
Molecular Recognition, Transient Chirality and Sulfur Hydrogen Bonding in the Benzyl Mercaptan Dimer

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
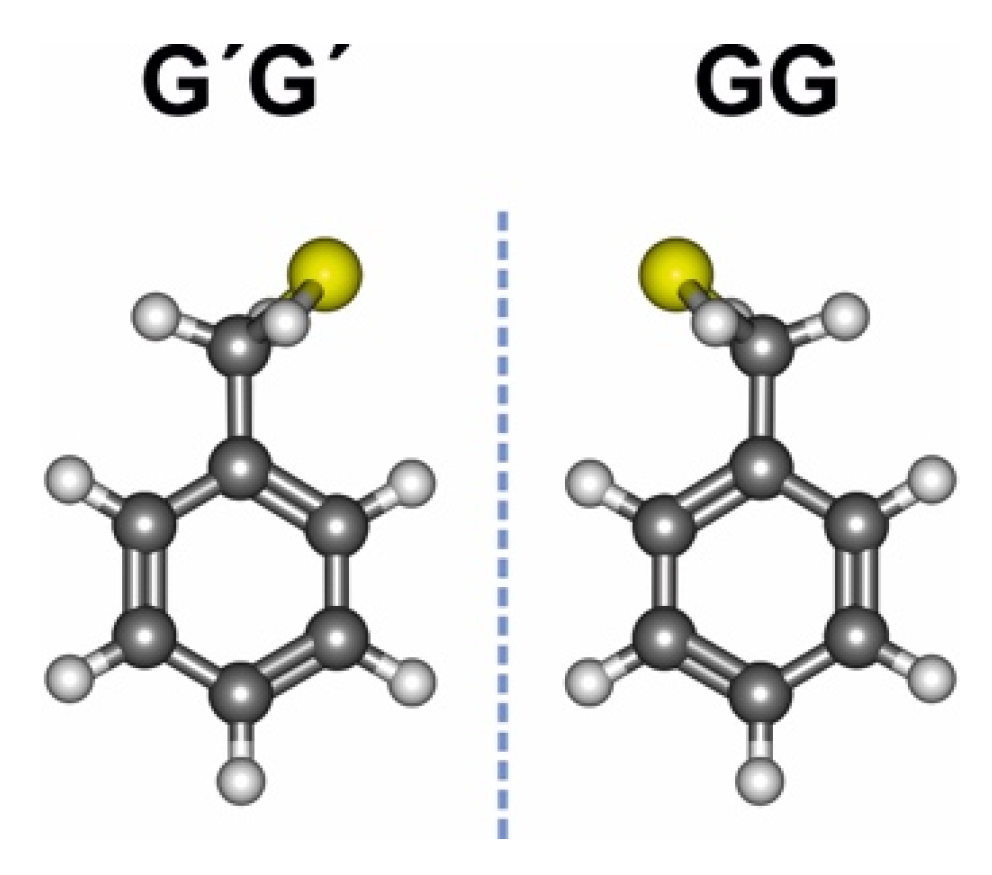
3.1. Benzyl Mercaptan Monomer
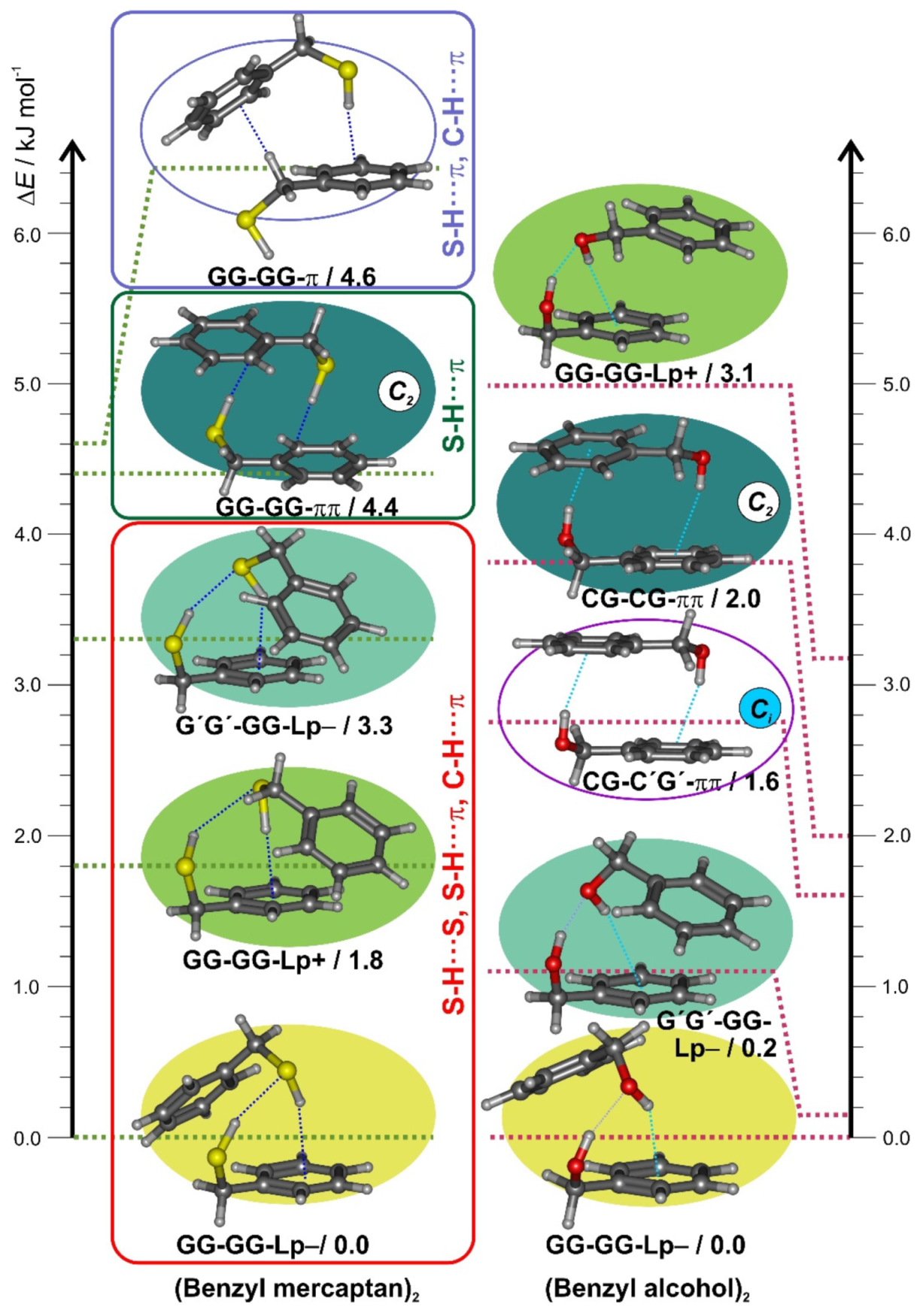
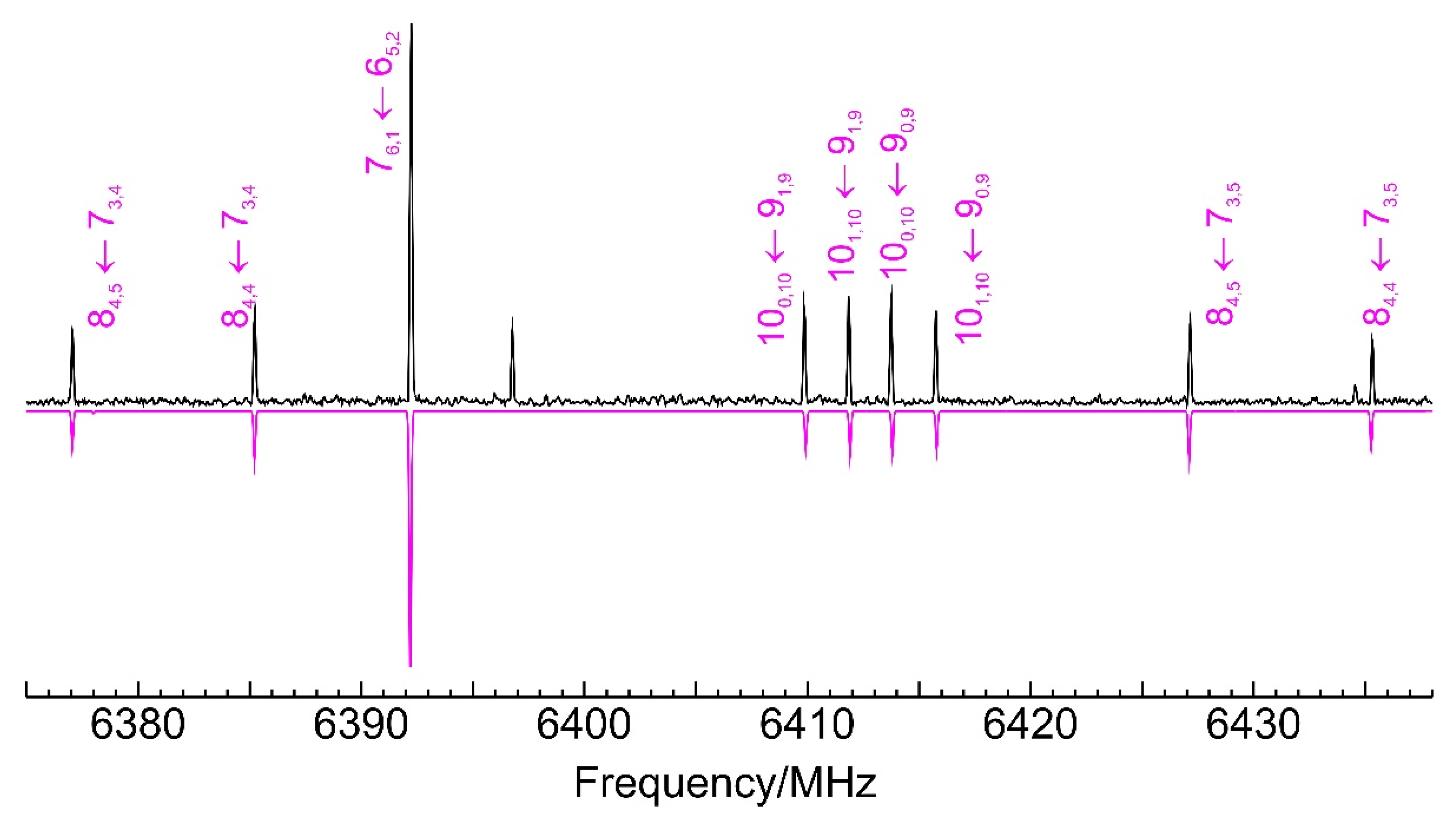
3.2. Benzyl Mercaptan Homodimer
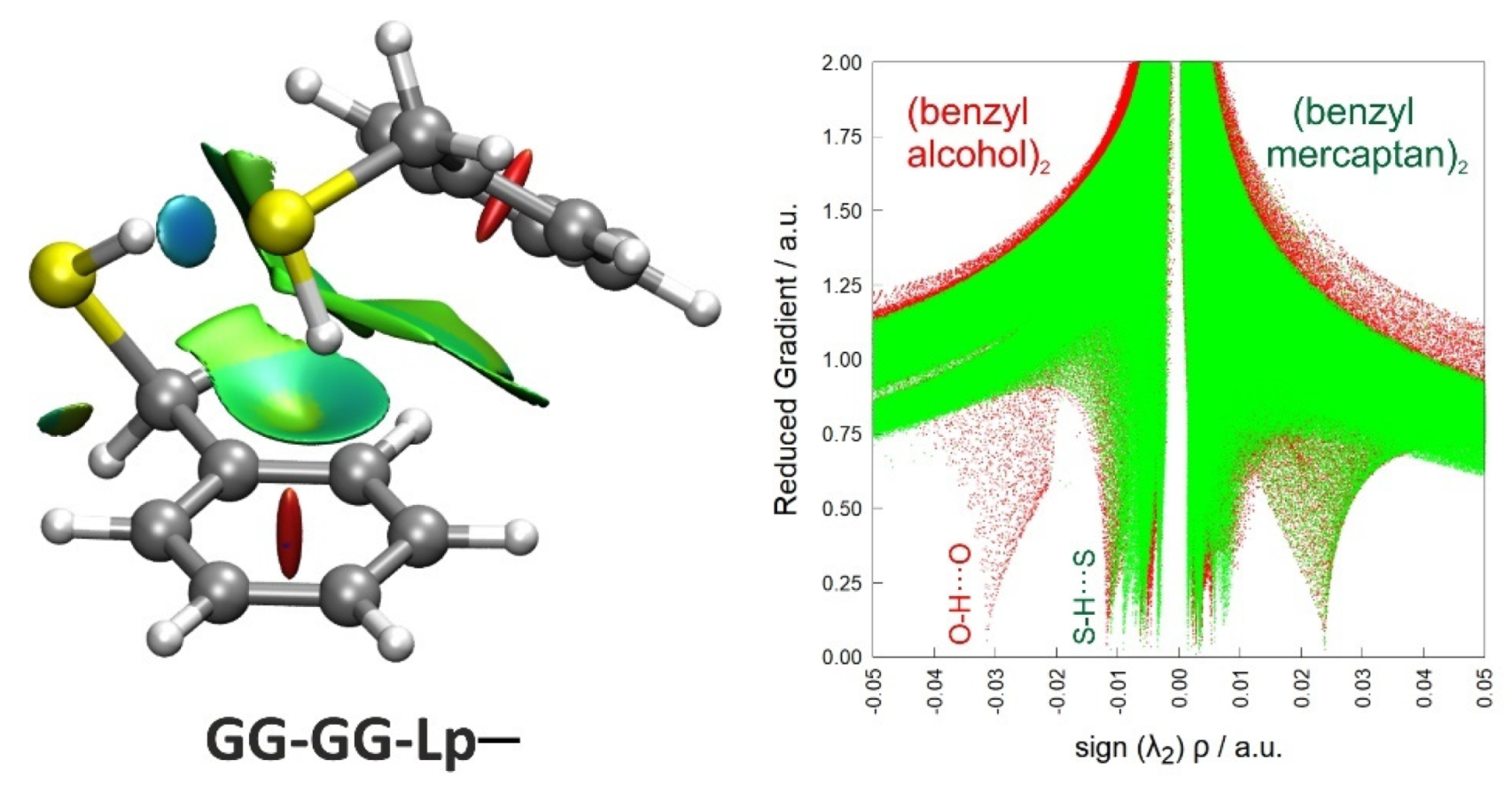
3.3. Non-Covalent Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | ΔEelect | ΔEdisp | ΔEind | ΔEexch | ΔEtotal | ΔES22 i |
|---|---|---|---|---|---|---|
| (Benzyl mercaptan)2 a | −39.3[34.4%] h | −61.1[53.6%] | −13.6[12.0%] | 78.7 | −35.3 | |
| (Thiophenol)2 PD1-trans b | −24.9[31.0%] | −47.9[59.5%] | −7.7[9.5%] | 54.6 | −25.9 | |
| (Thiophenol)2 PD2-cis | −26.0[29.4%] | −53.8[60.9%] | −8.4[9.6%] | 61.3 | −27.0 | |
| (H2S)2 c | −12.1[49.0%] | −7.8[31.7%] | −4.7[19.3%] | 19.2 | −5.4 | |
| (Benzyl alcohol)2 d | −58.7[44.5%] | −54.6[41.4%] | −18.6[14.1%] | 89.8 | −42.1 | |
| (Phenol)2 e | −41.8[48.3%] | −28.8[33.3%] | −15.9[18.4%] | 58.9 | −27.6 | −29.5 |
| (H2O)2 f | −35.7[63.5%] | −9.5[16.8%] | −11.1[19.8%] | 37.7 | −18.6 | −21.0 |
| Pyridine-methane g | −3.0[20.6%] | −10.9[74.6%] | −0.7[4.8%] | 9.4 | −5.2 |
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, M.; Zhang, L.; Wang, T. Supramolecular chirality in self-Assembled systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.R.; Salerno, F.; Fuchter, M.J. The added value of small-molecule chirality in technological applications. Nat. Rev. Chem. 2017, 1. [Google Scholar] [CrossRef]
- Schermann, J.-P. Spectroscopy and Modeling of Biomolecular Building Blocks; Elsevier: Amsterdam, The Netherlands, 2008; ISBN 9780444527080. [Google Scholar]
- Hobza, P.; Muller-Dethlefs, K. Non-Covalent Interactions; Hobza, P., Muller-Dethlefs, K., Eds.; Theoretical and Computational Chemistry Series; Royal Society of Chemistry: Cambridge, UK, 2009; ISBN 9781847558534. [Google Scholar]
- Scheiner, S. (Ed.) Challenges and Advances in Computational Chemistry and Physics. In Noncovalent Forces; Springer International Publishing: Cham, Switzerland, 2015; Volume 19, ISBN 9783319141626. [Google Scholar]
- Alkorta, I.; Elguero, J.; Frontera, A. Not only hydrogen bonds: Other noncovalent interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Juanes, M.; Saragi, R.T.; Caminati, W.; Lesarri, A. The Hydrogen Bond and Beyond: Perspectives for Rotational Investigations of Non-Covalent Interactions. Chem. A Eur. J. 2019, 25, 11402–11411. [Google Scholar] [CrossRef]
- Zehnacker, A.; Suhm, M.A. Chirality Recognition between Neutral Molecules in the Gas Phase. Angew. Chem. Int. Ed. 2008, 47, 6970–6992. [Google Scholar] [CrossRef]
- Zehnacker, A. (Ed.) Chiral Recognition in the Gas Phase; CRC Press: Boca Raton, FL, USA, 2010; ISBN 9781420082272. [Google Scholar]
- Pierini, M.; Troiani, A.; Speranza, M.; Piccirillo, S.; Bosman, C.; Toja, D.; Giardini-Guidoni, A. Gas-Phase Enantiodifferentiation of Chiral Molecules: Chiral Recognition of 1-Phenyl-1-propanol/2-Butanol Clusters by Resonance Enhanced Multiphoton Ionization Spectroscopy. Angew. Chem. Int. Ed. 1997, 36, 1729–1731. [Google Scholar] [CrossRef]
- Le Barbu, K.; Brenner, V.; Millié, P.; Lahmani, F.; Zehnacker-Rentien, A. An Experimental and Theoretical Study of Jet-Cooled Complexes of Chiral Molecules: The Role of Dispersive Forces in Chiral Discrimination. J. Phys. Chem. A 1998, 102, 128–137. [Google Scholar] [CrossRef]
- Borho, N.; Häber, T.; Suhm, M.A. Chiral self-recognition in the gas phase: The case of glycidol dimers. Phys. Chem. Chem. Phys. 2001, 3, 1945–1948. [Google Scholar] [CrossRef]
- Caminati, W.; Grabow, J.-U. Advancements in Microwave Spectroscopy. In Frontiers and Advances in Molecular Spectroscopy; Laane, J., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 569–598. ISBN 9780128112212. [Google Scholar]
- Portmann, S.; Inauen, A.; Lüthi, H.P.; Leutwyler, S. Chiral discrimination in hydrogen-bonded complexes. J. Chem. Phys. 2000, 113, 9577–9585. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Discrimination of hydrogen-bonded complexes with axial chirality. J. Chem. Phys. 2002, 117, 6463–6468. [Google Scholar] [CrossRef] [Green Version]
- Borho, N.; Xu, Y. Tailoring the key in a molecular lock-and-key model system: The propylene oxide⋯2-fluoroethanol complex. J. Am. Chem. Soc. 2008, 130, 5916–5921. [Google Scholar] [CrossRef] [PubMed]
- Hearn, J.P.I.; Cobley, R.V.; Howard, B.J. High-resolution spectroscopy of induced chiral dimers: A study of the dimers of ethanol by Fourier transform microwave spectroscopy. J. Chem. Phys. 2005, 123, 134324. [Google Scholar] [CrossRef] [PubMed]
- Loru, D.; Peña, I.; Sanz, M.E. Ethanol dimer: Observation of three new conformers by broadband rotational spectroscopy. J. Mol. Spectrosc. 2017, 335, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Snow, M.S.; Howard, B.J.; Evangelisti, L.; Caminati, W. From Transient to Induced Permanent Chirality in 2-Propanol upon Dimerization: A Rotational Study. J. Phys. Chem. A 2011, 115, 47–51. [Google Scholar] [CrossRef] [PubMed]
- King, A.K.; Howard, B.J. A microwave study of the hetero-chiral dimer of butan-2-ol. Chem. Phys. Lett. 2001, 348, 343–349. [Google Scholar] [CrossRef]
- Maris, A.; Giuliano, B.M.; Bonazzi, D.; Caminati, W. Molecular recognition of chiral conformers: A rotational study of the dimers of glycidol. J. Am. Chem. Soc. 2008, 130, 13860–13861. [Google Scholar] [CrossRef]
- Juanes, M.; Usabiaga, I.; León, I.; Evangelisti, L.; Fernández, J.A.; Lesarri, A. The Six Isomers of the Cyclohexanol Dimer: A Delicate Test for Dispersion Models. Angew. Chem. Int. Ed. 2020, 59, 14081–14085. [Google Scholar] [CrossRef]
- Liu, X.; Borho, N.; Xu, Y. Molecular self-recognition: Rotational spectra of the dimeric 2-fluoroethanol conformers. Chem. A Eur. J. 2009, 15, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Xu, Y. Chirality synchronization in trifluoroethanol dimer revisited: The missing heterochiral dimer. J. Phys. Chem. Lett. 2014, 5, 1850–1855. [Google Scholar] [CrossRef]
- Oswald, S.; Seifert, N.A.; Bohle, F.; Gawrilow, M.; Grimme, S.; Jäger, W.; Xu, Y.; Suhm, M.A. The Chiral Trimer and a Metastable Chiral Dimer of Achiral Hexafluoroisopropanol: A Multi-Messenger Study. Angew. Chem. Int. Ed. 2019, 58, 5080–5084. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Wategaonkar, S. ZEKE photoelectron spectroscopy of p-fluorophenol H2S/H2O complexes and dissociation energy measurement using the Birge-Sponer extrapolation method. J. Phys. Chem. A 2014, 118, 9386–9396. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Bhattacharyya, S.; Wategaonkar, S. Dissociation Energies of Sulfur-Centered Hydrogen-Bonded Complexes. J. Phys. Chem. A 2015, 119, 10863–10870. [Google Scholar] [CrossRef]
- Martin, D.E.; Robertson, E.G.; Thompson, C.D.; Morrison, R.J.S. Resonant 2-photon ionization study of the conformation and the binding of water molecules to 2-phenylethanethiol (PhC H2 C H2 SH). J. Chem. Phys. 2008, 128, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Lobo, I.A.; Robertson, P.A.; Villani, L.; Wilson, D.J.D.; Robertson, E.G. Thiols as Hydrogen Bond Acceptors and Donors: Spectroscopy of 2-Phenylethanethiol Complexes. J. Phys. Chem. A 2018, 122, 7171–7180. [Google Scholar] [CrossRef]
- Juanes, M.; Lesarri, A.; Pinacho, R.; Charro, E.; Rubio, J.E.; Enríquez, L.; Jaraíz, M. Sulfur Hydrogen Bonding in Isolated Monohydrates: Furfuryl Mercaptan versus Furfuryl Alcohol. Chem. A Eur. J. 2018, 24, 6564–6571. [Google Scholar] [CrossRef]
- Juanes, M.; Saragi, R.T.; Pinacho, R.; Rubio, J.E.; Lesarri, A. Sulfur hydrogen bonding and internal dynamics in the monohydrates of thenyl mercaptan and thenyl alcohol. Phys. Chem. Chem. Phys. 2020, 22, 12412–12421. [Google Scholar] [CrossRef] [PubMed]
- Wategaonkar, S.; Bhattacherjee, A. N-H···S Interaction Continues to Be an Enigma: Experimental and Computational Investigations of Hydrogen-Bonded Complexes of Benzimidazole with Thioethers. J. Phys. Chem. A 2018, 122, 4313–4321. [Google Scholar] [CrossRef] [PubMed]
- Cocinero, E.J.; Sánchez, R.; Blanco, S.; Lesarri, A.; López, J.C.; Alonso, J.L. Weak hydrogen bonds C–H⋯S and C–H⋯F–C in the thiirane–trifluoromethane dimer. Chem. Phys. Lett. 2005, 402, 4–10. [Google Scholar] [CrossRef]
- Ghosh, S.; Chopra, P.; Wategaonkar, S. C-H⋯S interaction exhibits all the characteristics of conventional hydrogen bonds. Phys. Chem. Chem. Phys. 2020, 22, 17482–17493. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Mandal, P.K.; Lovas, F.J.; Medcraft, C.; Walker, N.R.; Arunan, E. The H2S Dimer is Hydrogen-Bonded: Direct Confirmation from Microwave Spectroscopy. Angew. Chem. Int. Ed. 2018, 57, 15199–15203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saragi, R.T.; Juanes, M.; Pérez, C.; Pinacho, P.; Tikhonov, D.S.; Caminati, W.; Schnell, M.; Lesarri, A. Switching Hydrogen Bonding to π-Stacking: The Thiophenol Dimer and Trimer. J. Phys. Chem. Lett. 2021, 12, 1367–1373. [Google Scholar] [CrossRef]
- Mishra, K.K.; Borish, K.; Singh, G.; Panwaria, P.; Metya, S.; Madhusudhan, M.S.; Das, A. Observation of an Unusually Large IR Red-Shift in an Unconventional S–H···S Hydrogen-Bond. J. Phys. Chem. Lett. 2021, 12, 1228–1235. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Matsuda, Y.; Fujii, A.; Wategaonkar, S. Acid-base formalism in dispersion-stabilized S–H···Y (Y=O, S) hydrogen-bonding interactions. J. Phys. Chem. A 2015, 119, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chopra, P.; Wategaonkar, S.; Fujii, A. Electronic and Infrared Spectroscopy of Benzene-(H2S)n (n = 1 and 2): The Prototype of the SH-πInteraction. J. Phys. Chem. A 2019, 123, 7255–7260. [Google Scholar] [CrossRef] [PubMed]
- Biswal, H.S.; Bhattacharyya, S.; Bhattacherjee, A.; Wategaonkar, S. Nature and strength of sulfur-centred hydrogen bonds: Laser spectroscopic investigations in the gas phase and quantum-chemical calculations. Int. Rev. Phys. Chem. 2015, 34, 99–160. [Google Scholar] [CrossRef]
- Chand, A.; Sahoo, D.K.; Rana, A.; Jena, S.; Biswal, H.S. The Prodigious Hydrogen Bonds with Sulfur and Selenium in Molecular Assemblies, Structural Biology, and Functional Materials. Acc. Chem. Res. 2020, 53, 1580–1592. [Google Scholar] [CrossRef] [PubMed]
- Medel, R.; Suhm, M.A. Understanding benzyl alcohol aggregation by chiral modification: The pairing step. Phys. Chem. Chem. Phys. 2020, 22, 25538–25551. [Google Scholar] [CrossRef]
- Medel, R.; Camiruaga, A.; Saragi, R.T.; Pinacho, P.; Pérez, C.; Schnell, M.; Lesarri, A.; Suhm, M.A.; Fernandez, J.A. Rovibronic Signatures of Molecular Aggregation in the Gas Phase: Subtle Homochirality Trends in the Dimer, Trimer and Tetramer of Benzyl Alcohol. Phys. Chem. Chem. Phys. 2021, in press. [Google Scholar] [CrossRef]
- Neill, J.L.; Shipman, S.T.; Alvarez-Valtierra, L.; Lesarri, A.; Kisiel, Z.; Pate, B.H. Rotational spectroscopy of iodobenzene and iodobenzene-neon with a direct digital 2-8 GHz chirped-pulse Fourier transform microwave spectrometer. J. Mol. Spectrosc. 2011, 269, 21–29. [Google Scholar] [CrossRef]
- Shipman, S.T.; Pate, B.H. New Techniques in Microwave Spectroscopy. In Handbook of High-Resolution Spectroscopy; Major Reference Works; Merkt, F., Quack, M., Eds.; John Wiley & Sons, Ltd.: New York, NY, USA, 2011; pp. 801–828. ISBN 9780470749593. [Google Scholar]
- Grabow, J.-U. Fourier Transform Microwave Spectroscopy Measurement and Instrumentation. In Handbook of High-Resolution Spectroscopy; Merkt, F., Quack, M., Eds.; John Wiley & Sons, Ltd.: New York, NY, USA, 2011; pp. 723–799. ISBN 9780470749593. [Google Scholar]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Becke, A.D. A post-Hartree-Fock model of intermolecular interactions: Inclusion of higher-order corrections. J. Chem. Phys. 2006, 124, 174104. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press on Demand: Oxford, UK, 1997; ISBN 019509011X. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utzat, K.A.; Bohn, R.K.; Montgomery, J.A.; Michels, H.H.; Caminati, W. Rotational spectrum, tunneling motions, and potential barriers of benzyl alcohol. J. Phys. Chem. A 2010, 114, 6913–6916. [Google Scholar] [CrossRef] [PubMed]
- Saragi, R.T.; Juanes, M.; Caminati, W.; Lesarri, A.; Enríquez, L.; Jaraíz, M. Rotational Spectrum, Tunneling Motions, and Intramolecular Potential Barriers in Benzyl Mercaptan. J. Phys. Chem. A 2019, 123, 8435–8440. [Google Scholar] [CrossRef] [PubMed]
- Papousek, D.; Aliev, M.R. Molecular Vibrational-Rotational Spectra Studies in Physical and Theoretical Chemistry 17; Elsevier: Amsterdam, The Netherlands, 1982; ISBN 0444997377. [Google Scholar]
- Nayak, S.K.; Sathishkumar, R.; Row, T.N.G. Directing role of functional groups in selective generation of C–H⋯π interactions: In situ cryo-crystallographic studies on benzyl derivatives. CrystEngComm 2010, 12, 3112–3118. [Google Scholar] [CrossRef]
- Watson, J.K.G. Aspects of Quartic and Sextic Centrifugal Effects on Rotational Energy Levels. In Vibrational Spectra and Structure; Durig, J.R., Ed.; Elsevier B.V.: Amsterdam, The Netherlands, 1977; Volume 6, pp. 1–89. [Google Scholar]
- Godfrey, P.D.; Brown, R.D. Proportions of species observed in jet spectroscopy-vibrational-energy effects: Histamine tautomers and conformers. J. Am. Chem. Soc. 1998, 120, 10724–10732. [Google Scholar] [CrossRef]
- Florio, G.M.; Christie, R.A.; Jordan, K.D.; Zwier, T.S. Conformational preferences of jet-cooled melatonin: Probing trans- and cis-amide regions of the potential energy surface. J. Am. Chem. Soc. 2002, 124, 10236–10247. [Google Scholar] [CrossRef]
- Lesarri, A.; Pinacho, R.; Enríquez, L.; Rubio, J.E.; Jaraíz, M.; Abad, J.L.; Gigosos, M.A. Rotational spectra of tetracyclic quinolizidine alkaloids: Does a water molecule flip sparteine? Phys. Chem. Chem. Phys. 2017, 19, 17553–17559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Tian, F.; Lv, F.; Shang, Z. Geometric characteristics of hydrogen bonds involving sulfur atoms in proteins. Proteins Struct. Funct. Bioinform. 2009, 76, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Cukras, J.; Skóra, G.; Jankowska, J.; Lundell, J. Computational structures and SAPT interaction energies of HXeSH···H2Y (Y=O or S) complexes. Inorganics 2018, 6, 100. [Google Scholar] [CrossRef] [Green Version]
- Cukras, J.; Sadlej, J. Structure and energetics of weakly bound water-sulfur dioxide complexes. J. Mol. Struct. THEOCHEM 2007, 819, 41–51. [Google Scholar] [CrossRef]
- Seifert, N.A.; Steber, A.L.; Neill, J.L.; Pérez, C.; Zaleski, D.P.; Pate, B.H.; Lesarri, A. The interplay of hydrogen bonding and dispersion in phenol dimer and trimer: Structures from broadband rotational spectroscopy. Phys. Chem. Chem. Phys. 2013, 15, 11468–11477. [Google Scholar] [CrossRef]
- Mons, M.; Robertson, E.G.; Simons, J.P. Intra- and Intermolecular π-Type Hydrogen Bonding in Aryl Alcohols: UV and IR−UV Ion Dip Spectroscopy. J. Phys. Chem. A 2000, 104, 1430–1437. [Google Scholar] [CrossRef]
- Altnöder, J.; Bouchet, A.; Lee, J.J.; Otto, K.E.; Suhm, M.A.; Zehnacker-Rentien, A. Chirality-dependent balance between hydrogen bonding and London dispersion in isolated (±)-1-indanol clusters. Phys. Chem. Chem. Phys. 2013, 15, 10167–10180. [Google Scholar] [CrossRef] [PubMed]
- Mani, D.; Arunan, E. Rotational spectra of propargyl alcohol dimer: A dimer bound with three different types of hydrogen bonds. J. Chem. Phys. 2014, 141, 164311. [Google Scholar] [CrossRef]
- Longarte, A.; Redondo, C.; Fernández, J.A.; Castaño, F. IR/UV and UV/UV double-resonance study of guaiacol and eugenol dimers. J. Chem. Phys. 2005, 122, 164304. [Google Scholar] [CrossRef] [PubMed]
- Altnöder, J.; Lee, J.J.; Otto, K.E.; Suhm, M.A. Molecular Recognition in Glycolaldehyde, the Simplest Sugar: Two Isolated Hydrogen Bonds Win Over One Cooperative Pair. ChemistryOpen 2012, 1, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, L.; Spada, L.; Li, W.; Vazart, F.; Barone, V.; Caminati, W. The Borderline between Reactivity and Pre-reactivity of Binary Mixtures of Gaseous Carboxylic Acids and Alcohols. Angew. Chem. Int. Ed. 2017, 56, 3872–3875. [Google Scholar] [CrossRef] [Green Version]
- Asselin, P.; Madebène, B.; Soulard, P.; Georges, R.; Goubet, M.; Huet, T.R.; Pirali, O.; Zehnacker-Rentien, A. Competition between inter- and intra-molecular hydrogen bonding: An infrared spectroscopic study of jet-cooled amino-ethanol and its dimer. J. Chem. Phys. 2016, 145, 224313. [Google Scholar] [CrossRef] [Green Version]
- Seurre, N.; Le Barbu-Debus, K.; Lahmani, F.; Zehnacker-Rentien, A.; Sepioł, J. Electronic and vibrational spectroscopy of jet-cooled m-cyanophenol and its dimer: Laser-induced fluorescence and fluorescence-dip IR spectra in the S0 and S1 states. Chem. Phys. 2003, 295, 21–33. [Google Scholar] [CrossRef]
- Pérez, C.; León, I.; Lesarri, A.; Pate, B.H.; Martínez, R.; Millán, J.; Fernández, J.A. Isomerism of the Aniline Trimer. Angew. Chem. Int. Ed. 2018, 57, 15112–15116. [Google Scholar] [CrossRef] [Green Version]




| Experiment | Theory f | |||
|---|---|---|---|---|
| Isomer | 1 | 2 | 3 | |
| (GG-GG-Lp−) | (GG-GG-Lp+) | (GG-G′G′-Lp+) | ||
| A/MHz a | 490.79216(17) e | 496.24 | 538.12 | 509.23 |
| B/MHz | 344.12732(12) | 352.51 | 328.24 | 338.05 |
| C/MHz | 317.21115(11) | 324.58 | 299.94 | 319.40 |
| DJ/kHz | 0.03861(51) | 0.0326 | 0.0596 | 0.0507 |
| DJK/kHz | 0.0642(18) | 0.0608 | −0.0984 | −0.0040 |
| DK/kHz | −0.0718(20) | −0.0675 | 0.1748 | 0.0420 |
| d1/kHz | −0.00375(33) | −0.0026 | −0.0004 | −0.0031 |
| d2/kHz | 0.00089(13) | 0.0008 | −0.0005 | 0.0008 |
| |μa|/D | ++ | 1.6 | 0.4 | 1.0 |
| |μb|/D | ++ | 1.7 | 1.3 | 1.6 |
| |μc|/D | ++ | 1.4 | 0.8 | 0.7 |
| HBond donor b | ||||
| HS-CαCβ/deg | −41.3 | −50.6 | 42.8 | |
| SCα-CβC1/deg | −56.6 | −70.0 | 63.3 | |
| HBond acceptor | ||||
| HS-CαCβ/deg | −54.1 | −46.3 | −45.8 | |
| SCα-CβC1/deg | −65.7 | −56.1 | −68.9 | |
| r(S···H)/Å | 2.748 | 2.941 | 2.803 | |
| ∠(S-H···S)/deg | 162.9 | 137.5 | 158.6 | |
| r(S-H···centroid)/Å | 2.515 | 2.398 | 2.553 | |
| ΔEZPE/kJ mol−1 c | 0.0 | 1.7 | 3.4 | |
| ΔG/kJ mol−1 | 0.4 | 0.0 | 1.4 | |
| Ec/kJ mol−1 | −35.6 | −33.1 | −31.3 | |
| ΔEc/kJ mol−1 | 0.0 | 2.6 | 4.3 | |
| N d | 337 | |||
| σ/kHz | 8.5 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saragi, R.T.; Juanes, M.; Pinacho, R.; Rubio, J.E.; Fernández, J.A.; Lesarri, A. Molecular Recognition, Transient Chirality and Sulfur Hydrogen Bonding in the Benzyl Mercaptan Dimer. Symmetry 2021, 13, 2022. https://doi.org/10.3390/sym13112022
Saragi RT, Juanes M, Pinacho R, Rubio JE, Fernández JA, Lesarri A. Molecular Recognition, Transient Chirality and Sulfur Hydrogen Bonding in the Benzyl Mercaptan Dimer. Symmetry. 2021; 13(11):2022. https://doi.org/10.3390/sym13112022
Chicago/Turabian StyleSaragi, Rizalina Tama, Marcos Juanes, Ruth Pinacho, José Emiliano Rubio, José A. Fernández, and Alberto Lesarri. 2021. "Molecular Recognition, Transient Chirality and Sulfur Hydrogen Bonding in the Benzyl Mercaptan Dimer" Symmetry 13, no. 11: 2022. https://doi.org/10.3390/sym13112022
APA StyleSaragi, R. T., Juanes, M., Pinacho, R., Rubio, J. E., Fernández, J. A., & Lesarri, A. (2021). Molecular Recognition, Transient Chirality and Sulfur Hydrogen Bonding in the Benzyl Mercaptan Dimer. Symmetry, 13(11), 2022. https://doi.org/10.3390/sym13112022