Symmetry/Asymmetry of the NHN Hydrogen Bond in Protonated 1,8-Bis(dimethylamino)naphthalene

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compound and Synthesis
Bis(dimethylamino)naphthalene Sulfate
8-Bis(dimethylamino)naphthalene Sulfate-d2
2.2. Infrared Spectra Measurements
2.3. Incoherent Inelastic Neutron Scattering (IINS) Measurements
2.4. NMR Measurements
2.5. Static DFT and D3-DFT Calculations
2.6. Symmetry-Adapted Perturbation Theory Calculations
2.7. Car–Parrinello Molecular Dynamics Simulations
3. Results and Discussion
3.1. SAPT Analysis of the DMANH+ HSO4− Complex
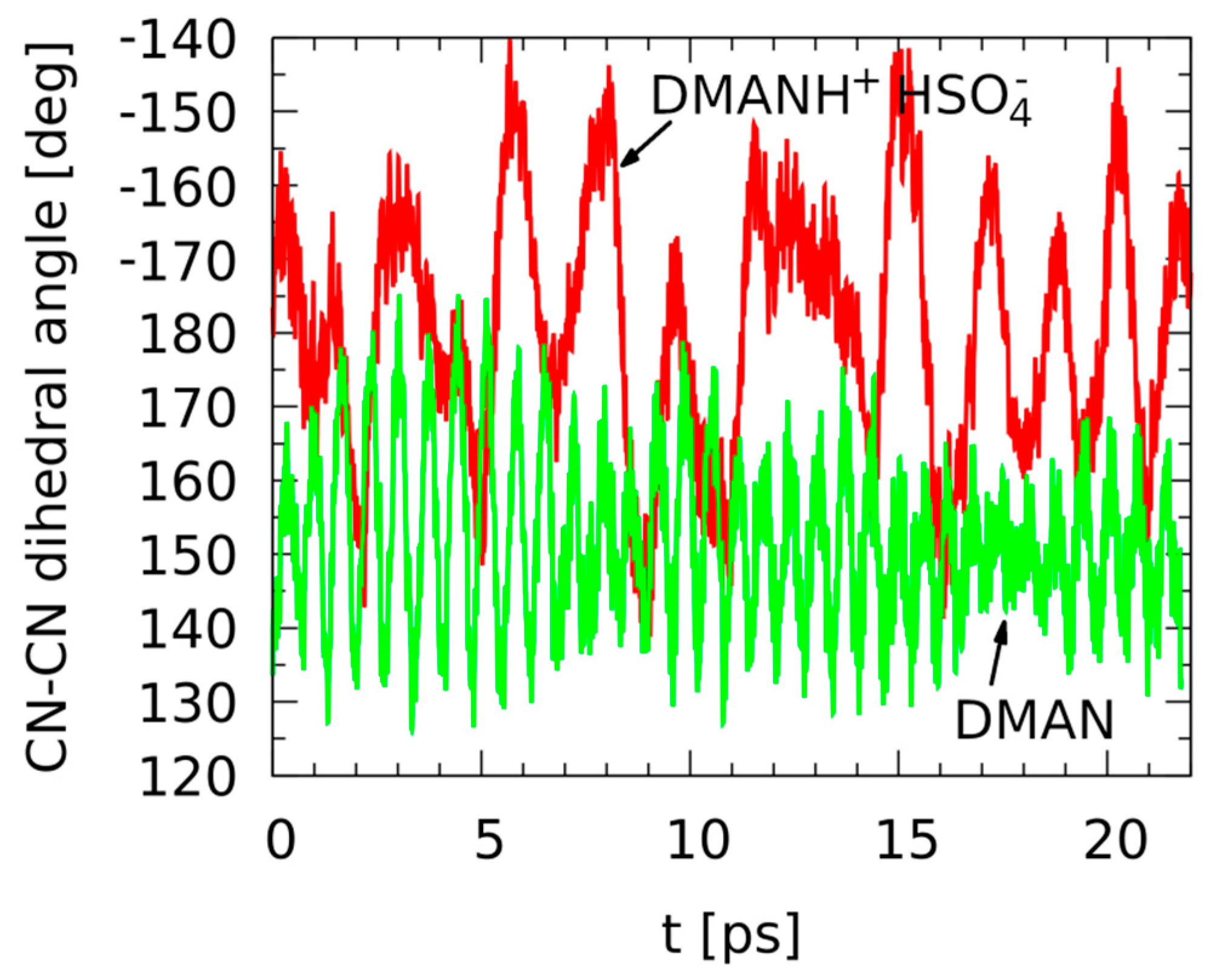
3.2. Metric Parameter Time-Evolution Analysis Based on Car–Parrinello Molecular Dynamics
3.3. DFT and D3-DFT Analysis of PES
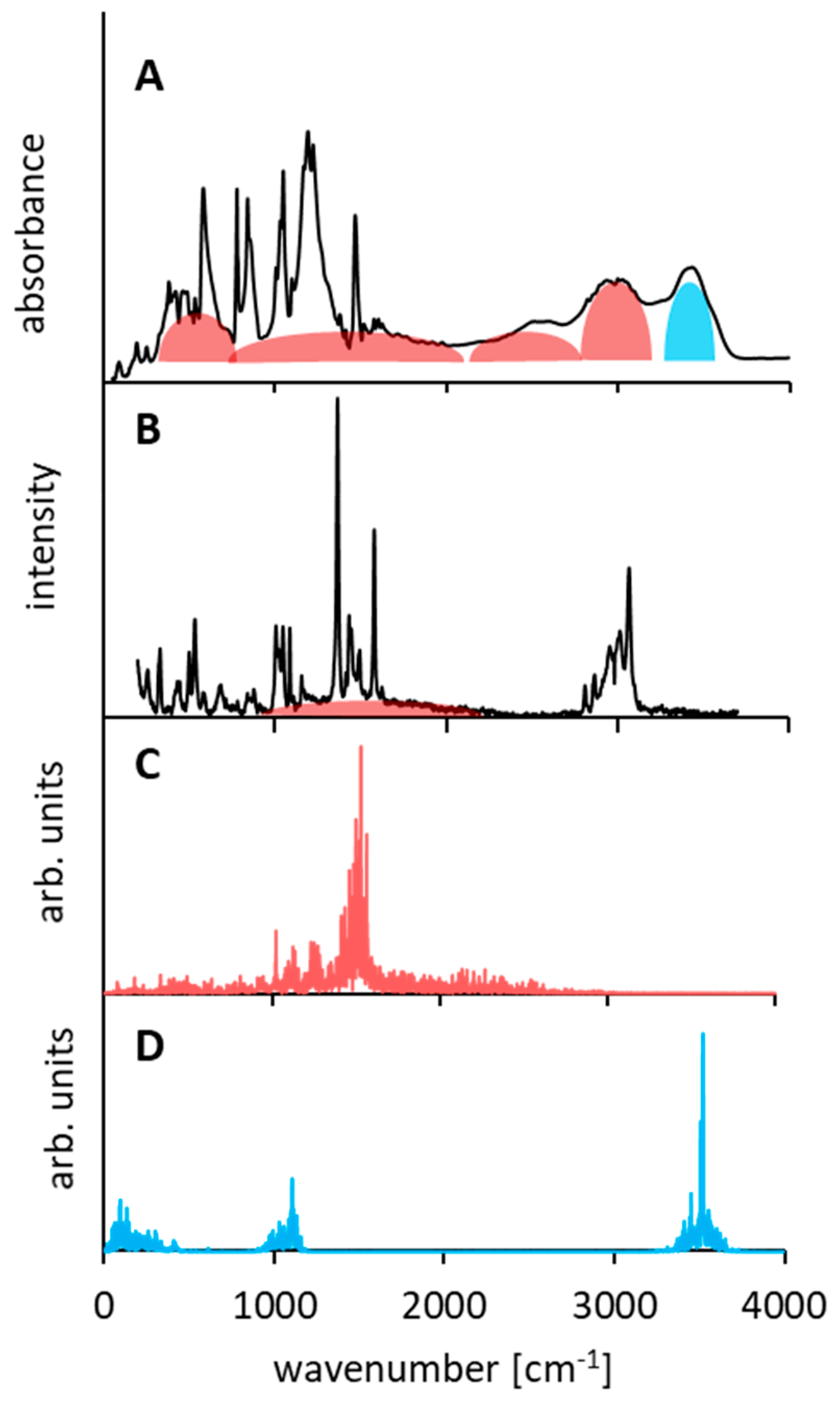
3.4. Comparison of Experimental IINS, IR, and Calculated CPMD Spectra
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode № | IR | IINS | Wavenumber [cm−1] | Intensities [km/mol] |
|---|---|---|---|---|
| exp. | exp. | calc. | calc. | |
| tr | 9, 14, 35, 46 | |||
| 1 | 56 | 51.74 | 0.52 | |
| 2 | 72 | |||
| 3 | 84 | 80 | ||
| 4 | 93 | 91 | 99.49 | 0.23 |
| 5 | 115.46 | 3.64 | ||
| 6 | 124 | 122 | 122.23 | 0.28 |
| 7 | 126.25 | 1.04 | ||
| 8 | 135 | 141 | 151.23 | 0.59 |
| 9 | 162 | 163 | 172.50 | 0.95 |
| 10 | 193 | 190 | ||
| 11 | 200 | 211.02 | 0.02 | |
| 12 | 243 | 222 | 231.19 | 0.68 |
| 13 | 263 | 259 | 261.03 | 4.05 |
| 14 | 286 | 277 | 295.65 | 1.52 |
| 15 | 304 | 296 | 303.45 | 1.05 |
| 16 | 310 | 312.48 | 4.87 | |
| 17 | 334 | 325.28 | 0.29 | |
| 18 | 351 | 351 | 350.55 | 1.38 |
| 19 | 363 | 361 | 367.12 | 4.21 |
| 20 | 382 | 385 | 385.57 | 3.15 |
| 21 | 443 | 446 | 442.71 | 1.12 |
| 22 | 468 | 474 | 478.21 | 5.77 |
| 23 | 484.85 | 2.56 | ||
| 24 | 525 | 528 | 535.68 | 5.75 |
| 25 | 536.21 | 1.67 | ||
| 26 | 538.78 | 0.01 | ||
| 27 | 577 | 551.01 | 1.67 | |
| 28 | 643 | 620 | 632.15 | 2.95 |
| 29 | 661 | 649 | 654.22 | 8.74 |
| 30 | 677.28 | 7.42 | ||
| 31 | 752 | 766 | 766.11 | 0.11 |
| 32 | 779 | 771.44 | 0.69 | |
| 33 | 775.50 | 79.16 | ||
| 34 | 793.09 | 3.25 | ||
| 35 | 818 | 839.55 | 27.81 | |
| 36 | 870 | 876.68 | 0.14 | |
| 37 | 887 | 892.78 | 1.88 | |
| 38 | 897.72 | 2.49 | ||
| 39 | 937 | 947.92 | 40.51 | |
| 40 | 956 | 971 | 973.02 | 0.65 |
| 41 | 980.44 | 0.31 | ||
| 42 | 1032 | 1041.07 | 95.25 | |
| 43 | 1056 | 1053.03 | 25.44 | |
| 44 | 1072.89 | 23.97 | ||
| 45 | 1092 | 1073.11 | 9.06 | |
| 46 | 1108.97 | 12.30 | ||
| 47 | 1113 | 1112.14 | 4.67 | |
| 48 | 1122.56 | 0.38 | ||
| 49 | 1136 | 1131.23 | 4.10 | |
| 50 | 1152 | 1158.87 | 0.66 | |
| 51 | 1162.50 | 44.36 | ||
| 52 | 1185 | 1178.07 | 1.90 | |
| 53 | 1197 | 1188.05 | 2.11 | |
| 54 | 1200 b | 1205.20 | 27.08 | |
| 55 | 1220.59 | 6.34 | ||
| 56 | 1226 | 1224.03 | 4.29 | |
| 57 | 1242.37 | 18.36 | ||
| 58 | 1248.75 | 14.89 | ||
| 59 | 1324 | 1333.17 | 15.30 | |
| 60 | 1343 | 1348.38 | 47.07 | |
| 61 | 1360.88 | 23.49 | ||
| 62 | 1384 | 1369.77 | 22.02 | |
| 63 | 1420 | 1407.98 | 74.73 | |
| 64 | 1448 | 1443.03 | 0.43 | |
| 65 | 1443.45 | 0.39 | ||
| 66 | 1456.69 | 21.91 | ||
| 67 | 1463 | 1466 b | 1464.24 | 5.54 |
| 68 | 1473.32 | 1.03 | ||
| 69 | 1479 | 1477.99 | 0.62 | |
| 70 | 1481.23 | 3.35 | ||
| 71 | 1485.18 | 9.10 | ||
| 72 | 1492.70 | 20.46 | ||
| 73 | 1493.08 | 30.41 | ||
| 74 | 1500.92 | 2.48 | ||
| 75 | 1511 | 1502.60 | 16.87 | |
| 76 | 1521.37 | 11.25 | ||
| 77 | 1524.98 | 22.79 | ||
| 78 | 1576 | 1542.86 | 22.63 | |
| 79 | 1605 | 1608.92 | 191.50 | |
| 80 | 1722 | 1632.70 | 1.17 | |
| 81 | 1641.10 | 11.45 | ||
| 82 | 2926.35 | 160.99 | ||
| 83 | 2777 | 2926.67 | 11.27 | |
| 84 | 2824 | 2939.86 | 267.58 | |
| 85 | 2852 | 2941.71 | 37.29 | |
| 86 | 2928 | 3051.65 | 27.31 | |
| 87 | 2971 | 3051.81 | 17.97 | |
| 88 | 3013 | 3056.51 | 12.52 | |
| 89 | 3048 | 3057.11 | 96.53 | |
| 90 | 3093 | 3101.58 | 16.29 | |
| 91 | 3101.61 | 24.04 | ||
| 92 | 3158.93 | 4.96 | ||
| 93 | 3159.18 | 17.51 | ||
| 94 | 3159.31 | 0.06 | ||
| 95 | 3161.34 | 1.11 | ||
| 96 | 3176.94 | 10.44 | ||
| 97 | 3180.00 | 43.23 | ||
| 98 | 3204.39 | 11.51 | ||
| 99 | 3204.41 | 8.22 |
| № | IR | IINS | Raman | Wavenumber [cm−1] | Intensities [km/mol] |
|---|---|---|---|---|---|
| exp. | exp. | exp. | calc. | calc. | |
| 1 | 14.57 | 0.03 | |||
| 2 | 17.59 | 1.10 | |||
| 3 | 36.81 | 1.37 | |||
| 4 | 42.72 | 0.88 | |||
| 5 | 60.97 | 1.86 | |||
| 6 | 83.84 | 25.22 | |||
| 7 | 90 | 90.41 | 5.08 | ||
| 8 | 93.29 | 0.17 | |||
| 9 | 147.02 | 43.61 | |||
| 10 | 152.75 | 31.05 | |||
| 11 | 162 | 161.14 | 3.03 | ||
| 12 | 185 | 185.56 | 9.32 | ||
| 13 | 185.81 | 15.51 | |||
| 14 | 194 | 198 | 198.61 | 0.12 | |
| 15 | 242.02 | 3.42 | |||
| 16 | 243.48 | 2.73 | |||
| 17 | 249 | 252.75 | 10.81 | ||
| 18 | 256.74 | 0.23 | |||
| 19 | 262 | 262 | 263.71 | 0.50 | |
| 20 | 274 | 288.84 | 3.22 | ||
| 21 | 325 | 325.96 | 0.01 | ||
| 22 | 329 | 332.45 | 8.04 | ||
| 23 | 378.53 | 3.30 | |||
| 24 | 382 | 383 | 384.15 | 6.38 | |
| 25 | 399.35 | 13.15 | |||
| 26 | 419 | 422.01 | 12.28 | ||
| 27 | 440 | 439 | 441.72 | 3.17 | |
| 28 | 466 | 470 | 481.60 | 0.24 | |
| 29 | 503 | 498.35 | 10.20 | ||
| 30 | 511 | 525.17 | 5.77 | ||
| 31 | 525.65 | 52.86 | |||
| 32 | 533 | 533 | 534 | 536.22 | 9.97 |
| 33 | 539.01 | 0.17 | |||
| 34 | 540.81 | 3.02 | |||
| 35 | 545.32 | 31.75 | |||
| 36 | 584 | 585 | 593 | 583.61 | 12.63 |
| 37 | 642 | 649.80 | 0.00 | ||
| 38 | 676 | 674.12 | 35.07 | ||
| 39 | 687 | 678.86 | 0.26 | ||
| 40 | 732.25 | 299.92 | |||
| 41 | 776.12 | 0.93 | |||
| 42 | 779 | 781.77 | 1.30 | ||
| 43 | 787 | 789.15 | 71.35 | ||
| 44 | 796.76 | 0.78 | |||
| 45 | 841 | 844 | 853.32 | 15.88 | |
| 46 | 846 | 877 | 866.23 | 13.02 | |
| 47 | 908.56 | 22.33 | |||
| 48 | 922.62 | 0.27 | |||
| 49 | 949 | 938.58 | 1.60 | ||
| 50 | 989.33 | 3.63 | |||
| 51 | 995.53 | 4.04 | |||
| 52 | 1002.12 | 1.26 | |||
| 53 | 1004.20 | 115.91 | |||
| 54 | 1007 | 1013 | 1007 | 1004.66 | 102.37 |
| 55 | 1031 | 1025 | 1047.32 | 17.60 | |
| 56 | 1049 | 1049 | 1047.85 | 18.21 | |
| 57 | 1087 | 1088.53 | 0.09 | ||
| 58 | 1099 | 1104.66 | 212.72 | ||
| 59 | 1108.80 | 8.72 | |||
| 60 | 1112 | 1110.39 | 10.96 | ||
| 61 | 1114 | 1114.95 | 4.13 | ||
| 62 | 1155 | 1170.56 | 30.47 | ||
| 63 | 1173 | 1175.17 | 65.76 | ||
| 64 | 1181.23 | 21.65 | |||
| 65 | 1193 | 1195.65 | 20.18 | ||
| 66 | 1196.71 | 26.24 | |||
| 67 | 1199.61 | 175.54 | |||
| 68 | 1200.30 | 166.15 | |||
| 69 | 1210 | 1217.08 | 170.92 | ||
| 70 | 1223 | 1228.08 | 9.32 | ||
| 71 | 1239.64 | 13.43 | |||
| 72 | 1245.05 | 26.67 | |||
| 73 | 1264.43 | 4.50 | |||
| 74 | 1305.30 | 11.88 | |||
| 75 | 1354.70 | 48.76 | |||
| 76 | 1365 | 1365.64 | 3.30 | ||
| 77 | 1379 | 1386.47 | 15.33 | ||
| 78 | 1414 | 1415 | 1436.10 | 1.65 | |
| 79 | 1431 | 1438.56 | 79.01 | ||
| 80 | 1443 | 1451.21 | 3.09 | ||
| 81 | 1469 | 1465.77 | 1.48 | ||
| 82 | 1480.31 | 7.45 | |||
| 83 | 1482.26 | 0.71 | |||
| 84 | 1488.20 | 15.25 | |||
| 85 | 1488.98 | 0.97 | |||
| 86 | 1490 | 1497.56 | 0.69 | ||
| 87 | 1495 | 1498.85 | 20.69 | ||
| 88 | 1503.20 | 8.53 | |||
| 89 | 1505.92 | 5.75 | |||
| 90 | 1511.50 | 82.43 | |||
| 91 | 1518 | 1514.96 | 23.30 | ||
| 92 | 1521.21 | 24.25 | |||
| 93 | 1578 | 1580 | 1551.47 | 22.12 | |
| 94 | 1603 | 1612.85 | 5.43 | ||
| 95 | 1626 | 1627 | 1617.33 | 8.56 | |
| 96 | 1644.36 | 9.08 | |||
| 97 | 1664.89 | 16.75 | |||
| 98 | 2600 | 2529.78 | 977.93 | ||
| 99 | 2809 | 2984.75 | 30.16 | ||
| 100 | 2940 | 2866 | 2989.37 | 66.45 | |
| 101 | 2952 | 3056.91 | 10.25 | ||
| 102 | 3015 | 3060.71 | 59.04 | ||
| 103 | 3065 | 3081.84 | 19.34 | ||
| 104 | 3082.70 | 19.25 | |||
| 105 | 3134.13 | 0.65 | |||
| 106 | 3137.65 | 8.34 | |||
| 107 | 3148.81 | 1.78 | |||
| 108 | 3150.19 | 9.61 | |||
| 109 | 3154.58 | 2.99 | |||
| 110 | 3160.22 | 69.10 | |||
| 111 | 3168.92 | 0.48 | |||
| 112 | 3172.93 | 0.06 | |||
| 113 | 3181.07 | 9.40 | |||
| 114 | 3186.35 | 13.84 | |||
| 115 | 3192.70 | 10.19 | |||
| 116 | 3198.99 | 4.40 | |||
| 117 | 3794.06 | 87.38 |
References
- Perrin, C.L. Symmetries of hydrogen bonds in solution. Science 1994, 266, 1665. [Google Scholar] [CrossRef] [PubMed]
- Perrin, C.L.; Nielson, J.B. Asymmetry of Hydrogen Bonds in Solutions of Monoanions of Dicarboxylic Acids. J. Am. Chem. Soc. 1997, 119, 12734–12741. [Google Scholar] [CrossRef]
- Chambron, J.-C.; Meyer, M. The ins and outs of proton complexation. Chem. Soc. Rev. 2009, 38, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Mavri, J.; Hodošček, M.; Hadži, D. Ab initio SCF and Møller-Plesset calculations on the hydrogen bond in hydrogen malonate: Effects of neighbour ions and polarizable medium. J. Mol. Struct. 1990, 209, 421–431. [Google Scholar] [CrossRef]
- Mavri, J.; Hadži, D. Influence of solvation on the hydrogen bond in hydrogen malonate an ab initio and semiempirical study. J. Mol. Struct. THEOCHEM 1998, 432, 257–262. [Google Scholar] [CrossRef]
- Staab, H.A.; Kriege, C.; Hieber, G.; Oberdorf, K. 1,8-Bis(dimethylamino)4,5-dihydroxynaphthalene, a Natural, Intramolecularly Protonated “Proton Sponge” with Zwitterionic Structure. Angew. Chem. Int. Ed. Engl. 1997, 36, 1884–1886. [Google Scholar] [CrossRef]
- Perrin, C.L.; Thoburn, J.D. Symmetries of hydrogen bonds in monoanions of dicarboxylic acids. J. Am. Chem. Soc. 1992, 114, 8559–8565. [Google Scholar] [CrossRef]
- Perrin, C.L.; Kim, Y.J. Symmetry of the Hydrogen Bond in Malonaldehyde Enol in Solution. J. Am. Chem. Soc. 1998, 120, 12641–12645. [Google Scholar] [CrossRef]
- Guo, J.; Tolstoy, P.M.; Koeppe, B.; Denisov, G.S.; Limbach, H.-H. NMR Study of Conformational Exchange and Geometries of Intramolecular Hydrogen Bonds in Monoanions of Succinic Acid and Derivatives. J. Phys. Chem. A 2011, 115, 9828–9836. [Google Scholar] [CrossRef]
- Guo, J.; Tolstoy, P.M.; Koeppe, B.; Golubev, N.S.; Denisov, G.S.; Smirnov, S.N.; Limbach, H.-H. Hydrogen Bond Geometries and Proton Tautomerism of Homo-Conjugated Anions of Carboxylic Acids Studied via H/D Isotope Effects on 13C NMR Chemical Shifts. J. Phys. Chem. A 2012, 116, 11180–11188. [Google Scholar] [CrossRef]
- Chmielewski, P.; Ozeryanskii, V.A.; Sobczyk, L.; Pozharskii, A.F. Primary 1 H/2 H isotope effect in the NMR chemical shift of HClO4 salts of 1,8-bis(dimethylamino)naphthalene derivatives. J. Phys. Org. Chem. 2007, 20, 643–648. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, L. Symmetry and 1 H NMR chemical shifts of short hydrogen bonds: Impact of electronic and nuclear quantum effects. Phys. Chem. Chem. Phys. 2020, 22, 4884–4895. [Google Scholar] [CrossRef] [PubMed]
- White, P.B.; Hong, M. 15N and 1 H Solid-State NMR Investigation of a Canonical Low-Barrier Hydrogen-Bond Compound: 1,8-Bis(dimethylamino)naphthalene. J. Phys. Chem. B 2015, 119, 11581–11589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin, C.L.; Ohta, B.K. Symmetry of N–H–N Hydrogen Bonds in 1,8-Bis(dimethylamino)naphthalene·H+ and 2,7-Dimethoxy-1,8-bis(dimethylamino)naphthalene·H+. J. Am. Chem. Soc. 2001, 123, 6520–6526. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, K.; Wilson, C.C.; Knight, K.S.; Jones, W.; Grech, E. Neutron Diffraction of a Complex of 1,8-Bis(dimethylamino)naphthalene with 1,2-Dichloromaleic Acid. Acta Cryst. B 1996, 52, 691–696. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.O.F.; Kallay, A.A.; Lloyd, H.; McIntyre, G.J.; Wilson, C.C.; Thomas, L.H. The Effect of Local Crystalline Environment on Hydrogen Atom Behavior in Molecular Complexes of a Proton Sponge. Cryst. Growth Des. 2016, 16, 2123–2129. [Google Scholar] [CrossRef] [Green Version]
- Gregorovic, A.; Apih, T.; Zagara, V.; Seliger, J. 14N NQR spectroscopy reveals the proton position in N–H N bonds: A case study with proton sponges. Phys. Chem. Chem. Phys. 2019, 21, 306–313. [Google Scholar] [CrossRef]
- Sabet-Sarvestani, H.; Izadyar, M.; Eshghi, H.; Noroozi-Shad, N.; Bakavoli, M. Proton sponge as a new efficient catalyst for carbon dioxide transformation to methanol: Theoretical approach. Fuel 2018, 221, 491–500. [Google Scholar] [CrossRef]
- Pawlukojć, A.; Natkaniec, I.; Grech, E.; Baran, J.; Malarski, Z.; Sobczyk, L. Incoherent inelastic neutron scattering, Raman and IR absorption studies on 1,8-bis(dimethylamino)naphthalene and its protonated forms. Spectrochim. Acta A 1998, 54, 439–448. [Google Scholar] [CrossRef]
- Brzeziński, B.; Głowiak, T.; Grech, E.; Malarski, Z.; Sobczyk, L. Structure and IR spectra of protonated 1,8-bis(dimethylamino)naphthalene proton sponge. Croat. Chem. Acta 1992, 65, 101–108. [Google Scholar]
- Lee, J.; Cheong, I.; Lee, S.-Y. Successful application of a neutral organic base, 1,8-bis(tetramethylguanidino)naphthalene (TMGN), for the radiosynthesis of [11C]raclopride. Appl. Rad. Isotop. 2016, 118, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.; Lorente, P.; Claramunt, R.M.; Marin, J.; Foces-Foces, C.; Llamas-Saiz, A.L.; Elguero, J.; Limbach, H.H. Localization of Hydrogen Bond Deuterons in Proton Sponges by Dipolar Solid State N-15 NMR Spectroscopy. Ber. Bunsenges. Phys. Chem. Chem. Phys. 1998, 102, 414–418. [Google Scholar] [CrossRef]
- Pietrzak, M.; Wehling, J.P.; Kong, S.; Tolstoy, P.M.; Shenderovich, I.G.; Lopez, C.; Claramunt, R.M.; Elguero, J.; Denisov, G.S.; Limbach, H.-H. Symmetrization of Cationic Hydrogen Bridges of Protonated Sponges Induced by Solvent and Counteranion Interactions as Revealed by NMR Spectroscopy. Chem. Eur. J. 2010, 16, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, M.; Wehling, J.; Limbach, H.-H.; Golubev, N.S.; Lopez, C.; Claramunt, R.M.; Elguero, J. 13C Detected Scalar Nitrogen-Nitrogen Couplings Across the Intramolecular Symmetric NHN Hydrogen Bond of Proton Sponge. J. Am. Chem. Soc. 2001, 123, 4338–4339. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.; Claramunt, R.M.; Llamas-Saiz, A.L.; Foces-Foces, C.; Elguero, J.; Sobrados, I.; Aguilar-Parrilla, F.; Limbach, H.H. X-ray diffraction and solid state NMR studies of 1,8-bis(dimethylamino)naphthalene and its complexes with picric and hexafluorophosphoric acids. New J. Chem. 1996, 20, 523–536. [Google Scholar]
- Saunders, M.; Telkowski, L.; Kates, M.R. NMR isotope shifts as a probe of electronic structure. J. Am. Chem. Soc. 1977, 99, 8070–8071. [Google Scholar] [CrossRef]
- Alder, R.W.; Bowman, P.S.; Steele, W.R.S.; Winterman, D.R. The remarkable basicity of 1,8-bis(dimethylamino)naphthalene. J. Chem. Soc. Chem. Commun. 1968, 723–724. [Google Scholar] [CrossRef]
- Alder, R.W.; East, S.P. In/Out Isomerism. Chem. Rev. 1996, 96, 2097–2111. [Google Scholar] [CrossRef]
- Staab, H.A.; Saupe, T. “Proton Sponges” and the Geometry of Hydrogen Bonds: Aromatic Nitrogen Bases with Exceptional Basicities. Angew. Chem. Int. Ed. Engl. 1988, 27, 865–879. [Google Scholar] [CrossRef]
- Pozharskii, A.F. Naphthalene “Proton Sponges”. Russ. Chem. Rev. 1998, 67, 1–27. [Google Scholar] [CrossRef]
- Pozharskii, A.F.; Ozeryanskii, V.A. Proton sponges. In The Chemistry of Anilines; Rappoport, Z., Ed.; J. Wiley & Sons: Chichester, UK, 2007; pp. 931–1026. [Google Scholar]
- Sobczyk, L. The specificity of the [NHN]+ hydrogen bonds in protonated naphthalene proton sponges. J. Mol. Struct. 2010, 972, 59–63. [Google Scholar] [CrossRef]
- Llamas-Saiz, A.L.; Foces-Foces, C.; Elguero, J. Proton sponges. J. Mol. Struct. 1994, 328, 297–323. [Google Scholar] [CrossRef]
- Kaljurand, I.; Saame, J.; Rodima, T.; Koppel, I.; Koppel, I.A.; Kögel, J.F.; Sundermeyer, J.; Köhn, U.; Coles, M.P.; Leito, I. Experimental Basicities of Phosphazene, Guanidinophosphazene, and Proton Sponge Superbases in the Gas Phase and Solution. J. Phys. Chem. A 2016, 120, 2591–2604. [Google Scholar] [CrossRef] [PubMed]
- Kçgel, J.F.; Xie, X.; Baal, E.; Gesevičius, D.; Oelkers, B.; Kovačević, B.; Sundermeyer, J. Superbasic Alkyl-Substituted Bisphosphazene Proton Sponges: Synthesis, Structural Features, Thermodynamic and Kinetic Basicity, Nucleophilicity and Coordination Chemistry. Chem. Eur. J. 2014, 20, 7670–7685. [Google Scholar] [CrossRef]
- Korzhenevskaya, N.G.; Schroeder, G.; Brzezinski, B.; Rybachenko, V.I. Concept of Superbasicity of 1,8-Bis(dialkylamino)naphthalenes ([Proton Sponges]). Rus. J. Org. Chem. 2001, 37, 1603–1610. [Google Scholar] [CrossRef]
- Raab, V.; Kipke, J.; Gschwind, R.M.; Sundermeyer, J. 1,8-Bis(tetramethylguanidino)naphthalene (TMGN): A New, Superbasic and Kinetically Active “Proton Sponge”. Chem. Eur. J. 2002, 8, 1682–1693. [Google Scholar] [CrossRef]
- Hibbert, F.; Robbins, H.J. Base-catalyzed proton transfer from an intramolecularly hydrogen-bonded naphthylammonium ion in 70% dimethyl sulfoxide-water (v/v). J. Am. Chem. Soc. 1978, 100, 8239−8244. [Google Scholar] [CrossRef]
- Hibbert, F.; Simpson, G.R. Acid–base properties of highly substituted diaminonaphthalenes. J. Chem. Soc. Perkin Trans. 1987, 2, 243–246. [Google Scholar] [CrossRef]
- Woźniak, K.; He, H.Y.; Klinowski, J.; Barr, T.L.; Milart, P. ESCA and Solid-State NMR Studies of Ionic Complexes of 1,8- Bis(dimethylamino)naphthalene. J. Phys. Chem. 1996, 100, 11420–11426. [Google Scholar] [CrossRef]
- Parkin, A.; Woźniak, K.; Wilson, C.C. From Proton Disorder to Proton Migration: A Continuum in the Hydrogen Bond of a Proton Sponge in the Solid State. Cryst. Grow. Des. 2007, 7, 1393–1398. [Google Scholar] [CrossRef]
- Hoser, A.A.; Dobrzycki, Ł.; Gutmann, M.J.; Woźniak, K. Charge Densities of Two Polymorphs of Hydrated 1,8-Bis(dimethylamino)naphthalene Hydrochloride; Similarities and Differences. Cryst. Grow. Des. 2010, 10, 5092–5104. [Google Scholar] [CrossRef]
- Mallinson, P.R.; Smith, G.T.; Wilson, C.C.; Grech, E.; Wozniak, K. From Weak Interactions to Covalent Bonds: A Continuum in the Complexes of 1,8-Bis(dimethylamino)naphthalene. J. Am. Chem. Soc. 2003, 125, 4259–4270. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, K.; Krygowski, T.M.; Kariuki, B.; Jones, W.; Grech, E. Crystallographic Studies on Sterically Affected Chemical-Species 0.2. Molecular and Crystal-Structure of 1,8-Bis(Dimethylamino)-Naphthalene Tetrafluoroborate—Analysis of Distortion of Geometry in the Aromatic Part Due to Intramolecular Hydrogen-Bonding. J. Mol. Struct. 1990, 240, 111–118. [Google Scholar] [CrossRef]
- Grech, E.; Klimkiewicz, J.; Nowicka-Scheibe, J.; Pietrzak, M.; Schilf, W.; Pozharski, A.F.; Ozeryanskii, V.A.; Bolvig, S.; Abildgaard, J.; Hansen, P.E. Deuterium isotope effects on 15N, 13C and 1 H chemical shifts of proton sponges. J. Mol. Struct. 2002, 615, 121–140. [Google Scholar] [CrossRef]
- Pawełka, Z.; Zeegers-Huyskens, T. The strange behaviour of the hydrogen bond complexes of 1,8-bis(dimethylamino) naphthalene in solution. J. Mol. Struct. THEOCHEM 1989, 200, 565–573. [Google Scholar] [CrossRef]
- Ozeryanskii, V.A.; Pozharskii, A.F.; Bieńko, A.J.; Sawka-Dobrowolska, W.; Sobczyk, L. [NHN]+ Hydrogen Bonding in Protonated 1,8-Bis(dimethylamino)-2,7-dimethoxynaphthalene. X-ray Diffraction, Infrared, and Theoretical ab Initio and DFT Studies. J. Phys. Chem. A 2005, 109, 1637–1642. [Google Scholar] [CrossRef]
- Ozeryanskii, V.A.; Pozharskii, A.F.; Głowiak, T.; Majerz, I.; Sobczyk, L.; Grech, I.; Nowicka-Szajbe, J. X-ray diffraction and IR-spectroscopic studies on protonated 4-amino-1,8-bis(dimethyloamino)naphthalene. J. Mol. Struct. 2002, 607, 1–8. [Google Scholar] [CrossRef]
- Brzezinski, B.; Schroeder, G.; Jarczewski, A.; Grech, E.; Nowicka-Scheibe, J.; Stefaniak, L.; Klimkiewicz, J. Proton transfer reactions from N-H acid to proton sponges in acetonitrile. Part 2. J. Mol. Struct. 1996, 377, 149–154. [Google Scholar]
- Baran, J.; Pawlukojc, A.; Majerz, I.; Malarski, Z.; Sobczyk, L.; Grech, E. Vibrational spectra of the adduct of 1,8-bis(dimethylamino)naphthalene with dichloromaleic acid (DMAN*DCM). Spectrochim. Acta A 2000, 56, 1801–1812. [Google Scholar] [CrossRef]
- Belding, L.; Stoyanov, P.; Dudding, T. Synthesis, Theoretical Analysis, and Experimental pKa Determination of a Fluorescent, Nonsymmetric, In−Out Proton Sponge. J. Org. Chem. 2016, 81, 6–13. [Google Scholar] [CrossRef]
- Antonov, A.S.; Pozharskii, A.F.; Tolstoy, P.M.; Filarowski, A.; Khoroshilova, O.V. 1,8-Bis(dimethylamino)naphthyl-2-ketimines: Inside vs outside protonation. Beilstein J. Org. Chem. 2018, 14, 2940–2948. [Google Scholar] [CrossRef] [PubMed]
- Ozeryanskii, V.A.; Pozharskii, A.F.; Antonov, A.S.; Filarowski, A. Out-Basicity of 1,8-bis(dimethylamino)naphthalene: The experimental and theoretical challenge. Org. Biomol. Chem. 2014, 12, 2360–2369. [Google Scholar] [CrossRef] [PubMed]
- Pozharskii, A.F.; Degtyarev, A.V.; Ryabtsova, O.V.; Ozeryanskii, V.A.; Kletskii, M.E.; Starikova, Z.A.; Sobczyk, L.; Filarowski, A. 2-α-hydroxyalkyl- and 2,7-di(α-hydroxyalkyl)-1,8-bis(dimethylamino)naphthalenes: Stabilization of nonconventional in/out conformers of “proton sponges” via N···H-O intramolecular hydrogen bonding. A remarkable kind of tandem nitrogen inversion. J. Org. Chem. 2007, 72, 3006–3019. [Google Scholar] [CrossRef] [PubMed]
- Belding, L.; Dudding, T. Synthesis and Theoretical Investigation of a 1,8-Bis(bis(diisopropylamino)cyclopropeniminyl)naphthalene Proton Sponge Derivative. Chem. Eur. J. 2014, 20, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Jezierska, A.; Panek, J.J. Theoretical study of intramolecular hydrogen bond in selected symmetric “proton sponges” on the basis of DFT and CPMD methods. J. Mol. Model. 2020, 26, 37. [Google Scholar] [CrossRef]
- Jezierska, A.; Panek, J.J. “Zwitterionic proton sponge” hydrogen bonding investigations on the basis of Car-Parrinello molecular dynamics. J. Chem. Inf. Model. 2015, 55, 1148–1157. [Google Scholar] [CrossRef]
- Masuda, Y.; Mori, Y.; Sakurai, K. Effects of Counterion and Solvent on Proton Location and Proton Transfer Dynamics of N−H···N Hydrogen Bond of Monoprotonated 1,8-Bis(dimethylamino)naphthalene. J. Phys. Chem. A 2013, 117, 10576–10587. [Google Scholar] [CrossRef]
- Majerz, I.; Olovsson, I. Proton transfer in the intramolecular NHN+ bonds in proton sponges with different hydrogen bridge flexibility. Phys. Chem. Chem. Phys. 2009, 11, 1297–1302. [Google Scholar] [CrossRef]
- Majerz, I.; Olovsson, I. The shape of the potential energy curves for NHN+ hydrogen bonds and the influence of non-linearity. Phys. Chem. Chem. Phys. 2008, 10, 3043–3051. [Google Scholar] [CrossRef]
- Horbatenko, Y.; Vyboishchikov, S.F. Hydrogen Motion in Proton Sponge Cations: A Theoretical Study. ChemPhysChem 2011, 12, 1118–1129. [Google Scholar] [CrossRef]
- Pozharskii, A.F.; Ryabtsova, O.V.; Ozeryanskii, V.A.; Degtyarev, A.V.; Kazheva, O.N.; Alexandrov, G.G.; Dyachenko, O.A. Organometallic Synthesis, Molecular Structure, and Coloration of 2,7-Disubstituted 1,8-Bis(dimethylamino)naphthalenes. How Significant Is the Influence of “Buttressing Effect” on Their Basicity? J. Org. Chem. 2003, 68, 10109–10122. [Google Scholar] [CrossRef]
- Alder, R.W.; Orpen, A.G.; Sessions, R.B. The structure of 1,6-diazabicyclo[4.4.4]tetradecane and of its inside protonated ion. J. Chem. Soc. Chem. Commun. 1983, 999–1000. [Google Scholar] [CrossRef]
- Alder, R.W.; Bryce, M.R.; Goode, N.C.; Miller, N.; Owen, J. Preparation of a range of NNN′N′-tetrasubstituted 1,8-diaminonaphthalenes. J. Chem. Soc. Perkin Trans. 1981, 1, 2840–2847. [Google Scholar] [CrossRef]
- Degtyarev, A.V.; Ryabtsova, O.V.; Pozharskii, A.F.; Ozeryanskii, V.A.; Starikova, Z.A.; Sobczyk, L.; Filarowski, A. 2,7-disubstituted proton sponges as borderline systems for investigating barrier-free intramolecular hydrogen bonds. Protonated 2,7-bis(trimethylsilyl)- and 2,7-di(hydroxymethyl)-1,8-bis(dimethylamino)naphthalenes. Tetrahedron 2008, 64, 6209–6214. [Google Scholar] [CrossRef]
- Pozharskii, A.F.; Ozeryanskii, V.A.; Starikova, Z.A. Molecular structure of 5,6-bis(dimethylamino)acenaphthene, 5,6- bis(dimethylamino)acenaphthylene, and their monohydrobromides: A comparison with some naphthalene proton sponges. J. Chem. Soc. Perkin Trans. 2002, 2, 318–322. [Google Scholar] [CrossRef]
- Barnett, G.H.; Hibbert, F. General base catalysis, isotope effects, activation parameters, and the mechanism of removal of the hydrogen-bonded proton from protonated 1,8-bis(diethylamino)-2,7-dimethoxynaphthalene. J. Am. Chem. Soc. 1984, 106, 2080–2084. [Google Scholar] [CrossRef]
- Filatova, E.A.; Gulevskaya, A.V.; Pozharskii, A.F.; Ermolenko, E.A.; Ozeryanskii, V.A.; Misharev, A.D. Synthesis of 2-Aryl- and 2,7-Diaryl-1,8-bis(dimethylamino)naphthalenes. Overview of the “Buttressing effect” in 2,7-Disubstituted Proton Sponges. ChemistrySelect 2020, 5, 9932–9945. [Google Scholar] [CrossRef]
- Randolph, C.E.; Fabijanczuk, K.C.; Blanksby, S.J.; McLuckey, S.A. Proton Transfer Reactions for the Gas-Phase Separation, Concentration, and Identification of Cardiolipins. Anal. Chem. 2020, 92, 10847–10855. [Google Scholar] [CrossRef]
- Swor, C.D.; Zakharov, L.N.; Tyler, D.R. A Colorimetric Proton Sponge. J. Org. Chem. 2010, 75, 6977–6979. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.C.H.; Parker, S.F.; Ramirez-Cuesta, A.J.; Tomkinson, J. Series on Neutron Techniques and Applications, Vibrational Spectroscopy with Neutrons; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2005. [Google Scholar]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- Mestrelab Research, S.L. Available online: https://mestrelab.com/software/mnova (accessed on 12 October 2020).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. et al. Gaussian 16, Revision С.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Soc. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided Mol. Design. 2000, 14, 123–134. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszyński, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 7, 1887–1930. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 2010, 133, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- CPMD 3.17.1; IBM Corp: 1990–2004; MPI für Festkörperforschung Stuttgart, 1997–2001.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gnuplot 4.2; Thomas Williams, Colin Kelley, 1986–1993, 1998, 2004, 2007.
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. 1991, B43, 1993–2006. [Google Scholar] [CrossRef]
- Hockney, R.W. The potential calculation and some applications. Meth. Comput. Phys. 1970, 9, 136–211. [Google Scholar]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. 1985, A31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef] [Green Version]
- Herschlag, D.; Pinney, M.M. Hydrogen Bonds: Simple after All? Biochemistry 2018, 57, 3338–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigala, P.A.; Ruben, E.A.; Liu, C.W.; Piccoli, P.M.B.; Hohenstein, E.G.; Martínez, T.J.; Schultz, A.J.; Herschlag, D. Determination of hydrogen bond structure in water versus aprotic environments to test the relationship between length and stability. J. Am. Chem. Soc. 2015, 137, 5730–5740. [Google Scholar] [CrossRef] [PubMed]
- Ashkar, R.; Bilheux, H.Z.; Bordallo, H.; Briber, R.; Callaway, D.J.E.; Cheng, X.; Chu, X.-Q.; Curtis, J.E.; Dadmun, M.; Fenimore, P.; et al. Neutron scattering in the biological sciences: Progress and prospects. Acta Cryst. 2018, D74, 1129–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, B.S. Inelastic Neutron Scattering: A Tool in Molecular Vibrational Spectroscopy and a Test of ab Initio Methods. Phys. Chem. A 2001, 105, 3949–3960. [Google Scholar] [CrossRef]
- Hudson, B.S. Vibrational spectroscopy using inelastic neutron scattering: Overview and outlook. Vibr. Spectr. 2006, 42, 25–32. [Google Scholar] [CrossRef]
- Pusztai, L. Neutron Scattering Methods in Chemistry. In Handbook of Nuclear Chemistry; Vértes, A., Nagy, S., Klencsár, Z., Lovas, R.G., Rösch, F., Eds.; Springer: Boston, MA, USA, 2011. [Google Scholar]
- Albers, P.W.; Parker, S.W. IINS Inelastic Incoherent Neutron Scattering in Catalysis Research. Adv. Catal. 2007, 51, 99–132. [Google Scholar]
- Tsapatsaris, N.; Kolesov, B.A.; Fischer, J.; Boldyreva, E.V.; Daemen, L.; Eckert, J.; Bordallo, H.N. Polymorphism of Paracetamol: A New Understanding of Molecular Flexibility through Local Methyl Dynamics. Mol. Pharm. 2014, 11, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Pawlukojć, A.; Prager, M.; Sawka-Dobrowolska, W.; Bator, G.; Sobczyk, L.; Ivanov, A.; Rols, S.; Grech, E.; Nowicka-Scheibe, J.; Unruh, T. The structure, methyl rotation reflected in elestic and quasielastic neutron scattering and vibrational spectra of 1,2,3,5-tetramethoxybenzene and its 2:1 complex with 1,2,4,5-tetracyanobenzene. J. Chem. Phys. 2008, 129, 154506–154512. [Google Scholar] [CrossRef] [PubMed]
- Bordallo, H.N.; Zakharov, B.A.; Boldyreva, E.V.; Johnson, M.R.; Koza, M.M.; Seydel, T.; Fischer, J. Application of Incoherent Inelastic Neutron Scattering in Pharmaceutical Analysis: Relaxation Dynamics in Phenacetin. Mol. Pharm. 2012, 9, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Bordallo, H.N.; Barthes, M.; Eckert, J. Vibrational dynamics of crystalline L-alanine. Phys. B 1998, 241-243, 1138–1140. [Google Scholar] [CrossRef] [Green Version]
- Pawlukojć, A.; Natkaniec, I.; Bator, G.; Sobczyk, L.; Grech, E.; Nowicka-Scheibe, J. Low frequency internal modes of 1,2,4,5-tetramethylbenzene, tetramethylpyrazine and tetramethyl-1,4-benzoquinone. INS, Raman, IR and theoretical DFT studies. Spectrochim. Acta A 2006, 63, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Prager, M.; Pawlukojc, A.; Wischnewski, A.; Wuttke, J. Inelastic neutron scattering study of methyl groups rotation in some methylxanthines. J. Chem. Phys. 2007, 127, 214509–214519. [Google Scholar] [CrossRef] [PubMed]
- Bator, G.; Sawka-Dobrowolska, W.; Sobczyk, L.; Grech, E.; Nowicka-Scheibe, J.; Pawlukojc, A.; Wuttke, J.; Baran, J.; Owczarek, M. 4,4’-, 5,5’-, and 6,6’-dimethyl-2,2’-bipyridyls: The structures, phase transitions, vibrations, and methyl group tunneling of their complexes with chloranilic acid. J. Chem. Phys. 2011, 135, 044509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Gangopadhyay, D.; Nandi, R.; Sharma, P.; Singh, R.K. Raman signatures of strong and weak hydrogen bonds in binary mixtures of phenol with acetonitrile, benzene and orthodichlorobenzene. Raman Spectr. 2016, 47, 712–719. [Google Scholar] [CrossRef]
- Abe, N.; Ito, M. Effects of hydrogen bonding on the Raman intensities of methanol, ethanol and water. Raman Spectr. 1978, 7, 161–167. [Google Scholar] [CrossRef]
- Zundel, G. The Hydrogen Bond. Recent Developments in Theory and Experiments; Schuster, P., Zundel, G., Sandorfy, C., Eds.; North-Holland: Amsterdam, The Netherlands, 1976; Volume 2, pp. 683–766. [Google Scholar]
- Zundel, G. Hydrogen bonds with large proton polarizability and proton transfer processes in electrochemistry and biology. Adv. Chem. Phys. 2000, 111, 1–217. [Google Scholar]
- Kwocz, A.; Panek, J.J.; Jezierska, A.; Hetmańczyk, Ł.; Pawlukojć, A.; Kochel, A.; Lipkowski, P.; Filarowski, A. A molecular roundabout: Triple cycle-arranged hydrogen bonds in light of experiment and theory. New J. Chem. 2018, 42, 19467–19477. [Google Scholar] [CrossRef]
- Jóźwiak, K.; Jezierska, A.; Panek, J.J.; Goremychkin, E.A.; Tolstoy, P.M.; Shenderovich, I.G.; Filarowski, A. Inter- vs. intra-molecular hydrogen bond patterns and proton dynamics in phthalic acid associates. Molecules 2020, 25, 4720. [Google Scholar] [CrossRef]






| Electrostatics | Exchange | Induction | Dispersion | Total HF | Total SAPT2 |
|---|---|---|---|---|---|
| −87.17 | 24.98 | −14.64 | −11.44 | −79.25 | −88.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piękoś, P.; Jezierska, A.; Panek, J.J.; Goremychkin, E.A.; Pozharskii, A.F.; Antonov, A.S.; Tolstoy, P.M.; Filarowski, A. Symmetry/Asymmetry of the NHN Hydrogen Bond in Protonated 1,8-Bis(dimethylamino)naphthalene. Symmetry 2020, 12, 1924. https://doi.org/10.3390/sym12111924
Piękoś P, Jezierska A, Panek JJ, Goremychkin EA, Pozharskii AF, Antonov AS, Tolstoy PM, Filarowski A. Symmetry/Asymmetry of the NHN Hydrogen Bond in Protonated 1,8-Bis(dimethylamino)naphthalene. Symmetry. 2020; 12(11):1924. https://doi.org/10.3390/sym12111924
Chicago/Turabian StylePiękoś, Patrycja, Aneta Jezierska, Jarosław J. Panek, Eugene A. Goremychkin, Alexander F. Pozharskii, Alexander S. Antonov, Peter M. Tolstoy, and Aleksander Filarowski. 2020. "Symmetry/Asymmetry of the NHN Hydrogen Bond in Protonated 1,8-Bis(dimethylamino)naphthalene" Symmetry 12, no. 11: 1924. https://doi.org/10.3390/sym12111924
APA StylePiękoś, P., Jezierska, A., Panek, J. J., Goremychkin, E. A., Pozharskii, A. F., Antonov, A. S., Tolstoy, P. M., & Filarowski, A. (2020). Symmetry/Asymmetry of the NHN Hydrogen Bond in Protonated 1,8-Bis(dimethylamino)naphthalene. Symmetry, 12(11), 1924. https://doi.org/10.3390/sym12111924