Phage Display Libraries for Antibody Therapeutic Discovery and Development

 , and
, and
Abstract
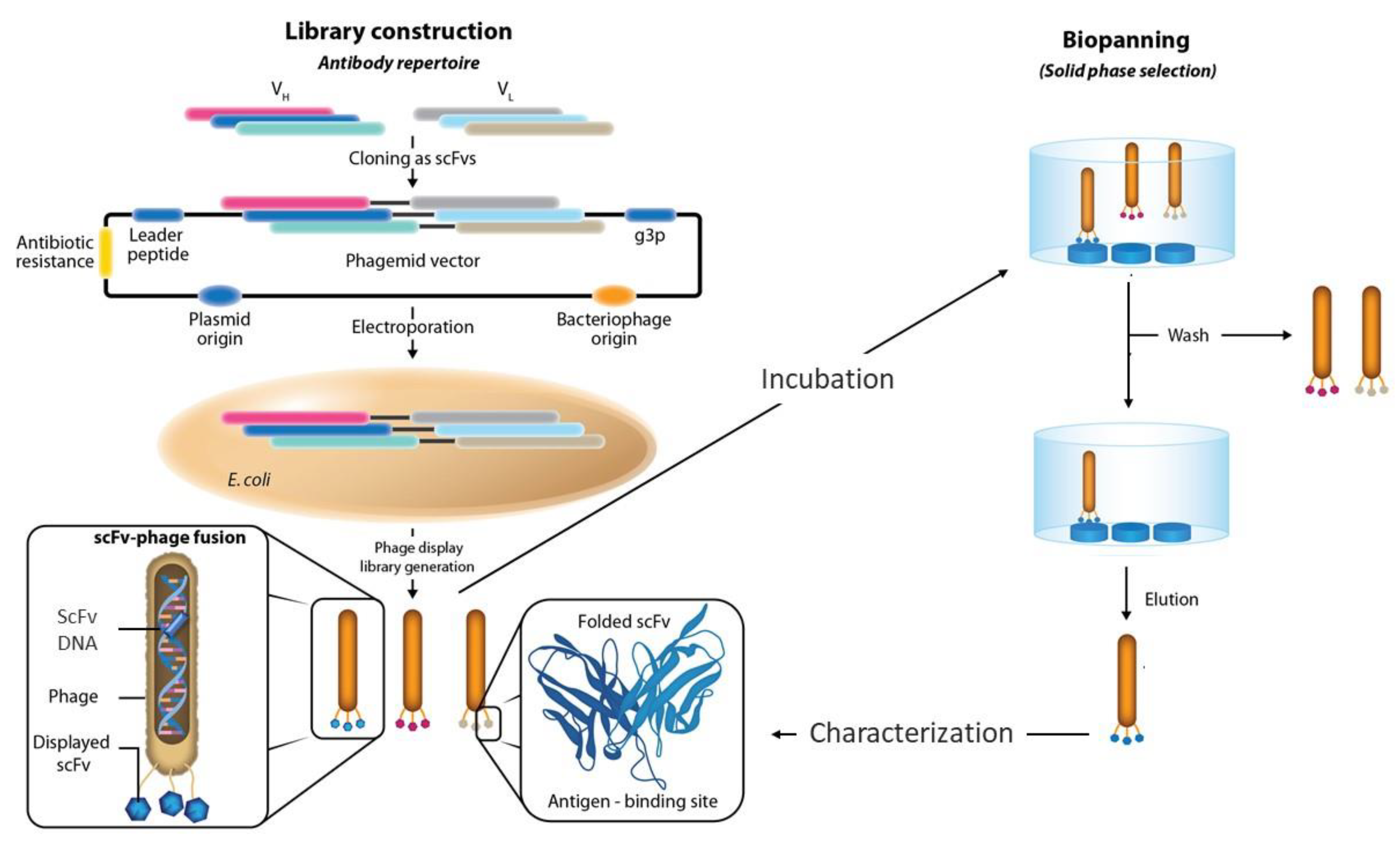
:1. Introduction
2. Size of the Phage Display Antibody Libraries
3. Effective Size of the Phage Display Antibody Libraries
4. Types of Antibody Repertoires
4.1. Naïve Libraries
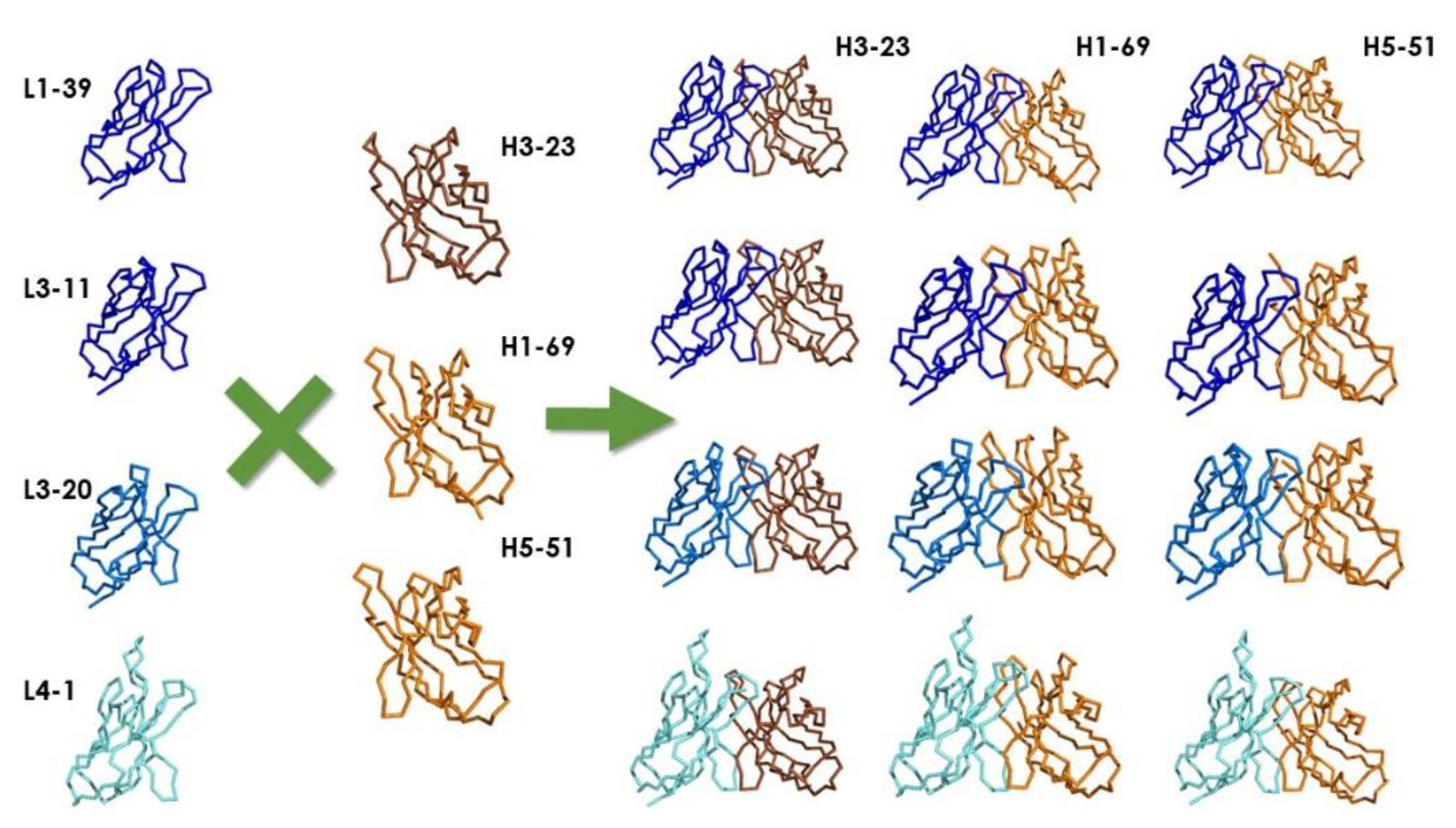
4.2. Synthetic Libraries
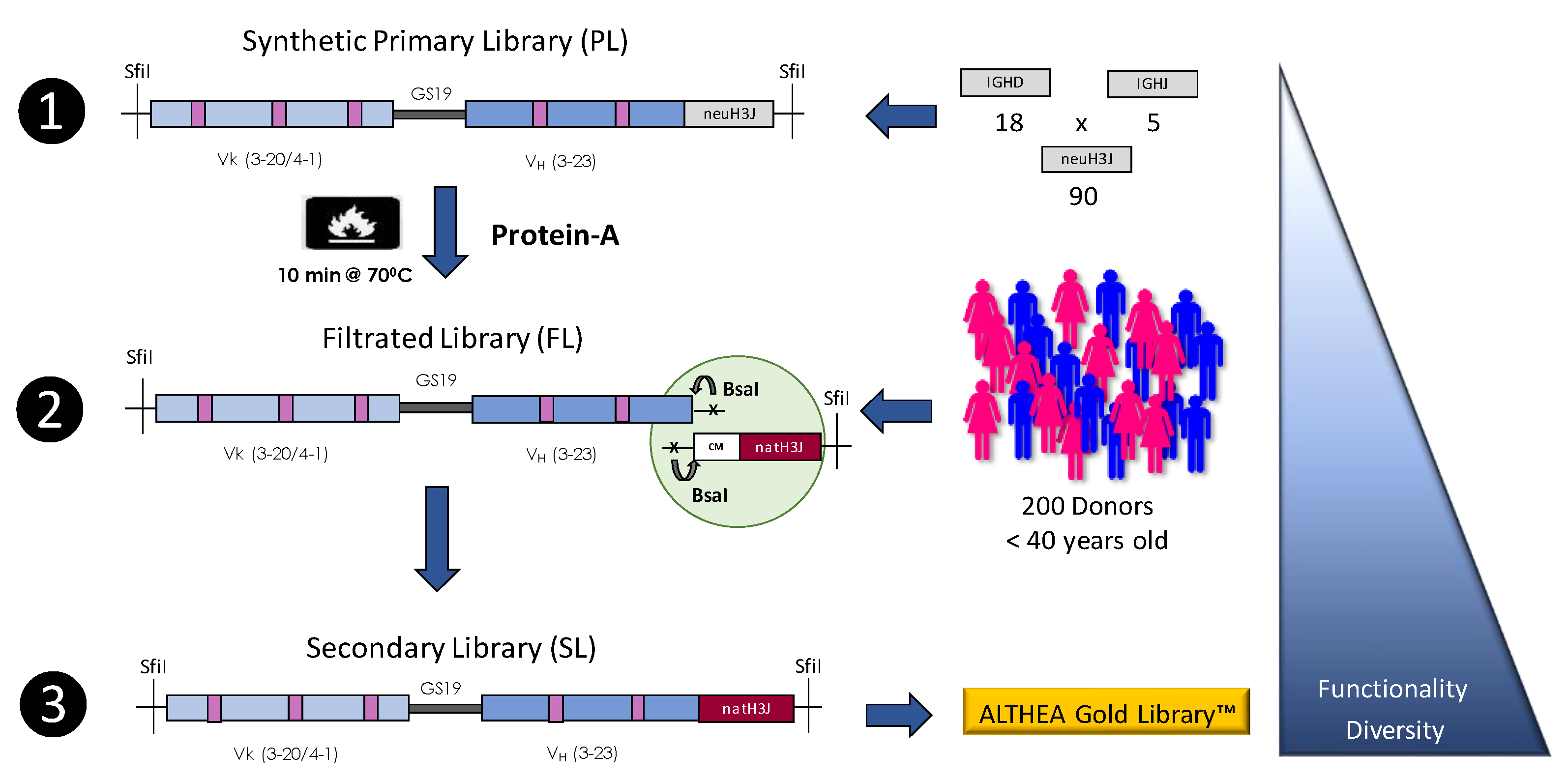
4.3. Semisynthetic Libraries
5. Panning Protocols and Targets
6. Enrichment with Positive Clones and Hit Rate
7. Affinity of the Selected Antibodies
8. Developability
9. Current Opportunities and Challenges
10. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, G. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Parmley, S.F.; Smith, G.P. Antibody-selectable filamentous fd phage vectors: Affinity purification of target genes. Gene 1988, 73, 305–318. [Google Scholar] [CrossRef]
- Scott, J.K.; Smith, G.P. Searching for peptide ligands with an epitope library. Science 1990, 249, 386–390. [Google Scholar] [CrossRef]
- Cwirla, S.E.; Peters, E.A.; Barrett, R.W.; Dower, W.J. Peptides on phage: A vast library of peptides for identifying ligands. Proc. Natl. Acad. Sci. USA 1990, 87, 6378–6382. [Google Scholar] [CrossRef]
- Devlin, J.; Panganiban, L.; Devlin, P. Random peptide libraries: A source of specific protein binding molecules. Science 1990, 249, 404–406. [Google Scholar] [CrossRef]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef]
- Amit, A.G.; Mariuzza, R.A.; Phillips, S.E.V.; Poljak, R.J. Three-dimensional structure of an antigen–antibody complex at 6 Å resolution. Nature 1985, 313, 156–158. [Google Scholar] [CrossRef]
- Bass, S.; Greene, R.; Wells, J.A. Hormone phage: An enrichment method for variant proteins with altered binding properties. Proteins Struct. Funct. Bioinform. 1990, 8, 309–314. [Google Scholar] [CrossRef]
- Marks, J.D.; Hoogenboom, H.R.; Bonnert, T.P.; McCafferty, J.; Griffiths, A.D.; Winter, G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J. Mol. Boil. 1991, 222, 581–597. [Google Scholar] [CrossRef]
- Barbas, C.F.; Bain, J.D.; Hoekstra, D.M.; Lerner, R.A. Semisynthetic combinatorial antibody libraries: A chemical solution to the diversity problem. Proc. Natl. Acad. Sci. USA 1992, 89, 4457–4461. [Google Scholar] [CrossRef]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Almagro, J.C.; Daniels-Wells, T.R.; Perez-Tapia, S.M.; Penichet, M.L. Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front. Immunol. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Li, S.; Kussie, P.; Ferguson, K.M. Structural Basis for EGF Receptor Inhibition by the Therapeutic Antibody IMC-11F8. Structure 2008, 16, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Osbourn, J.; Groves, M.; Vaughan, T. From rodent reagents to human therapeutics using antibody guided selection. Methods 2005, 36, 61–68. [Google Scholar] [CrossRef]
- Frenzel, A.; Kügler, J.; Helmsing, S.; Meier, D.; Schirrmann, T.; Hust, M.; Dübel, S. Designing Human Antibodies by Phage Display. Transfus. Med. Hemotherapy 2017, 44, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. mAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, A.; Williams, S.; Hartley, O.; Tomlinson, I.; Waterhouse, P.; Crosby, W.; Kontermann, R.; Jones, P.; Low, N.; Allison, T. Isolation of high affinity human antibodies directly from large synthetic repertoires. EMBO J. 1994, 13, 3245–3260. [Google Scholar] [CrossRef]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Prediction of Aggregation Prone Regions of Therapeutic Proteins. J. Phys. Chem. B 2010, 114, 6614–6624. [Google Scholar] [CrossRef]
- Jain, T.; Sun, T.; Durand, S.; Hall, A.; Houston, N.R.; Nett, J.H.; Sharkey, B.; Bobrowicz, B.; Caffry, I.; Yu, Y.; et al. Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. USA 2017, 114, 944–949. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.; Griffiths, A.D.; Hawkins, R.E.; Hoogenboom, H.R. Making Antibodies by Phage Display Technology. Annu. Rev. Immunol. 1994, 12, 433–455. [Google Scholar] [CrossRef]
- Sidhu, S.S. Phage display in pharmaceutical biotechnology. Curr. Opin. Biotechnol. 2000, 11, 610–616. [Google Scholar] [CrossRef]
- Finlay, W.J.J.; Almagro, J.C. Natural and man-made V-gene repertoires for antibody discovery. Front. Immunol. 2012, 3, 342. [Google Scholar] [CrossRef] [Green Version]
- Nixon, A.E.; Sexton, D.J.; Ladner, R.C. Drugs derived from phage display: From candidate identification to clinical practice. mAbs 2014, 6, 73–85. [Google Scholar] [CrossRef]
- Hoogenboom, H.R. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005, 23, 1105–1116. [Google Scholar] [CrossRef]
- Geyer, C.R.; McCafferty, J.; Dübel, S.; Bradbury, A.R.M.; Sidhu, S.S. Recombinant Antibodies and In Vitro Selection Technologies. Adv. Struct. Saf. Stud. 2012, 901, 11–32. [Google Scholar]
- Schwimmer, L.J.; Huang, B.; Giang, H.; Cotter, R.L.; Chemla-Vogel, D.S.; Dy, F.V.; Tam, E.M.; Zhang, F.; Toy, P.; Bohmann, D.J.; et al. Discovery of diverse and functional antibodies from large human repertoire antibody libraries. J. Immunol. Methods 2013, 391, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Kügler, J.; Wilke, S.; Meier, D.; Tomszak, F.; Frenzel, A.; Schirrmann, T.; Dübel, S.; Garritsen, H.; Hock, B.; Toleikis, L.; et al. Generation and analysis of the improved human HAL9/10 antibody phage display libraries. BMC Biotechnol. 2015, 15, 551. [Google Scholar] [CrossRef]
- Kim, S.; Park, I.; Park, S.G.; Cho, S.; Kim, J.H.; Ipper, N.S.; Choi, S.S.; Lee, E.S.; Hong, H.J. Generation, Diversity Determination, and Application to Antibody Selection of a Human Naïve Fab Library. Mol. Cells 2017, 40, 655–666. [Google Scholar]
- Shi, L.; Wheeler, J.C.; Sweet, R.W.; Lu, J.; Luo, J.; Tornetta, M.; Whitaker, B.; Reddy, R.; Brittingham, R.; Borozdina, L.; et al. De Novo Selection of High-Affinity Antibodies from Synthetic Fab Libraries Displayed on Phage as pIX Fusion Proteins. J. Mol. Boil. 2010, 397, 385–396. [Google Scholar] [CrossRef]
- Prassler, J.; Thiel, S.; Pracht, C.; Polzer, A.; Peters, S.; Bauer, M.; Nörenberg, S.; Stark, Y.; Kölln, J.; Popp, A.; et al. HuCAL PLATINUM, a Synthetic Fab Library Optimized for Sequence Diversity and Superior Performance in Mammalian Expression Systems. J. Mol. Boil. 2011, 413, 261–278. [Google Scholar] [CrossRef]
- Tiller, T.; Schuster, I.; Deppe, D.; Siegers, K.; Strohner, R.; Herrmann, T.; Berenguer, M.; Poujol, D.; Stehle, J.; Stark, Y.; et al. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. mAbs 2013, 5, 445–470. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Bujak, E.; Putelli, A.; Villa, A.; Matasci, M.; Gualandi, L.; Hemmerle, T.; Wulhfard, S.; Neri, D. A Highly Functional Synthetic Phage Display Library Containing over 40 Billion Human Antibody Clones. PLoS ONE 2014, 9, e100000. [Google Scholar] [CrossRef]
- Valadon, P.; Pérez-Tapia, S.M.; Nelson, R.S.; Guzmán-Bringas, O.U.; Arrieta-Oliva, H.I.; Gómez-Castellano, K.M.; Almagro, J.C. ALTHEA Gold Libraries™: Antibody libraries for therapeutic antibody discovery. mAbs 2019, 11, 516–531. [Google Scholar] [CrossRef]
- Perelson, A.S.; Oster, G.F. Theoretical studies of clonal selection: Minimal antibody repertoire size and reliability of self-non-self discrimination. J. Theor. Boil. 1979, 81, 645–670. [Google Scholar] [CrossRef]
- Perelson, A.S. Immune network theory. Immunol. Rev. 1989, 110, 5–36. [Google Scholar] [CrossRef]
- Kabat, E.A. Sequences of Proteins of Immunological Interest: Tabulation and Analysis of Amino Acid and Nucleic Acid Sequences of Precursors, V-Regions, C-Regions, J-Chain, T-Cell Receptors for Antigenm T-Cell Surface Antigens, [beta]2-Microglobulins, Major Histocompatibility Antigens, Thy-1, Complement, C-Reactive Protein, Thymopoietin, Integrins, Post-Gamme Globulin, [alpha]2-Macroglobulins, and Other Related Proteins, 5th ed.; NIH Publication: Bethesda, MD, USA, 1991. [Google Scholar]
- Raghunathan, G.; Smart, J.; Williams, J.; Almagro, J.C. Antigen-binding site anatomy and somatic mutations in antibodies that recognize different types of antigens. J. Mol. Recognit. 2012, 25, 103–113. [Google Scholar] [CrossRef]
- Knappik, A.; Ge, L.; Honegger, A.; Pack, P.; Fischer, M.; Wellnhofer, G.; Hoess, A.; Wölle, J.; Plückthun, A.; Virnekäs, B. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J. Mol. Boil. 2000, 296, 57–86. [Google Scholar] [CrossRef]
- Lin, B.; Renshaw, M.W.; Autote, K.; Smith, L.M.; Calveley, P.; Bowdish, K.S.; Frederickson, S. A step-wise approach significantly enhances protein yield of a rationally-designed agonist antibody fragment in E. coli. Protein Expr. Purif. 2008, 59, 55–63. [Google Scholar] [CrossRef]
- Almagro, J.C. Identification of differences in the specificity-determining residues of antibodies that recognize antigens of different size: Implications for the rational design of antibody repertoires. J. Mol. Recognit. 2004, 17, 132–143. [Google Scholar] [CrossRef]
- Zadeh, A.S.; Grässer, A.; Dinter, H.; Hermes, M.; Schindowski, K. Efficient Construction and Effective Screening of Synthetic Domain Antibody Libraries. Methods Protoc. 2019, 2, 17. [Google Scholar] [CrossRef]
- Clackson, T.; Hoogenboom, H.R.; Griffiths, A.D.; Winter, G. Making antibody fragments using phage display libraries. Nature 1991, 352, 624–628. [Google Scholar] [CrossRef]
- Barbas, C.F.; Kang, A.S.; Lerner, R.A.; Benkovic, S.J. Assembly of combinatorial antibody libraries on phage surfaces: The gene III site. Proc. Natl. Acad. Sci. USA 1991, 88, 7978–7982. [Google Scholar] [CrossRef]
- Chan, S.K.; Lim, T.S. Immune Human Antibody Libraries for Infectious Diseases. Pharm. Biotechnol. 2017, 61–78. [Google Scholar]
- Klarenbeek, A.; El Mazouari, K.; Desmyter, A.; Blanchetot, C.; Hultberg, A.; De Jonge, N.; Roovers, R.C.; Cambillau, C.; Spinelli, S.; Del-Favero, J.; et al. Camelid Ig V genes reveal significant human homology not seen in therapeutic target genes, providing for a powerful therapeutic antibody platform. mAbs 2015, 7, 693–706. [Google Scholar] [CrossRef]
- Van der Woning, B.; De Boeck, G.; Blanchetot, C.; Bobkov, V.; Klarenbeek, A.; Saunders, M.; De Haard, H. DNA immunization combined with scFv phage display identifies antagonistic GCGR specific antibodies and reveals new epitopes on the small extracellular loops. mAbs 2016, 8, 1126–1135. [Google Scholar] [CrossRef] [Green Version]
- Söderlind, E.; Strandberg, L.; Jirholt, P.; Kobayashi, N.; Alexeiva, V.; Åberg, A.-M.; Nilsson, A.; Jansson, B.; Ohlin, M.; Wingren, C.; et al. Recombining germline-derived CDR sequences for creating diverse single-framework antibody libraries. Nat. Biotechnol. 2000, 18, 852–856. [Google Scholar] [CrossRef]
- Hoet, R.M.; Cohen, E.H.; Kent, R.B.; Rookey, K.; Schoonbroodt, S.; Hogan, S.; Rem, L.; Frans, N.; Daukandt, M.; Pieters, H.; et al. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat. Biotechnol. 2005, 23, 344–348. [Google Scholar] [CrossRef]
- Rothe, C.; Urlinger, S.; Löhning, C.; Prassler, J.; Stark, Y.; Jäger, U.; Hubner, B.; Bardroff, M.; Pradel, I.; Boss, M.; et al. The Human Combinatorial Antibody Library HuCAL GOLD Combines Diversification of All Six CDRs According to the Natural Immune System with a Novel Display Method for Efficient Selection of High-Affinity Antibodies. J. Mol. Boil. 2008, 376, 1182–1200. [Google Scholar] [CrossRef]
- Pini, A.; Viti, F.; Santucci, A.; Carnemolla, B.; Zardi, L.; Neri, P.; Neri, D. Design and Use of a Phage Display Library: Human Antibodies with Subnanomolar Affinity against a Marker of Angiogenesis Eluted from a Two-Dimensional Gel. J. Boil. Chem. 1998, 273, 21769–21776. [Google Scholar] [CrossRef]
- Silacci, M.; Brack, S.; Schirru, G.; Mårlind, J.; Ettorre, A.; Merlo, A.; Viti, F.; Neri, D. Design, construction, and characterization of a large synthetic human antibody phage display library. Proteomics 2005, 5, 2340–2350. [Google Scholar] [CrossRef]
- Villa, A.; Lovato, V.; Bujak, E.; Wulhfard, S.; Pasche, N.; Neri, D. A novel synthetic naïve human antibody library allows the isolation of antibodies against a new epitope of oncofetal fibronectin. mAbs 2011, 3, 264–272. [Google Scholar] [CrossRef]
- Glanville, J.; Zhai, W.; Berka, J.; Telman, D.; Huerta, G.; Mehta, G.R.; Ni, I.; Mei, L.; Sundar, P.D.; Day, G.M.R.; et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc. Natl. Acad. Sci. USA 2009, 106, 20216–20221. [Google Scholar] [CrossRef] [Green Version]
- Watson, C.T.; Steinberg, K.M.; Huddleston, J.; Warren, R.L.; Malig, M.; Schein, J.; Willsey, A.J.; Joy, J.B.; Scott, J.K.; Graves, T.A.; et al. Complete Haplotype Sequence of the Human Immunoglobulin Heavy-Chain Variable, Diversity, and Joining Genes and Characterization of Allelic and Copy-Number Variation. Am. J. Hum. Genet. 2013, 92, 530–546. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, F.; Ishii, K.; Bourvagnet, P.; Kuma, K.-I.; Hayashida, H.; Miyata, T.; Honjo, T. The Complete Nucleotide Sequence of the Human Immunoglobulin Heavy Chain Variable Region Locus. J. Exp. Med. 1998, 188, 2151–2162. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, C.; Lowe, D.; Edwards, B.; Welsh, F.; Dilks, T.; Hardman, C.; Vaughan, T. Modelling the human immune response: Performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng. Des. Sel. 2009, 22, 159–168. [Google Scholar] [CrossRef]
- Bhat, N.M.; Bieber, M.M.; Teng, N.N. Cytotoxicity of Murine B Lymphocytes Induced by Human VH4-34 (VH4.21) Gene-Encoded Monoclonal Antibodies. Clin. Immunol. Immunopathol. 1997, 84, 283–289. [Google Scholar] [CrossRef]
- Barbié, V.; Lefranc, M.-P. The Human Immunoglobulin Kappa Variable (IGKV) Genes and Joining (IGKJ) Segments. Exp. Clin. Immunogenet. 1998, 15, 171–183. [Google Scholar] [CrossRef]
- Zemlin, M.; Klinger, M.; Link, J.; Zemlin, C.; Bauer, K.; Engler, J.A.; Schroeder, H.W.; Kirkham, P.M. Expressed Murine and Human CDR-H3 Intervals of Equal Length Exhibit Distinct Repertoires that Differ in their Amino Acid Composition and Predicted Range of Structures. J. Mol. Boil. 2003, 334, 733–749. [Google Scholar] [CrossRef]
- Lefranc, M.P. IMGT, the International ImMunoGeneTics Information System. Cold Spring Harb. Protoc. 2011, 2011, 115. [Google Scholar] [CrossRef]
- Sondek, J.; Shortle, D. A general strategy for random insertion and substitution mutagenesis: Substoichiometric coupling of trinucleotide phosphoramidites. Proc. Natl. Acad. Sci. USA 1992, 89, 3581–3585. [Google Scholar] [CrossRef]
- Chothia, C.; Lesk, A.M. Canonical structures for the hypervariable regions of immunoglobulins. J. Mol. Boil. 1987, 196, 901–917. [Google Scholar] [CrossRef]
- Al-Lazikani, B.; Lesk, A.M.; Chothia, C. Standard conformations for the canonical structures of immunoglobulins. J. Mol. Boil. 1997, 273, 927–948. [Google Scholar] [CrossRef]
- Morea, V.; Tramontano, A.; Rustici, M.; Chothia, C.; Lesk, A. Antibody structure, prediction and redesign. Biophys. Chem. 1997, 68, 9–16. [Google Scholar] [CrossRef]
- Martin, A.C.; Thornton, J.M. Structural Families in Loops of Homologous Proteins: Automatic Classification, Modelling and Application to Antibodies. J. Mol. Boil. 1996, 263, 800–815. [Google Scholar] [CrossRef]
- Vargas-Madrazo, E.; Almagro, J.C.; Lara-Ochoa, F. Structural repertoire in VH pseudogenes of immunoglobulins: Comparison with human germline genes and human amino acid sequences. J. Mol. Biol. 1995, 246, 74–81. [Google Scholar] [CrossRef]
- North, B.; Lehmann, A.; Dunbrack, R.J. A New Clustering of Antibody CDR loop conformations. J. Mol. Biol. 2011, 406, 228–256. [Google Scholar] [CrossRef]
- Brulle, J.V.D.; Fischer, M.; Langmann, T.; Horn, G.; Waldmann, T.; Arnold, S.; Fuhrmann, M.; Schatz, O.; O’Connell, T.; O’Connell, D.; et al. A novel solid phase technology for high-throughput gene synthesis. Biotechniques 2008, 45, 340–343. [Google Scholar] [CrossRef]
- Vargas-Madrazo, E.; Lara-Ochoa, F.; Almagro, J.C. Canonical Structure Repertoire of the Antigen-binding Site of Immunoglobulins Suggests Strong Geometrical Restrictions Associated to the Mechanism of Immune Recognition. J. Mol. Boil. 1995, 254, 497–504. [Google Scholar] [CrossRef]
- Lara-Ochoa, F.; Almagro, J.C.; Vargas-Madrazo, E.; Conrad, M. Antibody-antigen recognition: A canonical structure paradigm. J. Mol. Evol. 1996, 43, 678–684. [Google Scholar] [CrossRef]
- Rojas, G.; Almagro, J.C.; Acevedo, B.; Gavilondo, J.V. Phage antibody fragments library combining a single human light chain variable region with immune mouse heavy chain variable regions. J. Biotechnol. 2002, 94, 287–298. [Google Scholar] [CrossRef]
- Almagro, J.C.; Quintero-Hernández, V.; Ortiz-León, M.; Velandia, A.; Smith, S.L.; Becerril, B.; Quintero-Hernández, V.; Ortíz-León, M.; Quintero-Hernández, V.; Ortiz-León, M. Design and validation of a synthetic VH repertoire with tailored diversity for protein recognition. J. Mol. Recognit. 2006, 19, 413–422. [Google Scholar] [CrossRef]
- Cobaugh, C.W.; Almagro, J.C.; Pogson, M.; Iverson, B.; Georgiou, G. Synthetic Antibody Libraries Focused Towards Peptide Ligands. J. Mol. Boil. 2008, 378, 622–633. [Google Scholar] [CrossRef] [Green Version]
- De Wildt, R.M.; van Venrooij, W.J.; Winter, G.; Hoet, R.M.; Tomlinson, I.M. Somatic insertions and deletions shape the human antibody repertoire. J. Mol. Biol. 1999, 294, 701–710. [Google Scholar] [CrossRef]
- Teplyakov, A.; Obmolova, G.; Malia, T.J.; Luo, J.; Muzammil, S.; Sweet, R.; Almagro, J.C.; Gilliland, G.L. Structural diversity in a human antibody germline library. mAbs 2016, 8, 1045–1063. [Google Scholar] [CrossRef] [Green Version]
- Almagro, J.C.; Domínguez-Martinez, V.; Lara-Ochoa, F.; Vargas-Madrazo, E. Structural repertoire in human VL pseudogenes of immunoglobulins: Comparison with functional germline genes and amino acid sequences. Immunogenetics 1996, 43, 92–96. [Google Scholar]
- Teplyakov, A.; Obmolova, G.; Malia, T.J.; Raghunathan, G.; Martínez, C.; Fransson, J.; Edwards, W.; Connor, J.; Husovsky, M.; Beck, H.; et al. Structural insights into humanization of anti-tissue factor antibody 10H10. mAbs 2018, 10, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Bourne, P.E.; Westbrook, J.; Zardecki, C. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Gilliland, G.L.; Luo, J.; Vafa, O.; Almagro, J.C. Leveraging SBDD in protein therapeutic development: Antibody engineering. Methods Mol. Biol. 2012, 841, 321–349. [Google Scholar]
- Almagro, J.C.; Beavers, M.P.; Hernandez-Guzman, F.; Maier, J.; Shaulsky, J.; Butenhof, K.; Luo, J. Antibody modeling assessment. Proteins 2011, 79, 3050–3066. [Google Scholar] [CrossRef]
- Almagro, J.C.; Teplyakov, A.; Luo, J.; Sweet, R.W.; Kodangattil, S.; Hernandez-Guzman, F.; Gilliland, G.L.; Hernandez-Guzman, F. Second antibody modeling assessment (AMA-II). Proteins Struct. Funct. Bioinform. 2014, 82, 1553–1562. [Google Scholar] [CrossRef]
- Hillson, J.L. The structural basis of germline-encoded VH3 immunoglobulin binding to staphylococcal protein A. J. Exp. Med. 1993, 178, 331–336. [Google Scholar] [CrossRef]
- Graille, M.; Stura, E.; Corper, A.L.; Sutton, B.J.; Taussig, M.J.; Charbonnier, J.-B.; Silverman, G.J. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: Structural basis for recognition of B-cell receptors and superantigen activity. Proc. Natl. Acad. Sci. USA 2000, 97, 5399–5404. [Google Scholar] [CrossRef]
- Wu, S.J.; Luo, J.; O’Neil, K.T.; Kang, J.; Lacy, E.R.; Canziani, G.; Baker, A.; Huang, M.; Tang, Q.M.; Raju, T.; et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng. Des. Sel. 2010, 23, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Bethea, D.; Wu, S.-J.; Luo, J.; Hyun, L.; Lacy, E.R.; Teplyakov, A.; Jacobs, S.A.; O’Neil, K.T.; Gilliland, G.L.; Feng, Y. Mechanisms of self-association of a human monoclonal antibody CNTO607. Protein Eng. Des. Sel. 2012, 25, 531–538. [Google Scholar] [CrossRef] [Green Version]
- De Wildt, R.M.; Mundy, C.R.; Gorick, B.D.; Tomlinson, I.M. Antibody arrays for high-throughput screening of antibody-antigen interactions. Nat. Biotechnol. 2000, 18, 989–994. [Google Scholar] [CrossRef]
- Munke, A.; Persson, J.; Weiffert, T.; De Genst, E.; Meisl, G.; Arosio, P.; Carnerup, A.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J.; et al. Phage display and kinetic selection of antibodies that specifically inhibit amyloid self-replication. Proc. Natl. Acad. Sci. USA 2017, 114, 6444–6449. [Google Scholar] [CrossRef] [Green Version]
- Ossysek, K.; Uchański, T.; Kulesza, M.; Bzowska, M.; Klaus, T.; Woś, K.; Bereta, J. A new expression vector facilitating production and functional analysis of scFv antibody fragments selected from Tomlinson I+J phagemid libraries. Immunol. Lett. 2015, 167, 95–102. [Google Scholar]
- Raybould, M.I.J.; Marks, C.; Krawczyk, K.; Taddese, B.; Nowak, J.; Lewis, A.P.; Bujotzek, A.; Shi, J.; Deane, C.M. Five computational developability guidelines for therapeutic antibody profiling. Proc. Natl. Acad. Sci. USA 2019, 116, 4025–4030. [Google Scholar] [CrossRef] [Green Version]
- Iezzi, M.E.; Policastro, L.; Werbajh, S.; Podhajcer, O.; Canziani, G.A. Single-Domain Antibodies and the Promise of Modular Targeting in Cancer Imaging and Treatment. Front. Immunol. 2018, 9, 273. [Google Scholar] [CrossRef] [Green Version]
- Jespers, L.; Schon, O.; Famm, K.; Winter, G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat. Biotechnol. 2004, 22, 1161–1165. [Google Scholar] [CrossRef]
- Arbabi-Ghahroudi, M. Camelid Single-Domain Antibodies: Historical Perspective and Future Outlook. Front. Immunol. 2017, 8, 1589. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Bian, H.; Wu, X.; Fu, T.; Fu, Y.; Hong, J.; Ho, M. Construction and next-generation sequencing analysis of a large phage-displayed V. Antib. Ther. 2019, 2, 1–11. [Google Scholar] [CrossRef]
- Duggan, S. Caplacizumab: First Global Approval. Drugs 2018, 78, 1639–1642. [Google Scholar] [CrossRef] [Green Version]
- Hanes, J.; Plückthun, A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. USA 1997, 94, 4937–4942. [Google Scholar] [CrossRef]
- Francisco, J.A.; Campbell, R.; Iverson, B.L.; Georgiou, G. Production and fluorescence-activated cell sorting of Escherichia coli expressing a functional antibody fragment on the external surface. Proc. Natl. Acad. Sci. USA 1993, 90, 10444–10448. [Google Scholar] [CrossRef]
- Beerli, R.R.; Bauer, M.; Buser, R.B.; Gwerder, M.; Muntwiler, S.; Maurer, P.; Saudan, P.; Bachmann, M.F. Isolation of human monoclonal antibodies by mammalian cell display. Proc. Natl. Acad. Sci. USA 2008, 105, 14336–14341. [Google Scholar] [CrossRef] [Green Version]
- Cherf, G.M.; Cochran, J.R. Applications of yeast surface display for protein engineering. Adv. Struct. Saf. Stud. 2015, 1319, 155–175. [Google Scholar]
- Boder, E.T.; Midelfort, K.S.; Wittrup, K.D. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl. Acad. Sci. USA 2000, 97, 10701–10705. [Google Scholar] [CrossRef] [Green Version]
- Doerner, A.; Rhiel, L.; Zielonka, S.; Kolmar, H. Therapeutic antibody engineering by high efficiency cell screening. FEBS Lett. 2014, 588, 278–287. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library Name | Company/Laboratory | Repertoire | Display Format | Size (a) | Reference |
|---|---|---|---|---|---|
| XFab1 | Xoma | Naïve | Fab | 3.1 × 1011 | [26] |
| XscFv2 | Xoma | Naïve | scFv | 3.6 × 1011 | [26] |
| HAL9/10 | TU-IB (b) | Naïve | scFv | 1.5 × 1010 | [27] |
| KNU-Fab | KNU (c) | Naïve | Fab | 3.0 × 1010 | [28] |
| pIX V3.0 | Janssen Bio | Synthetic | Fab | 3.0 × 1010 | [29] |
| HuCAL PLATINUM | MorphoSys | Synthetic | Fab | 4.5 × 1010 | [30] |
| Ylanthia | MorphoSys | Synthetic | Fab | 1.3 × 1011 | [31] |
| PHILODiamond | ETH Zurich | Synthetic | scFv | 4.1 × 1010 | [32] |
| ALTHEA Gold Libraries | GlobalBio/ADL | Semisynthetic | scFv | 2.1 × 1010 | [33] |
| Library Name | ORF (%) | Display (%) | ||
|---|---|---|---|---|
| Kappa | Lambda | Kappa | Lambda | |
| XFab1 | 76 | 85 | 70 | 85 |
| XscFv2 | 74 | 66 | 71 | 58 |
| HAL9/10 | - | - | - | - |
| KNU-Fab | 91 | N/A | - | N/A |
| pIX V3.0 | 46 (28–72) | N/A | 77 (51–90) | N/A |
| HuCAL PLATINUM | 85–97 | 75–93 | - | - |
| Ylanthia | 82 | 82 | - | - |
| PHILODiamond | 93 | 93 | 90 | 90 |
| ALTHEA Gold Libraries™ | 85 | N/A | 83–85 | N/A |
| Ylanthia | pIX V3.0 | PHILODiamond | ALTHEA Gold Libraries | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| VH | Kappa | Lambda | VH | Kappa | Lambda | VH | Kappa | Lambda | VH | Kappa | Lambda |
| 1–18 | 1–6 | 1–40 | 1–69 | 1–39 | N/A | 3–23 | 3–20 | 3–19 | 3–23 | 3–20 | N/A |
| 1–46 | 1–9 | 1–47 | 3–23 | 3–11 | 4–1 | ||||||
| 1–69 | 1–12 | 1–51 | 5–51 | 3–20 | |||||||
| 3–7 | 1–15 | 2–11 | 4–1 | ||||||||
| 3–11 | 1–27 | 2–23 | |||||||||
| 3–15 | 1–39 | 3–1 | |||||||||
| 3–21 | 3–15 | ||||||||||
| 3–23 | 3–20 | ||||||||||
| 3–53 | |||||||||||
| 3–74 | |||||||||||
| 5–51 | |||||||||||
| 6–1 | |||||||||||
| HCDR3 (loop lengths) | |||||||||||
| 4–25 | 3–15 | 4–7 | Natural (1–25) | ||||||||
| Library | Panning | Screening | KD (nM) | ||||
|---|---|---|---|---|---|---|---|
| Number of Targets | Selection Method (a) | Rounds | Number of Assayed Clones | Positive Clones (%) | Hit Rate (%) | ||
| XFab1 | 7 | PS, SP | 3 | 757 (465–930) | 28 (10–16) | 7 (3–11) | 0.90 (0.02–2.10) |
| XscFv2 | 7 | PS, SP | 3 | 797 (558–930) | 58 (16–88) | 10 (5–17) | 0.46 (0.01–1.50) |
| HAL9/10 | 440 (b) | - | - | 20,000 | - | 17 (c) | - |
| KNU-Fab | 10 | PS, SP | 3–4 | 94 | - | 5 (2–9) | 37.80 (1.70–130.00) |
| pIX V2/3 | 6 | SP | 4 | 94 | - | 11 (5–18) | 1.00 (0.20–20.00) |
| HuCAL PLATINUM | 6 | PS, SP | 3 | 10,000 | - | 15 (10–30) | 0.24 (0.002–10.00) |
| Ylanthia | 9 | PS, SP | 3 | 4050 (2900–5200) | 1–49 (range) | 1–37 (range) | 0.70–190 (Fab) |
| PHILODiamond | 15 | PS, SP | 2–3 | 94 | 22 (1–61) | - | 9.00–150.00 (scFv) |
| ALTHEA Gold Libraries™ | 7 | PS, SP | 3–4 | 43–90 | 20 (10–46) | 10 (2–40) | 1.00 (0.09–20.00) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almagro, J.C.; Pedraza-Escalona, M.; Arrieta, H.I.; Pérez-Tapia, S.M. Phage Display Libraries for Antibody Therapeutic Discovery and Development. Antibodies 2019, 8, 44. https://doi.org/10.3390/antib8030044
Almagro JC, Pedraza-Escalona M, Arrieta HI, Pérez-Tapia SM. Phage Display Libraries for Antibody Therapeutic Discovery and Development. Antibodies. 2019; 8(3):44. https://doi.org/10.3390/antib8030044
Chicago/Turabian StyleAlmagro, Juan C., Martha Pedraza-Escalona, Hugo Iván Arrieta, and Sonia Mayra Pérez-Tapia. 2019. "Phage Display Libraries for Antibody Therapeutic Discovery and Development" Antibodies 8, no. 3: 44. https://doi.org/10.3390/antib8030044
APA StyleAlmagro, J. C., Pedraza-Escalona, M., Arrieta, H. I., & Pérez-Tapia, S. M. (2019). Phage Display Libraries for Antibody Therapeutic Discovery and Development. Antibodies, 8(3), 44. https://doi.org/10.3390/antib8030044