. Res. 2018, 2018, 2386187. [Google Scholar] [CrossRef]
- Chan, T.; Gallagher, J.; Cheng, N.-L.; Carvajal-Borda, F.; Plummer, J.; Govekung, A.; Barrett, J.A.; Khare, P.D.; Cooper, L.J.N.; Shah, R.R. CD19-specific chimeric antigen receptor-modified T cells with safety switch produced under “point-of-care” using the sleeping beauty system for the very rapid manufacture and treatment of B-cell malignancies. Blood 2017, 130, 1324. [Google Scholar]
- Ghassemi, S.; Prachi, P.; Scholler, J.; Nunez-Cruz, S.; Barrett, D.M.; Bedoya, F.; Fraietta, J.A.; Lacey, S.F.; Levine, B.L.; Grupp, S.A.; et al. Minimally ex vivo manipulated gene-modified T cells display enhanced tumor control. Blood 2016, 128, 4549. [Google Scholar]









| Type | Clinical Stage | Total | ||
|---|---|---|---|---|
| Phase I/II | Phase III | Approved | ||
| T-cell or NK cell-redirecting bispecific Abs | 59 | 0 | 2 ** | 61 |
| Autologous CAR-T, CAR-NK, CAR-NKT cells | 207 | 2 | 2 | 211 |
| Allogeneic CAR-T, CAR-NK, CAR-NKT cells | 14 | 0 | 0 | 14 |
| Allogeneic NK or Autologous T cells engineered with Fc RIIIa for binding therapeutic antibodies | 3 | 0 | 0 | 3 |
| Total of CAR-T, T-cell or NK cell redirected killing of tumor cells | 283 | 2 | 4 | 289 |
| Bispecific T- or NK-Cell Redirecting Antibody Format *** | Clinical Stage | Total | ||
|---|---|---|---|---|
| Phase I/II | Phase III | Approved | ||
| Short half-life bivalent fragments (e.g., BiTE®s, DART®s, ImTACs, other bivalent fragments) | 15 | 0 | 1 | 16 |
| Half-life extended bivalent fragments (e.g., DART®-Fc, Extended half-life BiTE®s, TriTAC) | 11 | 0 | 0 | 11 |
| Asymmetric bivalent IgG-like (e.g., Trion, BEAT, Xencor H/A platform, Duobodies, other asymmetric platforms) | 21 | 0 | 1 **** | 22 |
| Roche TCB 2:1, Chugai ART-Ig®-scFv and Teneobio 2:1 platforms (two binding sites for target cell, one for CD3) | 4 | 0 | 0 | 4 |
| ADAPTIR® and TandAb platforms (tetravalent platforms) | 4 | 0 | 0 | 4 |
| Chemically conjugated IgGs (tetravalent; two IgGs) | 4 | 0 | 0 | 4 |
| Total | 59 | 0 | 2 | 61 |
| CAR-T Format | Clinical Stage | Total | ||
|---|---|---|---|---|
| Phase I/II | Phase III | Approved | ||
| Autologous CAR-T **—Single CAR | 176 | 2 | 2 | 180 |
| Autologous CAR-T **—Multiple CARs for different targets or multiple CAR-Ts dosed in combination | 30 | 0 | 0 | 30 |
| Autologous CAR-NKT | 1 | 0 | 0 | 1 |
| Allogeneic CAR-T | 11 | 0 | 0 | 11 |
| Allogeneic CAR-NK or -NKT | 3 | 0 | 0 | 3 |
| Total | 221 | 2 | 2 | 225 |
| Primary Target | Primary Indications | Therapeutic Format | Total | ||
|---|---|---|---|---|---|
| TRBAs | CAR-T/NKs | rCells Expressing FcγRIIIa | |||
| CD19 | B-cell cancer (NHL, etc.) | 2 | 88 | 0 | 90 |
| BCMA | MM | 7 | 26 | 1 | 34 |
| CD123 | AML | 5 | 8 | 0 | 13 |
| Mesothelin | Solid tumors | 1 | 12 | 0 | 13 |
| GD2 | Solid and neurological tumors | 2 | 10 | 0 | 12 |
| CD20 | B-cell cancer (NHL, etc.) | 5 | 4 | 2 | 11 |
| CD33 | AML | 6 | 4 | 0 | 10 |
| HER2 | Solid tumors | 3 | 6 | 0 | 9 |
| CD22 | B-cell cancer (NHL, etc.) | 0 | 8 | 0 | 8 |
| CD30 | HL | 1 | 5 | 6 | |
| PSMA | Solid tumor (prostate) | 4 | 2 | 0 | 6 |
| EGFRvIII | Neurological tumors | 2 | 4 | 0 | 6 |
| EGFR | Solid tumors | 1 | 3 | 0 | 4 |
| CD38 | MM | 2 | 2 | 0 | 4 |
| EpCAM | Solid tumors | 2 | 2 | 0 | 4 |
| PSCA | Solid tumor (prostate) | 1 | 3 | 0 | 4 |
| CEA (CEACAM5) | Solid tumors | 2 | 1 | 0 | 3 |
| HIV | Virus | 1 | 1 | 0 | 2 |
| Glypican-3 | Solid tumors | 1 | 1 | 0 | 2 |
| Flt3 | AML | 1 | 1 | 0 | 2 |
| NKG2D ligands | Solid tumors | 0 | 2 | 0 | 2 |
| Claudin 18.2 | Solid tumors | 0 | 2 | 0 | 2 |
| DLL3 | SCLC | 1 | 1 | 0 | 2 |
| CS1 (SLAMF7) | MM | 0 | 2 | 0 | 2 |
| MUC16 | Solid tumors | 1 | 1 | 0 | 2 |
| Lewis-Y | Solid tumors | 0 | 2 | 0 | 2 |
| cMet | Solid tumors | 0 | 2 | 0 | 2 |
| Others with single candidate | Mostly solid tumors | 10 | 16 | 0 | 26 |
| Undisclosed/other | Unknown | 0 | 6 | 0 | 6 |
| Total | -- | 61 | 225 | 3 | 289 |
| Property | T-Cell Redirecting Biologic Drug | |||||||
|---|---|---|---|---|---|---|---|---|
| Kymriah® | Blincyto® | Kymriah® | Yescarta® | Liso-cel | Blincyto® | bb2121 | AMG420 | |
| Sponsor | Novartis | Amgen | Novartis | Gilead (Kite) | Juno/Celgene | Amgen | Bluebird/Celgene | Amgen |
| Format | CAR-T; 4-1BB CS | TRBA (BiTE®) | CAR-T; 4-1BB CS | CAR-T; CD28 CS | CAR-T; 4-1BB CS | TRBA (BiTE®) | CAR-T; 4-1BB CS | TRBA (BiTE®) |
| Trial | EL | TW | JU | ZU | TC | Phase 1 | Phase 1 | Phase 1 |
| Target | CD19 | CD19 | CD19 | CD19 | CD19 | CD19 | BCMA | BCMA |
| Indication | B-ALL | B-ALL (PCN) | DLBCL | DLBCL | DLBCL | DLBCL | MM | MM |
| # Number of Patients | 63 | 271 | 93 | 101 | 73 | 11 | 33 | 42 |
| ORR | ND | ND | 52% | 83% | 80% | 55% | 85% | 31% |
| CR/CR * | 83% | 34% | 40% | 58% | 59% | 36% | 45% | 17% |
| PR | 20% | ND | 12% | 25% | 21% | 18% | 39% | 10% |
| Median response duration time | NR | 7.7 mo | 11.7 mo | 11.1 mo | 10.2 mo NR | 13.3 mo | 11.8 mo | NR |
| Grade 3+ AEs | ND | ND | 89% | 98% | 16% | 90% | ND | ND |
| CRS incidence | 77% | 15% | 58% | 58% | 37% | ND | 76% | 38% |
| Grade 3+ CRS | ND | nk | 22% | 11% | 1% | ND | 6% | ND |
| Neurotoxicity | ND | 65% | 21% | 64% | 25% | 71% | ND | ND |
| Grade 3+ Neurotoxicity | ND | 13% | 12% | 32% | 15% | 20% | ND | 0% |
| Elimination half-life | 21.7 d RP; 2.7 d NRP | NA | 91.3 d RP; 15.4 d NRP | ND | ND | NA | ND | NA |
| References | [290] | [384,405] | [290,406] | [291,407] | [408] | [393,409] | [295,410] | [411,412] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strohl, W.R.; Naso, M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41. https://doi.org/10.3390/antib8030041
Strohl WR, Naso M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies. 2019; 8(3):41. https://doi.org/10.3390/antib8030041
Chicago/Turabian StyleStrohl, William R., and Michael Naso. 2019. "Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells" Antibodies 8, no. 3: 41. https://doi.org/10.3390/antib8030041
APA StyleStrohl, W. R., & Naso, M. (2019). Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies, 8(3), 41. https://doi.org/10.3390/antib8030041