
Symmetric and Asymmetric Synapses Driving Neurodegenerative Disorders

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Ischemic Stroke
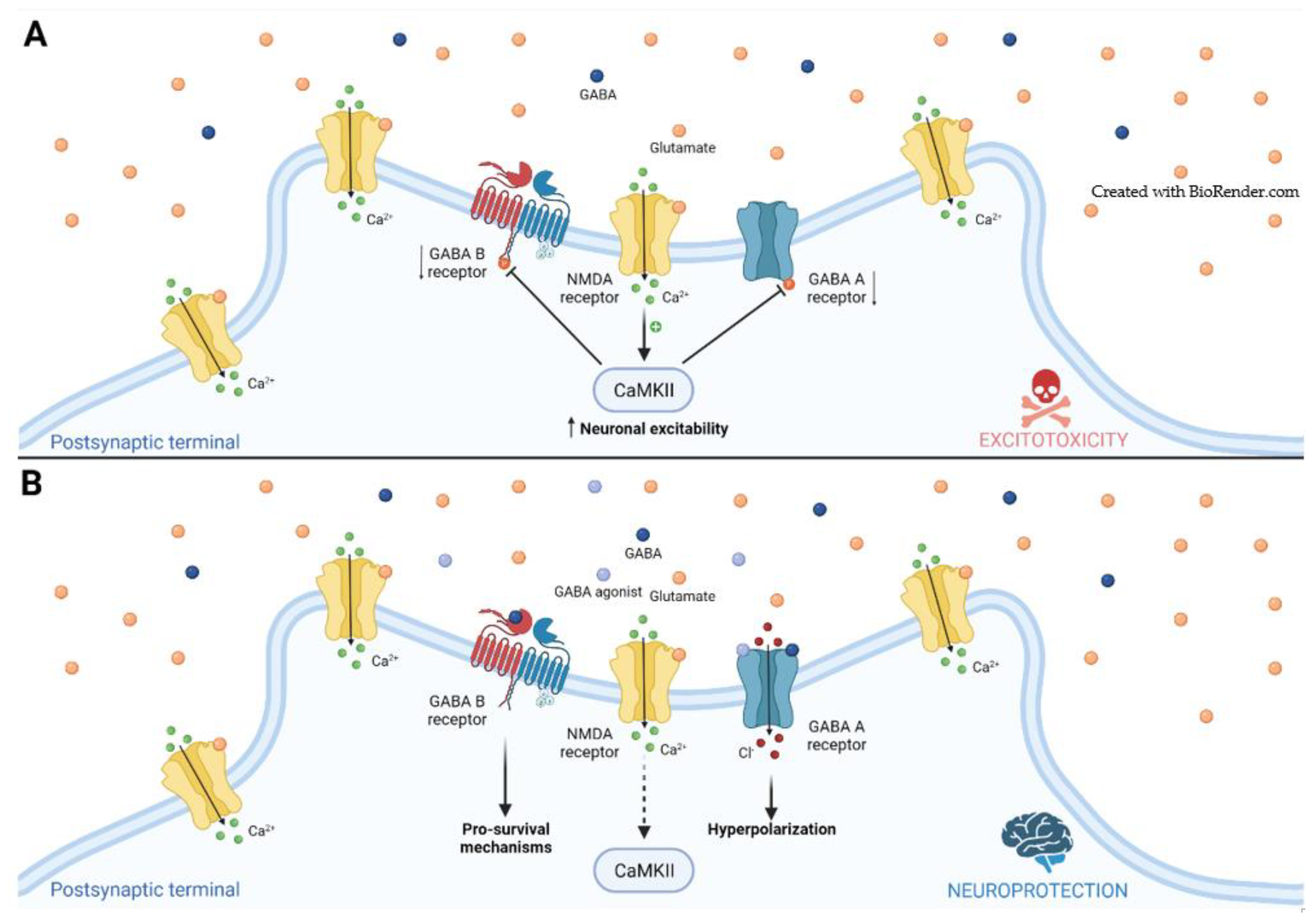
2.1. GABA Receptors
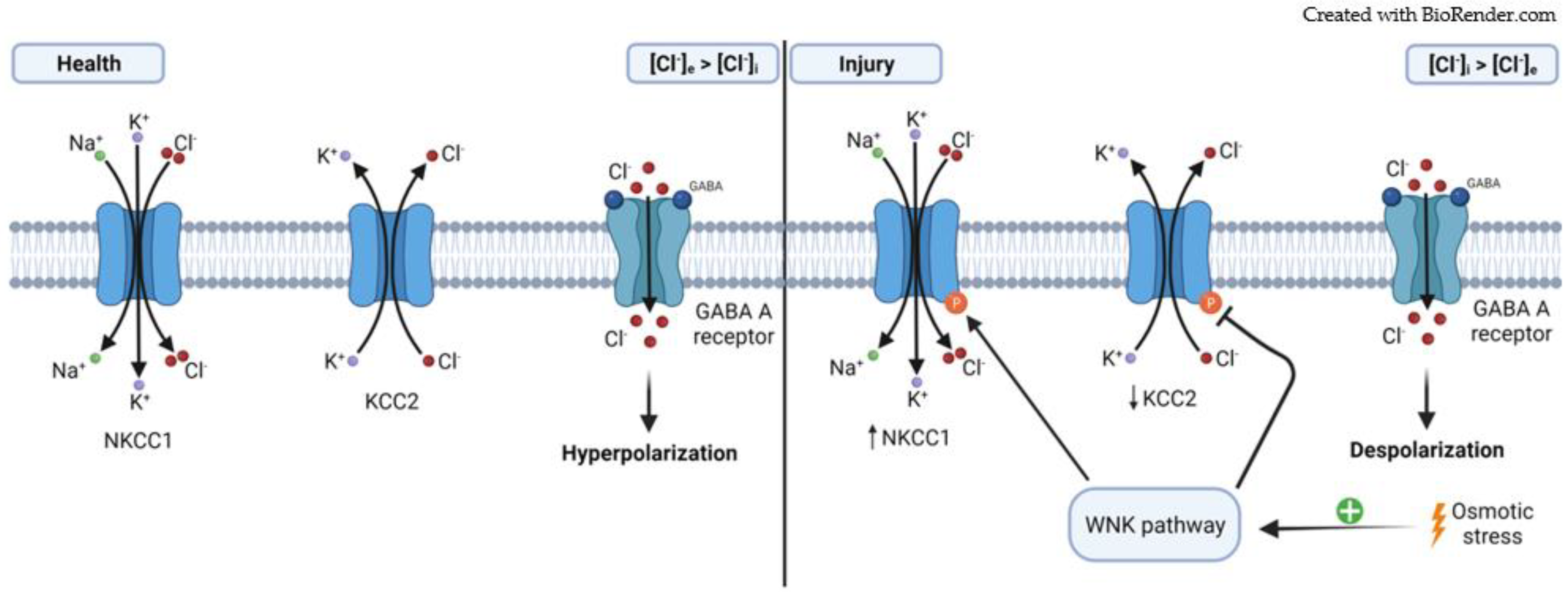
2.2. Cation–Chloride Cotransporters
3. Alzheimer’s Disease
3.1. Physiological Tau in Synapses
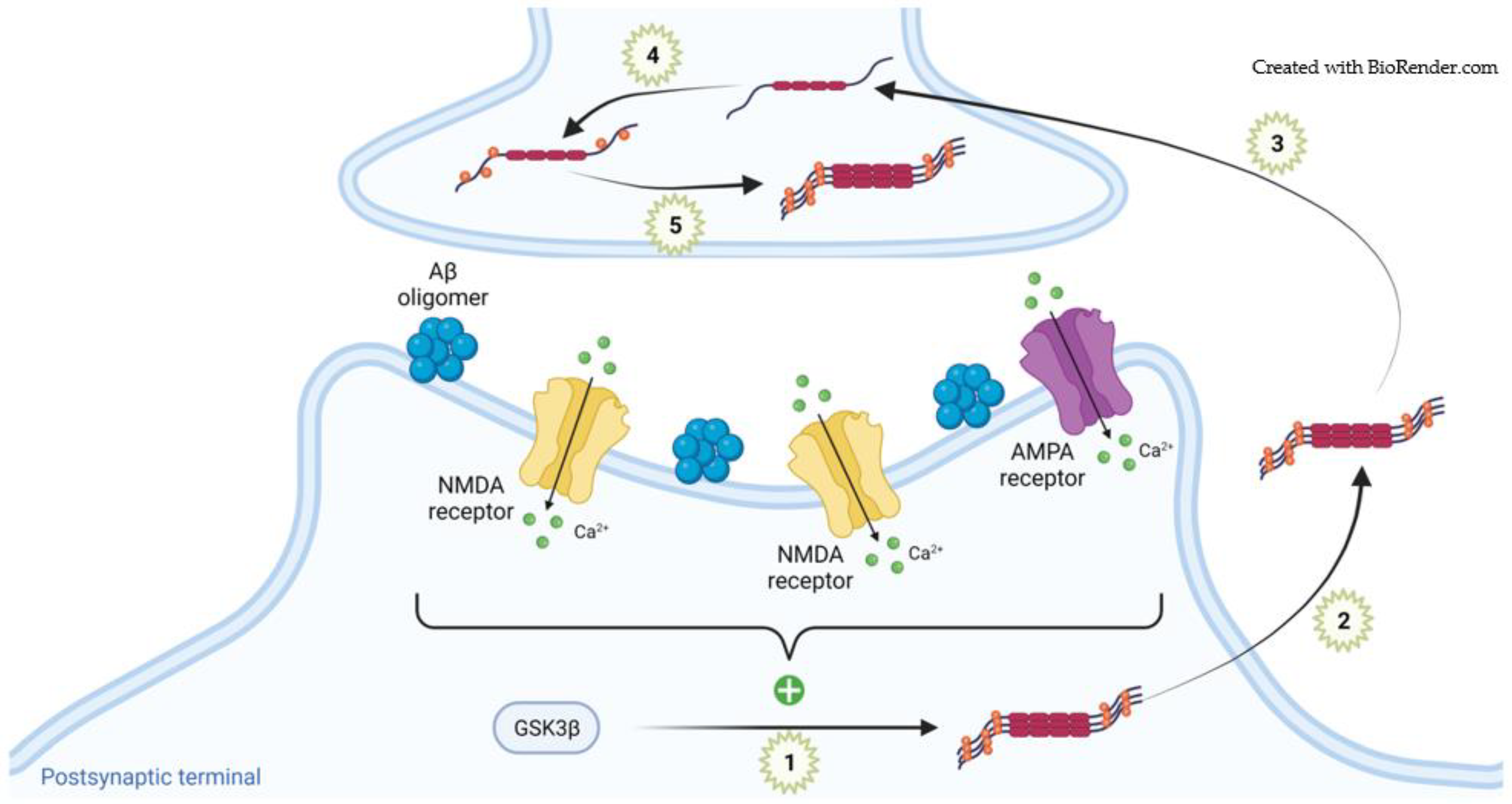
3.2. Pathogenic Tau in Synapses
4. Parkinson’s Disease
4.1. Symmetric Synapses
4.1.1. The Striatum—STR
4.1.2. The External Segment of the Globus Pallidus—GPe
4.1.3. The Substantia Nigra Pars Reticulate—SNr
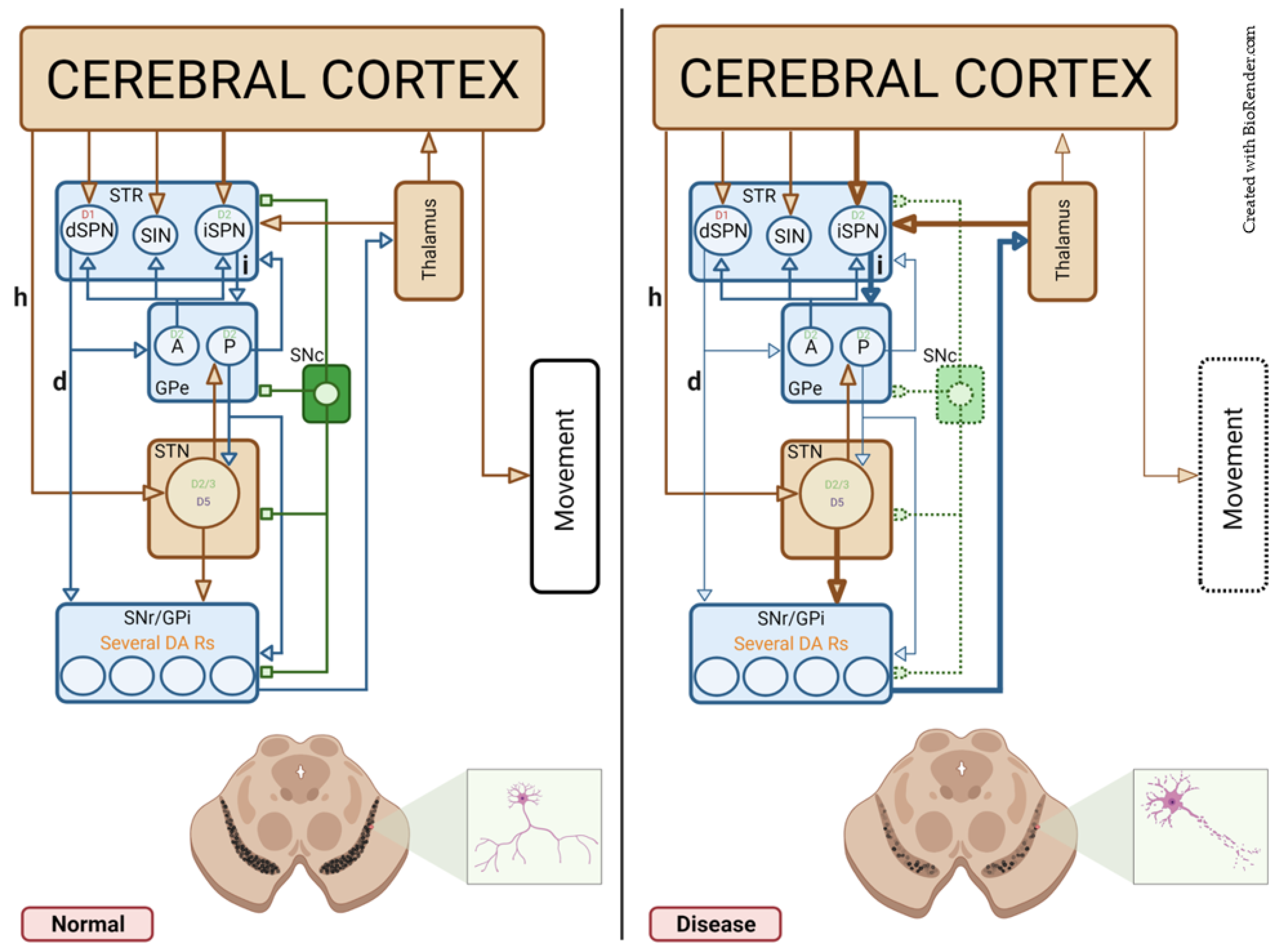
4.1.4. Healthy and Pathological DA Modulation in Symmetric Synapses
4.2. Asymmetric Synapses
4.2.1. The STR
4.2.2. The Subthalamic Nucleus—STN
4.2.3. Healthy and Pathological DA Modulation in Asymmetric Synapses
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gray, E.G. Axo-somatic and axo-dendritic synapses of the cerebral cortex: An electron microscope study. J. Anat. 1959, 93, 420–433. [Google Scholar] [PubMed]
- Colonnier, M. Synaptic patterns on different cell types in the different laminae of the cat visual cortex. An electron microscope study. Brain Res. 1968, 9, 268–287. [Google Scholar] [CrossRef]
- Klemann, C.J.; Roubos, E.W. The gray area between synapse structure and function-Gray’s synapse types I and II revisited. Synapse 2011, 65, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Siekevitz, P. The postsynaptic density: A possible role in long-lasting effects in the central nervous system. Proc. Natl. Acad. Sci. USA 1985, 82, 3494–3498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parato, J.; Bartolini, F. The microtubule cytoskeleton at the synapse. Neurosci. Lett. 2021, 753, 135850. [Google Scholar] [CrossRef] [PubMed]
- Moraes, B.J.; Coelho, P.; Fão, L.; Ferreira, I.L.; Rego, A.C. Modified Glutamatergic Postsynapse in Neurodegenerative Disorders. Neuroscience 2021, 454, 116–139. [Google Scholar] [CrossRef] [PubMed]
- Smart, T.G.; Paoletti, P. Synaptic neurotransmitter-gated receptors. Cold Spring Harb. Perspect. Biol. 2012, 4, a009662. [Google Scholar] [CrossRef]
- Sheng, M.; Kim, E. The postsynaptic organization of synapses. Cold Spring Harb. Perspect. Biol. 2011, 3, a005678. [Google Scholar] [CrossRef] [Green Version]
- Rodzli, N.A.; Lockhart-Cairns, M.P.; Levy, C.W.; Chipperfield, J.; Bird, L.; Baldock, C.; Prince, S.M. The Dual PDZ Domain from Postsynaptic Density Protein 95 Forms a Scaffold with Peptide Ligand. Biophys J. 2020, 119, 667–689. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Sheng, M. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 2004, 5, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Luscher, B.; Fuchs, T.; Kilpatrick, C.L. GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 2011, 70, 385–409. [Google Scholar] [CrossRef] [Green Version]
- Tyagarajan, S.K.; Fritschy, J.M. Gephyrin: A master regulator of neuronal function? Nat. Rev. Neurosci. 2014, 15, 141–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzarelli, R.; Griguoli, M.; Zacchi, P.; Petrini, E.M.; Barberis, A.; Cattaneo, A.; Cherubini, E. Tuning GABAergic Inhibition: Gephyrin Molecular Organization and Functions. Neuroscience 2020, 439, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Abraham, W.C.; Jones, O.D.; Glanzman, D.L. Is plasticity of synapses the mechanism of long-term memory storage? NPJ Sci. Learn. 2019, 4, 9. [Google Scholar] [CrossRef]
- Madadi Asl, M.; Vahabie, A.H.; Valizadeh, A. Dopaminergic Modulation of Synaptic Plasticity, Its Role in Neuropsychiatric Disorders, and Its Computational Modeling. Basic Clin. Neurosci. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Malenka, R.C. Synaptic plasticity in the hippocampus: LTP and LTD. Cell 1994, 78, 535–538. [Google Scholar] [CrossRef]
- Bloodgood, B.L.; Giessel, A.J.; Sabatini, B.L. Biphasic synaptic Ca influx arising from compartmentalized electrical signals in dendritic spines. PLoS Biol. 2009, 7, e1000190. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.E.; Chiu, C.Q.; Carroll, R.C. Long-term plasticity at inhibitory synapses. Curr. Opin. Neurobiol. 2011, 21, 328–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadim, F.; Bucher, D. Neuromodulation of neurons and synapses. Curr. Opin. Neurobiol. 2014, 29, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinta, S.J.; Andersen, J.K. Dopaminergic neurons. Int. J. Biochem. Cell Biol. 2005, 37, 942–946. [Google Scholar] [CrossRef]
- Tritsch, N.X.; Sabatini, B.L. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 2012, 76, 33–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedrosa, V.; Clopath, C. The Role of Neuromodulators in Cortical Plasticity. A Computational Perspective. Front Synaptic. Neurosci. 2017, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Béjot, Y.; Bailly, H.; Durier, J.; Giroud, M. Epidemiology of stroke in Europe and trends for the 21st century. Presse Med. 2016, 45, e391–e398. [Google Scholar] [CrossRef] [PubMed]
- Hay, B.; Yi, S.; Patel, P. Cerebral Ischemia. In Gupta and Gelb’s Essentials of Neuroanesthesia and Neurointensive Care, 2nd ed.; Gupta, A., Gelb, A., Duane, D., Adapa, R., Eds.; Cambridge University Press: Cambridge, UK, 2018; pp. 39–47. [Google Scholar] [CrossRef]
- Martín-Aragón Baudel, M.A.; Poole, A.V.; Darlison, M.G. Chloride co-transporters as possible therapeutic targets for stroke. J. Neurochem. 2017, 140, 195–209. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.A.; Amruta, N.; Pinteaux, E.; Bix, G.J. Neurogenesis After Stroke: A Therapeutic Perspective. Transl. Stroke Res. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Clarkson, A.N.; Huang, B.S.; Macisaac, S.E.; Mody, I.; Carmichael, S.T. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 2010, 468, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiu, T.; Farzampour, Z.; Paz, J.T.; Wang, E.H.; Badgely, C.; Olson, A.; Micheva, K.D.; Wang, G.; Lemmens, R.; Tran, K.V.; et al. Enhanced phasic GABA inhibition during the repair phase of stroke: A novel therapeutic target. Brain 2016, 139, 468–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mele, M.; Costa, R.O.; Duarte, C.B. Alterations in GABAA-Receptor Trafficking and Synaptic Dysfunction in Brain Disorders. Front Cell Neurosci. 2019, 13, 77. [Google Scholar] [CrossRef]
- Tanaka, E.; Ogawa, Y.; Fujii, R.; Shimonaka, T.; Sato, Y.; Hamazaki, T.; Nagamura-Inoue, T.; Shintaku, H.; Tsuji, M. Metabolomic analysis and mass spectrometry imaging after neonatal stroke and cell therapies in mouse brains. Sci. Rep. 2020, 10, 21881. [Google Scholar] [CrossRef] [PubMed]
- Mele, M.; Leal, G.; Duarte, C.B. Role of GABAA R trafficking in the plasticity of inhibitory synapses. J. Neurochem. 2016, 139, 997–1018. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, S.T. Brain excitability in stroke: The yin and yang of stroke progression. Arch. Neurol. 2012, 69, 161–167. [Google Scholar] [CrossRef]
- Alia, C.; Spalletti, C.; Lai, S.; Panarese, A.; Micera, S.; Caleo, M. Reducing GABAA-mediated inhibition improves forelimb motor function after focal cortical stroke in mice. Sci. Rep. 2016, 6, 37823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemann, G.; Redecker, C.; Neumann-Haefelin, T.; Freund, H.J.; Witte, O.W. Increased long-term potentiation in the surround of experimentally induced focal cortical infarction. Ann. Neurol. 1998, 44, 255–258. [Google Scholar] [CrossRef]
- Cramer, S.C. Repairing the human brain after stroke: I. Mechanisms of spontaneous recovery. Ann. Neurol. 2008, 63, 272–287. [Google Scholar] [CrossRef]
- Wang, Y.C.; Dzyubenko, E.; Sanchez-Mendoza, E.H.; Sardari, M.; Silva de Carvalho, T.; Doeppner, T.R.; Kaltwasser, B.; Machado, P.; Kleinschnitz, C.; Bassetti, C.L.; et al. Postacute Delivery of GABAA α5 Antagonist Promotes Postischemic Neurological Recovery and Peri-infarct Brain Remodeling. Stroke 2018, 49, 2495–2503. [Google Scholar] [CrossRef] [PubMed]
- van Nieuwenhuijzen, P.S.; Parker, K.; Liao, V.; Houlton, J.; Kim, H.L.; Johnston, G.A.R.; Hanrahan, J.R.; Chebib, M.; Clarkson, A.N. Targeting GABAC Receptors Improves Post-Stroke Motor Recovery. Brain Sci. 2021, 11, 315. [Google Scholar] [CrossRef]
- Chalifoux, J.R.; Carter, A.G. GABAB receptor modulation of synaptic function. Curr. Opin. Neurobiol. 2011, 21, 339–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mele, M.; Ribeiro, L.; Inácio, A.R.; Wieloch, T.; Duarte, C.B. GABA(A) receptor dephosphorylation followed by internalization is coupled to neuronal death in in vitro ischemia. Neurobiol. Dis. 2014, 65, 220–232. [Google Scholar] [CrossRef] [Green Version]
- Hausrat, T.J.; Muhia, M.; Gerrow, K.; Thomas, P.; Hirdes, W.; Tsukita, S.; Heisler, F.F.; Herich, L.; Dubroqua, S.; Breiden, P.; et al. Radixin regulates synaptic GABAA receptor density and is essential for reversal learning and short-term memory. Nat. Commun. 2015, 6, 6872. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.T.; Mele, M.; Baptista, M.S.; Gomes, J.R.; Ruscher, K.; Nobre, R.J.; de Almeida, L.P.; Wieloch, T.; Duarte, C.B. Gephyrin Cleavage in In Vitro Brain Ischemia Decreases GABAA Receptor Clustering and Contributes to Neuronal Death. Mol. Neurobiol. 2016, 53, 3513–3527. [Google Scholar] [CrossRef] [PubMed]
- Benke, D.; Balakrishnan, K.; Zemoura, K. Regulation of cell surface GABA(B) receptors: Contribution to synaptic plasticity in neurological diseases. Adv. Pharm. 2015, 73, 41–70. [Google Scholar] [CrossRef]
- Huang, L.; Li, Q.; Wen, R.; Yu, Z.; Li, N.; Ma, L.; Feng, W. Rho-kinase inhibitor prevents acute injury against transient focal cerebral ischemia by enhancing the expression and function of GABA receptors in rats. Eur. J. Pharm. 2017, 797, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Zagrean, A.M.; Grigoras, I.F.; Iesanu, M.I.; Ionescu, R.B.; Chitimus, D.M.; Haret, R.M.; Ianosi, B.; Ceanga, M.; Zagrean, L. Neuronal Transmembrane Chloride Transport Has a Time-Dependent Influence on Survival of Hippocampal Cultures to Oxygen-Glucose Deprivation. Brain. Sci. 2019, 9, 360. [Google Scholar] [CrossRef] [Green Version]
- Zemoura, K.; Balakrishnan, K.; Grampp, T.; Benke, D. Ca2+/Calmodulin-Dependent Protein Kinase II (CaMKII) β-Dependent Phosphorylation of GABAB1 Triggers Lysosomal Degradation of GABAB Receptors via Mind Bomb-2 (MIB2)-Mediated Lys-63-Linked Ubiquitination. Mol. Neurobiol. 2019, 56, 1293–1309. [Google Scholar] [CrossRef] [Green Version]
- García-Berrocoso, T.; Llombart, V.; Colàs-Campàs, L.; Hainard, A.; Licker, V.; Penalba, A.; Ramiro, L.; Simats, A.; Bustamante, A.; Martínez-Saez, E.; et al. Single Cell Immuno-Laser Microdissection Coupled to Label-Free Proteomics to Reveal the Proteotypes of Human Brain Cells After Ischemia. Mol. Cell. Proteom. 2018, 17, 175–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramiro, L.; García-Berrocoso, T.; Briansó, F.; Goicoechea, L.; Simats, A.; Llombart, V.; Gonzalo, R.; Hainard, A.; Martínez-Saez, E.; Canals, F.; et al. Integrative Multi-omics Analysis to Characterize Human Brain Ischemia. Mol. Neurobiol. 2021, 58, 4107–4121. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Leone, G.; Saulle, E.; Pisani, F.; Bernardi, G.; Calabresi, P. Coactivation of GABA(A) and GABA(B) receptor results in neuroprotection during in vitro ischemia. Stroke 2004, 35, 596–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.J.; Ye, Z.; Yu, H.; Chen, Y.; Wang, Y.W.; Zhao, J.H.; Sun, J.F.; Xu, L.M. Shrm4 contributes to autophagy inhibition and neuroprotection following ischemic stroke by mediating GABAB receptor activation. FASEB J. 2020, 34, 15837–15848. [Google Scholar] [CrossRef]
- Lyden, P.D.; Hedges, B. Protective effect of synaptic inhibition during cerebral ischemia in rats and rabbits. Stroke 1992, 23, 1463–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shames, J.L.; Ring, H. Transient reversal of anoxic brain injury-related minimally conscious state after zolpidem administration: A case report. Arch. Phys. Med. Rehabil. 2008, 89, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.D.; Yamawaki, N.; Fisher, A.E.; Clauss, R.P.; Woodhall, G.L.; Stanford, I.M. GABA(A) alpha-1 subunit mediated desynchronization of elevated low frequency oscillations alleviates specific dysfunction in stroke--a case report. Clin. Neurophysiol. 2010, 121, 549–555. [Google Scholar] [CrossRef]
- Hummel, F.C.; Cohen, L.G. Non-invasive brain stimulation: A new strategy to improve neurorehabilitation after stroke? Lancet Neurol. 2006, 5, 708–712. [Google Scholar] [CrossRef]
- Kokinovic, B.; Medini, P. Loss of GABAB -mediated interhemispheric synaptic inhibition in stroke periphery. J. Physiol. 2018, 596, 1949–1964. [Google Scholar] [CrossRef] [Green Version]
- Cirillo, J.; Mooney, R.A.; Ackerley, S.J.; Barber, P.A.; Borges, V.M.; Clarkson, A.N.; Mangold, C.; Ren, A.; Smith, M.C.; Stinear, C.M.; et al. Neurochemical balance and inhibition at the subacute stage after stroke. J. Neurophysiol. 2020, 123, 1775–1790. [Google Scholar] [CrossRef]
- Schulte, J.T.; Wierenga, C.J.; Bruining, H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev. 2018, 90, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Romaus-Sanjurjo, D.; Rodicio, M.C.; Barreiro-Iglesias, A. Gamma-aminobutyric acid (GABA) promotes recovery from spinal cord injury in lampreys: Role of GABA receptors and perspective on the translation to mammals. Neural Regen. Res. 2019, 14, 1695–1696. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 2017, 40, 536–554. [Google Scholar] [CrossRef]
- Wang, G.; Huang, H.; He, Y.; Ruan, L.; Huang, J. Bumetanide protects focal cerebral ischemia-reperfusion injury in rat. Int. J. Clin. Exp. Pathol. 2014, 7, 1487–1494. [Google Scholar] [PubMed]
- Zhang, J.; Gao, G.; Begum, G.; Wang, J.; Khanna, A.R.; Shmukler, B.E.; Daubner, G.M.; de Los Heros, P.; Davies, P.; Varghese, J.; et al. Functional kinomics establishes a critical node of volume-sensitive cation-Cl− cotransporter regulation in the mammalian brain. Sci. Rep. 2016, 6, 35986. [Google Scholar] [CrossRef]
- Josiah, S.S.; Meor Azlan, N.F.; Zhang, J. Targeting the WNK-SPAK/OSR1 Pathway and Cation-Chloride Cotransporters for the Therapy of Stroke. Int. J. Mol. Sci. 2021, 22, 1232. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Dempsey, R.J.; Flemmer, A.; Forbush, B.; Sun, D. Inhibition of Na(+)-K(+)-Cl(-) cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 2003, 961, 22–31. [Google Scholar] [CrossRef]
- Jaenisch, N.; Witte, O.W.; Frahm, C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke 2010, 41, e151–e159. [Google Scholar] [CrossRef]
- Begum, G.; Yuan, H.; Kahle, K.T.; Li, L.; Wang, S.; Shi, Y.; Shmukler, B.E.; Yang, S.S.; Lin, S.H.; Alper, S.L.; et al. Inhibition of WNK3 Kinase Signaling Reduces Brain Damage and Accelerates Neurological Recovery After Stroke. Stroke 2015, 46, 1956–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, X.P.; Wang, H.B.; Cheng, X.; Yang, L.; Sun, X.Y.; Qu, H.L.; Zhao, S.S.; Zhou, Z.K.; Liu, T.T.; Xiao, T.; et al. Inhibition of Nkcc1 promotes axonal growth and motor recovery in ischemic rats. Neuroscience 2017, 365, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Glykys, J.; Dzhala, V.; Egawa, K.; Kahle, K.T.; Delpire, E.; Staley, K. Chloride Dysregulation, Seizures, and Cerebral Edema: A Relationship with Therapeutic Potential. Trends Neurosci. 2017, 40, 276–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Mu, X.; Wang, H.; Song, C.; Ma, W.; Jolkkonen, J.; Zhao, C. Chloride Co-transporter NKCC1 Inhibitor Bumetanide Enhances Neurogenesis and Behavioral Recovery in Rats After Experimental Stroke. Mol. Neurobiol. 2017, 54, 2406–2414. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Song, S.; Banerjee, S.; Jiang, T.; Zhang, J.; Kahle, K.T.; Sun, D.; Zhang, Z. The WNK-SPAK/OSR1 Kinases and the Cation-Chloride Cotransporters as Therapeutic Targets for Neurological Diseases. Aging Dis. 2019, 10, 626–636. [Google Scholar] [CrossRef] [Green Version]
- Bhuiyan, M.I.H.; Song, S.; Yuan, H.; Begum, G.; Kofler, J.; Kahle, K.T.; Yang, S.S.; Lin, S.H.; Alper, S.L.; Subramanya, A.R.; et al. WNK-Cab39-NKCC1 signaling increases the susceptibility to ischemic brain damage in hypertensive rats. J. Cereb. Blood Flow Metab. 2017, 37, 2780–2794. [Google Scholar] [CrossRef]
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab. 2017, 25, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Bhuiyan, M.I.H.; Zhang, T.; Karimy, J.K.; Wu, Z.; Fiesler, V.M.; Zhang, J.; Huang, H.; Hasan, M.N.; Skrzypiec, A.E.; et al. Modulation of brain cation-Cl− cotransport via the SPAK kinase inhibitor ZT-1a. Nat. Commun. 2020, 11, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pin-Barre, C.; Constans, A.; Brisswalter, J.; Pellegrino, C.; Laurin, J. Effects of High-Versus Moderate-Intensity Training on Neuroplasticity and Functional Recovery After Focal Ischemia. Stroke 2017, 48, 2855–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khirug, S.; Soni, S.; Saez Garcia, M.; Tessier, M.; Zhou, L.; Kulesskaya, N.; Rauvala, H.; Lindholm, D.; Ludwig, A.; Molinari, F.; et al. Protective Role of Low Ethanol Administration Following Ischemic Stroke via Recovery of KCC2 and p75NTR Expression. Mol. Neurobiol. 2021, 58, 1145–1161. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 369, 20130144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan, P.; Piers, T.; Yi, J.H.; Kim, D.H.; Huh, S.; Park, S.J.; Ryu, J.H.; Whitcomb, D.J.; Cho, K. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J. Neurosci. 2015, 35, 4804–4812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejanovic, B.; Huntley, M.A.; De Mazière, A.; Meilandt, W.J.; Wu, T.; Srinivasan, K.; Jiang, Z.; Gandham, V.; Friedman, B.A.; Ngu, H.; et al. Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 2018, 100, 1322–1336.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijalkov, M.; Volpe, G.; Fernaud-Espinosa, I.; DeFelipe, J.; Pereira, J.B.; Merino-Serrais, P. Dendritic spines are lost in clusters in Alzheimer’s disease. Sci. Rep. 2021, 11, 12350. [Google Scholar] [CrossRef]
- Sun, X.Y.; Li, L.J.; Dong, Q.X.; Zhu, J.; Huang, Y.R.; Hou, S.J.; Yu, X.L.; Liu, R.T. Rutin prevents tau pathology and neuroinflammation in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2021, 18, 131. [Google Scholar] [CrossRef] [PubMed]
- Biundo, F.; Del Prete, D.; Zhang, H.; Arancio, O.; D’Adamio, L. A role for tau in learning, memory and synaptic plasticity. Sci. Rep. 2018, 8, 3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisman, J.; Buzsáki, G.; Eichenbaum, H.; Nadel, L.; Ranganath, C.; Redish, A.D. Viewpoints: How the hippocampus contributes to memory, navigation and cognition. Nat. Neurosci. 2017, 20, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Kosik, K.S.; Finch, E.A. MAP2 and tau segregate into dendritic and axonal domains after the elaboration of morphologically distinct neurites: An immunocytochemical study of cultured rat cerebrum. J. Neurosci. 1987, 7, 3142–3153. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondragón-Rodríguez, S.; Trillaud-Doppia, E.; Dudilot, A.; Bourgeois, C.; Lauzon, M.; Leclerc, N.; Boehm, J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 2012, 287, 32040–32053. [Google Scholar] [CrossRef] [Green Version]
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Götz, J. Somatodendritic accumulation of Tau in Alzheimer’s disease is promoted by Fyn-mediated local protein translation. EMBO J. 2017, 36, 3120–3138. [Google Scholar] [CrossRef]
- Li, X.; Kumar, Y.; Zempel, H.; Mandelkow, E.M.; Biernat, J.; Mandelkow, E. Novel diffusion barrier for axonal retention of Tau in neurons and its failure in neurodegeneration. EMBO J. 2011, 30, 4825–4837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frandemiche, M.L.; De Seranno, S.; Rush, T.; Borel, E.; Elie, A.; Arnal, I.; Lanté, F.; Buisson, A. Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J. Neurosci. 2014, 34, 6084–6097. [Google Scholar] [CrossRef]
- de Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calafate, S.; Buist, A.; Miskiewicz, K.; Vijayan, V.; Daneels, G.; de Strooper, B.; de Wit, J.; Verstreken, P.; Moechars, D. Synaptic Contacts Enhance Cell-to-Cell Tau Pathology Propagation. Cell Rep. 2015, 11, 1176–1183. [Google Scholar] [CrossRef] [Green Version]
- DeVos, S.L.; Corjuc, B.T.; Oakley, D.H.; Nobuhara, C.K.; Bannon, R.N.; Chase, A.; Commins, C.; Gonzalez, J.A.; Dooley, P.M.; Frosch, M.P.; et al. Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain. Front. Neurosci. 2018, 12, 267. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Zhou, Z.; Zhang, L.; Wang, Y.; Zhang, Y.W.; Zhong, M.; Xu, S.C.; Chen, C.H.; Li, L.; Yu, Z.P. Tau protein is involved in morphological plasticity in hippocampal neurons in response to BDNF. Neurochem. Int. 2012, 60, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Voelzmann, A.; Okenve-Ramos, P.; Qu, Y.; Chojnowska-Monga, M.; Del Caño-Espinel, M.; Prokop, A.; Sanchez-Soriano, N. Tau and spectraplakins promote synapse formation and maintenance through Jun kinase and neuronal trafficking. Elife 2016, 5, e14694. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron 2018, 99, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Christensen, N.R.; Čalyševa, J.; Fernandes, E.F.A.; Lüchow, S.; Clemmensen, L.S.; Haugaard-Kedström, L.M.; Strømgaard, K. PDZ Domains as Drug Targets. Adv. Ther. 2019, 2, 1800143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, L.; Hochrainer, K.; Hattori, Y.; Ahn, S.J.; Anfray, A.; Wang, G.; Uekawa, K.; Seo, J.; Palfini, V.; Blanco, I.; et al. Tau induces PSD95-neuronal NOS uncoupling and neurovascular dysfunction independent of neurodegeneration. Nat. Neurosci. 2020, 23, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Shipton, O.A.; Leitz, J.R.; Dworzak, J.; Acton, C.E.; Tunbridge, E.M.; Denk, F.; Dawson, H.N.; Vitek, M.P.; Wade-Martins, R.; Paulsen, O.; et al. Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J. Neurosci. 2011, 31, 1688–1692. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Bhattacharyya, S.; Ma, C.; Morishita, W.; Fang, S.; Rizo, J.; Malenka, R.C. Calcium binding to PICK1 is essential for the intracellular retention of AMPA receptors underlying long-term depression. J. Neurosci. 2010, 30, 16437–16452. [Google Scholar] [CrossRef] [Green Version]
- Yagishita, S.; Murayama, M.; Ebihara, T.; Maruyama, K.; Takashima, A. Glycogen Synthase Kinase 3β-mediated Phosphorylation in the Most C-terminal Region of Protein Interacting with C Kinase 1 (PICK1) Regulates the Binding of PICK1 to Glutamate Receptor Subunit GluA2. J. Biol. Chem. 2015, 290, 29438–29448. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Kimura, T. Microtubule-associated tau contributes to intra-dendritic trafficking of AMPA receptors in multiple ways. Neurosci. Lett. 2017, 653, 276–282. [Google Scholar] [CrossRef]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 2021, 11, 595532. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Pires, G.; MacMurray, C.; Askenazi, M.; Nayak, S.; Bourdon, M.; Safar, J.; Ueberheide, B.; Wisniewski, T. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain 2020, 143, 2803–2817. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.S.; Witton, J.; Johnson, J.D.; Ahmed, Z.; Ward, M.; Randall, A.D.; Hutton, M.L.; Isaac, J.T.; O’Neill, M.J.; Ashby, M.C. Altered Synapse Stability in the Early Stages of Tauopathy. Cell Rep. 2017, 18, 3063–3068. [Google Scholar] [CrossRef] [Green Version]
- Torres, A.K.; Jara, C.; Olesen, M.A.; Tapia-Rojas, C. Pathologically phosphorylated tau at S396/404 (PHF-1) is accumulated inside of hippocampal synaptic mitochondria of aged Wild-type mice. Sci. Rep. 2021, 11, 4448. [Google Scholar] [CrossRef]
- Pallas-Bazarra, N.; Draffin, J.; Cuadros, R.; Antonio Esteban, J.; Avila, J. Tau is required for the function of extrasynaptic NMDA receptors. Sci. Rep. 2019, 9, 9116. [Google Scholar] [CrossRef]
- Gladding, C.M.; Raymond, L.A. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol. Cell Neurosci. 2011, 48, 308–320. [Google Scholar] [CrossRef]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef] [Green Version]
- Jo, J.; Whitcomb, D.J.; Olsen, K.M.; Kerrigan, T.L.; Lo, S.C.; Bru-Mercier, G.; Dickinson, B.; Scullion, S.; Sheng, M.; Collingridge, G.; et al. Aβ(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat. Neurosci. 2011, 14, 545–547. [Google Scholar] [CrossRef]
- Zhao, X.; Kotilinek, L.A.; Smith, B.; Hlynialuk, C.; Zahs, K.; Ramsden, M.; Cleary, J.; Ashe, K.H. Caspase-2 cleavage of tau reversibly impairs memory. Nat. Med. 2016, 22, 1268–1276. [Google Scholar] [CrossRef]
- Regan, P.; Mitchell, S.J.; Kim, S.C.; Lee, Y.; Yi, J.H.; Barbati, S.A.; Shaw, C.; Cho, K. Regulation of synapse weakening through interactions of the microtubule associated protein tau with PACSIN1. J. Neurosci. 2021, 41, 7162–7170. [Google Scholar] [CrossRef]
- Zempel, H.; Dennissen, F.J.A.; Kumar, Y.; Luedtke, J.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture. J. Biol. Chem. 2017, 292, 12192–12207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, M.; Gopinath, P.; Govindaraju, T. Role of Post-translational Modifications in Alzheimer’s Disease. Chembiochem 2020, 21, 1052–1079. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef]
- Marzi, S.J.; Leung, S.K.; Ribarska, T.; Hannon, E.; Smith, A.R.; Pishva, E.; Poschmann, J.; Moore, K.; Troakes, C.; Al-Sarraj, S.; et al. A histone acetylome-wide association study of Alzheimer’s disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat. Neurosci. 2018, 21, 1618–1627. [Google Scholar] [CrossRef] [Green Version]
- Klein, H.U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 2019, 22, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Lester, E.; Ooi, F.K.; Bakkar, N.; Ayers, J.; Woerman, A.L.; Wheeler, J.; Bowser, R.; Carlson, G.A.; Prusiner, S.B.; Parker, R. Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron 2021, 109, 1675–1691.e9. [Google Scholar] [CrossRef]
- Regan, M.C.; Grant, T.; McDaniel, M.J.; Karakas, E.; Zhang, J.; Traynelis, S.F.; Grigorieff, N.; Furukawa, H. Structural Mechanism of Functional Modulation by Gene Splicing in NMDA Receptors. Neuron 2018, 98, 521–529.e3. [Google Scholar] [CrossRef] [Green Version]
- d’Errico, P.; Meyer-Luehmann, M. Mechanisms of Pathogenic Tau and Aβ Protein Spreading in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 265. [Google Scholar] [CrossRef]
- Fani, G.; Mannini, B.; Vecchi, G.; Cascella, R.; Cecchi, C.; Dobson, C.M.; Vendruscolo, M.; Chiti, F. Aβ Oligomers Dysregulate Calcium Homeostasis by Mechanosensitive Activation of AMPA and NMDA Receptors. ACS Chem. Neurosci. 2021, 12, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Sayas, C.L.; Ávila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.C.; Teravskis, P.J.; Dummer, B.W.; Zhao, X.; Huganir, R.L.; Liao, D. Tau phosphorylation and tau mislocalization mediate soluble Aβ oligomer-induced AMPA glutamate receptor signaling deficits. Eur. J. Neurosci. 2014, 39, 1214–1224. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.Y.; Kuo, P.C.; Wang, Y.T.; Lin, H.T.; Roe, A.D.; Wang, B.Y.; Han, C.L.; Hyman, B.T.; Chen, Y.J.; Tai, H.C. β-Amyloid Induces Pathology-Related Patterns of Tau Hyperphosphorylation at Synaptic Terminals. J. Neuropathol. Exp. Neurol. 2018, 77, 814–826. [Google Scholar] [CrossRef]
- Mirbaha, H.; Chen, D.; Morazova, O.A.; Ruff, K.M.; Sharma, A.M.; Liu, X.; Goodarzi, M.; Pappu, R.V.; Colby, D.W.; Mirzaei, H.; et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife 2018, 7, e36584. [Google Scholar] [CrossRef]
- Furman, J.L.; Vaquer-Alicea, J.; White, C.L., 3rd; Cairns, N.J.; Nelson, P.T.; Diamond, M.I. Widespread tau seeding activity at early Braak stages. Acta Neuropathol. 2017, 133, 91–100. [Google Scholar] [CrossRef]
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, D.L.; Corjuc, B.T.; et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 2020, 26, 1256–1263. [Google Scholar] [CrossRef]
- Wegmann, S.; Bennett, R.E.; Delorme, L.; Robbins, A.B.; Hu, M.; McKenzie, D.; Kirk, M.J.; Schiantarelli, J.; Tunio, N.; Amaral, A.C.; et al. Experimental evidence for the age dependence of tau protein spread in the brain. Sci. Adv. 2019, 5, eaaw6404. [Google Scholar] [CrossRef] [Green Version]
- Swanson, E.; Breckenridge, L.; McMahon, L.; Som, S.; McConnell, I.; Bloom, G.S. Extracellular Tau Oligomers Induce Invasion of Endogenous Tau into the Somatodendritic Compartment and Axonal Transport Dysfunction. J. Alzheimers Dis. 2017, 58, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Ondrejcak, T.; Klyubin, I.; Corbett, G.T.; Fraser, G.; Hong, W.; Mably, A.J.; Gardener, M.; Hammersley, J.; Perkinton, M.S.; Billinton, A.; et al. Cellular Prion Protein Mediates the Disruption of Hippocampal Synaptic Plasticity by Soluble Tau In Vivo. J. Neurosci. 2018, 38, 10595–10606. [Google Scholar] [CrossRef] [Green Version]
- Tracy, T.E.; Gan, L. Acetylated tau in Alzheimer’s disease: An instigator of synaptic dysfunction underlying memory loss: Increased levels of acetylated tau blocks the postsynaptic signaling required for plasticity and promotes memory deficits associated with tauopathy. Bioessays 2017, 39, 1600224. [Google Scholar] [CrossRef] [Green Version]
- Trzeciakiewicz, H.; Tseng, J.H.; Wander, C.M.; Madden, V.; Tripathy, A.; Yuan, C.X.; Cohen, T.J. A Dual Pathogenic Mechanism Links Tau Acetylation to Sporadic Tauopathy. Sci. Rep. 2017, 7, 44102. [Google Scholar] [CrossRef]
- Caballero, B.; Bourdenx, M.; Luengo, E.; Diaz, A.; Sohn, P.D.; Chen, X.; Wang, C.; Juste, Y.R.; Wegmann, S.; Patel, B.; et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat. Commun. 2021, 12, 2238. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef]
- Meder, D.; Herz, D.M.; Rowe, J.B.; Lehéricy, S.; Siebner, H.R. The role of dopamine in the brain- lessons learned from Parkinson’s disease. Neuroimage 2019, 190, 79–93. [Google Scholar] [CrossRef]
- Grillner, S.; Robertson, B. The Basal Ganglia Over 500 Million Years. Curr. Biol. 2016, 26, R1088–R1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, S.; Shen, W.; Graves, S.M.; Surmeier, D.J. Dopaminergic modulation of striatal function and Parkinson’s disease. J. Neural. Transm. (Vienna) 2019, 126, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Mallet, N.; Delgado, L.; Chazalon, M.; Miguelez, C.; Baufreton, J. Cellular and Synaptic Dysfunctions in Parkinson’s Disease: Stepping out of the Striatum. Cells 2019, 8, 1005. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.S.; Surmeier, D.J.; Yung, W.H. Striatal information signaling and integration in globus pallidus: Timing matters. Neurosignals 2005, 14, 281–289. [Google Scholar] [CrossRef]
- Hallworth, N.E.; Bevan, M.D. Globus pallidus neurons dynamically regulate the activity pattern of subthalamic nucleus neurons through the frequency-dependent activation of postsynaptic GABAA and GABAB receptors. J. Neurosci. 2005, 25, 6304–6315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevan, M.D.; Hallworth, N.E.; Baufreton, J. GABAergic control of the subthalamic nucleus. Prog. Brain Res. 2007, 160, 173–188. [Google Scholar] [CrossRef]
- Raju, D.V.; Ahern, T.H.; Shah, D.J.; Wright, T.M.; Standaert, D.G.; Hall, R.A.; Smith, Y. Differential synaptic plasticity of the corticostriatal and thalamostriatal systems in an MPTP-treated monkey model of parkinsonism. Eur. J. Neurosci. 2008, 27, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Tepper, J.M.; Koós, T.; Ibanez-Sandoval, O.; Tecuapetla, F.; Faust, T.W.; Assous, M. Heterogeneity and Diversity of Striatal GABAergic Interneurons: Update 2018. Front. Neuroanat. 2018, 12, 91. [Google Scholar] [CrossRef] [Green Version]
- Milardi, D.; Quartarone, A.; Bramanti, A.; Anastasi, G.; Bertino, S.; Basile, G.A.; Buonasera, P.; Pilone, G.; Celeste, G.; Rizzo, G.; et al. The Cortico-Basal Ganglia-Cerebellar Network: Past, Present and Future Perspectives. Front. Syst. Neurosci. 2019, 13, 61. [Google Scholar] [CrossRef] [PubMed]
- Suarez, L.M.; Solis, O.; Sanz-Magro, A.; Alberquilla, S.; Moratalla, R. Dopamine D1 Receptors Regulate Spines in Striatal Direct-Pathway and Indirect-Pathway Neurons. Mov. Disord. 2020, 35, 1810–1821. [Google Scholar] [CrossRef] [PubMed]
- Roseberry, T.K.; Lee, A.M.; Lalive, A.L.; Wilbrecht, L.; Bonci, A.; Kreitzer, A.C. Cell-Type-Specific Control of Brainstem Locomotor Circuits by Basal Ganglia. Cell 2016, 164, 526–537. [Google Scholar] [CrossRef] [Green Version]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiyama, F.; Nakano, T.; Matsuda, W.; Furuta, T.; Udagawa, J.; Kaneko, T. A single-neuron tracing study of arkypallidal and prototypic neurons in healthy rats. Brain Struct. Funct. 2016, 221, 4733–4740. [Google Scholar] [CrossRef] [PubMed]
- Abecassis, Z.A.; Berceau, B.L.; Win, P.H.; García, D.; Xenias, H.S.; Cui, Q.; Pamukcu, A.; Cherian, S.; Hernández, V.M.; Chon, U.; et al. Npas1+-Nkx2.1+ Neurons Are an Integral Part of the Cortico-pallido-cortical Loop. J. Neurosci. 2020, 40, 743–768. [Google Scholar] [CrossRef]
- Baufreton, J.; Kirkham, E.; Atherton, J.F.; Menard, A.; Magill, P.J.; Bolam, J.P.; Bevan, M.D. Sparse but selective and potent synaptic transmission from the globus pallidus to the subthalamic nucleus. J. Neurophysiol. 2009, 102, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Sadek, A.R.; Magill, P.J.; Bolam, J.P. A single-cell analysis of intrinsic connectivity in the rat globus pallidus. J. Neurosci. 2007, 27, 6352–6362. [Google Scholar] [CrossRef] [PubMed]
- Ketzef, M.; Silberberg, G. Differential Synaptic Input to External Globus Pallidus Neuronal Subpopulations In Vivo. Neuron 2021, 109, 516–529.e4. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.E.; Woodhall, G.L.; Wilson, C.L.; Stanford, I.M. Functional characterization of GABAergic pallidopallidal and striatopallidal synapses in the rat globus pallidus in vitro. Eur. J. Neurosci. 2008, 28, 2401–2408. [Google Scholar] [CrossRef]
- Atherton, J.F.; Menard, A.; Urbain, N.; Bevan, M.D. Short-term depression of external globus pallidus-subthalamic nucleus synaptic transmission and implications for patterning subthalamic activity. J. Neurosci. 2013, 33, 7130–7144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connelly, W.M.; Schulz, J.M.; Lees, G.; Reynolds, J.N. Differential short-term plasticity at convergent inhibitory synapses to the substantia nigra pars reticulata. J. Neurosci. 2010, 30, 14854–14861. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.H.; Farries, M.A.; Fee, M.S. Basal ganglia output to the thalamus: Still a paradox. Trends Neurosci. 2013, 36, 695–705. [Google Scholar] [CrossRef]
- Kase, D.; Uta, D.; Ishihara, H.; Imoto, K. Inhibitory synaptic transmission from the substantia nigra pars reticulata to the ventral medial thalamus in mice. Neurosci. Res. 2015, 97, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.K.; Yung, K.K. Distinct cellular distribution of GABA(B)R1 and GABA(A)alpha1 receptor immunoreactivity in the rat substantia nigra. Neuroscience 2000, 99, 65–76. [Google Scholar] [CrossRef]
- Ng, T.K.; Yung, K.K. Subpopulations of neurons in rat substantia nigra display GABA(B)R2 receptor immunoreactivity. Brain Res. 2001, 920, 210–216. [Google Scholar] [CrossRef]
- Higgs, M.H.; Wilson, C.J. Unitary synaptic connections among substantia nigra pars reticulata neurons. J. Neurophysiol. 2016, 115, 2814–2829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Hernández, T.; Rodríguez, M. Compartmental organization and chemical profile of dopaminergic and GABAergic neurons in the substantia nigra of the rat. J. Comp. Neurol. 2000, 421, 107–135. [Google Scholar] [CrossRef]
- Kha, H.T.; Finkelstein, D.I.; Tomas, D.; Drago, J.; Pow, D.V.; Horne, M.K. Projections from the substantia nigra pars reticulata to the motor thalamus of the rat: Single axon reconstructions and immunohistochemical study. J. Comp. Neurol. 2001, 440, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.; Tanimura, A.; Graves, S.M.; Shen, W.; Surmeier, D.J. Striatal synapses, circuits, and Parkinson’s disease. Curr. Opin. Neurobiol. 2018, 48, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Chiken, S.; Takada, M.; Nambu, A. Altered Dynamic Information Flow through the Cortico-Basal Ganglia Pathways Mediates Parkinson’s Disease Symptoms. Cereb. Cortex 2021, 16, bhab164. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.M.; Doig, N.M.; Brimblecombe, K.R.; Lopes, E.F.; Siddorn, R.E.; Threlfell, S.; Connor-Robson, N.; Bengoa-Vergniory, N.; Pasternack, N.; Wade-Martins, R.; et al. GABA uptake transporters support dopamine release in dorsal striatum with maladaptive downregulation in a parkinsonism model. Nat. Commun. 2020, 11, 4958. [Google Scholar] [CrossRef] [PubMed]
- Escande, M.V.; Taravini, I.R.; Zold, C.L.; Belforte, J.E.; Murer, M.G. Loss of Homeostasis in the Direct Pathway in a Mouse Model of Asymptomatic Parkinson’s Disease. J. Neurosci. 2016, 36, 5686–5698. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Zhao, L.; Xia, H.; Xu, C.; Wang, D.; Liu, K.; Lin, L.; Li, X.; Yan, Z.; Gao, H. NMR-Based Metabolomics Reveal a Recovery from Metabolic Changes in the Striatum of 6-OHDA-Induced Rats Treated with Basic Fibroblast Growth Factor. Mol. Neurobiol. 2016, 53, 6690–6697. [Google Scholar] [CrossRef]
- Ryan, M.B.; Bair-Marshall, C.; Nelson, A.B. Aberrant Striatal Activity in Parkinsonism and Levodopa-Induced Dyskinesia. Cell Rep. 2018, 23, 3438–3446.e5. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.G.; Marshall, J.D.; Ahanonu, B.; Wu, Y.W.; Kim, T.H.; Grewe, B.F.; Zhang, Y.; Li, J.Z.; Ding, J.B.; Ehlers, M.D.; et al. Diametric neural ensemble dynamics in parkinsonian and dyskinetic states. Nature 2018, 557, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.Y. Synaptic and cellular plasticity in Parkinson’s disease. Acta Pharm. Sin. 2020, 41, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Hoover, B.R.; Marshall, J.F. Molecular, chemical, and anatomical characterization of globus pallidus dopamine D2 receptor mRNA-containing neurons. Synapse 2004, 52, 100–113. [Google Scholar] [CrossRef]
- Baufreton, J.; Bevan, M.D. D2-like dopamine receptor-mediated modulation of activity-dependent plasticity at GABAergic synapses in the subthalamic nucleus. J. Physiol. 2008, 586, 2121–2142. [Google Scholar] [CrossRef] [PubMed]
- Kita, H.; Kita, T. Role of Striatum in the Pause and Burst Generation in the Globus Pallidus of 6-OHDA-Treated Rats. Front. Syst. Neurosci. 2011, 5, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharott, A.; Vinciati, F.; Nakamura, K.C.; Magill, P.J. A Population of Indirect Pathway Striatal Projection Neurons Is Selectively Entrained to Parkinsonian Beta Oscillations. J. Neurosci. 2017, 37, 9977–9998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, K.Y.; Baufreton, J.; Surmeier, D.J.; Chan, C.S.; Bevan, M.D. Proliferation of external globus pallidus-subthalamic nucleus synapses following degeneration of midbrain dopamine neurons. J. Neurosci. 2012, 32, 13718–13728. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.Y.; Atherton, J.F.; Wokosin, D.; Surmeier, D.J.; Bevan, M.D. Heterosynaptic regulation of external globus pallidus inputs to the subthalamic nucleus by the motor cortex. Neuron 2015, 85, 364–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.S.; Glajch, K.E.; Gertler, T.S.; Guzman, J.N.; Mercer, J.N.; Lewis, A.S.; Goldberg, A.B.; Tkatch, T.; Shigemoto, R.; Fleming, S.M.; et al. HCN channelopathy in external globus pallidus neurons in models of Parkinson’s disease. Nat. Neurosci. 2011, 14, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Glajch, K.E.; Kelver, D.A.; Hegeman, D.J.; Cui, Q.; Xenias, H.S.; Augustine, E.C.; Hernández, V.M.; Verma, N.; Huang, T.Y.; Luo, M.; et al. Npas1+ Pallidal Neurons Target Striatal Projection Neurons. J. Neurosci. 2016, 36, 5472–5488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, A.J.; Stanford, I.M. Dopamine D2 receptor mediated presynaptic inhibition of striatopallidal GABA(A) IPSCs in vitro. Neuropharmacology 2001, 41, 62–71. [Google Scholar] [CrossRef]
- Bugaysen, J.; Bar-Gad, I.; Korngreen, A. Continuous modulation of action potential firing by a unitary GABAergic connection in the globus pallidus in vitro. J. Neurosci. 2013, 33, 12805–12809. [Google Scholar] [CrossRef]
- Stefani, A.; Spadoni, F.; Martorana, A.; Lavaroni, F.; Martella, G.; Sancesario, G.; Bernardi, G. D2-mediated modulation of N-type calcium currents in rat globus pallidus neurons following dopamine denervation. Eur. J. Neurosci. 2002, 15, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Ciliax, B.J.; Nash, N.; Heilman, C.; Sunahara, R.; Hartney, A.; Tiberi, M.; Rye, D.B.; Caron, M.G.; Niznik, H.B.; Levey, A.I. Dopamine D(5) receptor immunolocalization in rat and monkey brain. Synapse 2000, 37, 125–145. [Google Scholar] [CrossRef]
- Rivera, A.; Trías, S.; Peñafiel, A.; Angel Narváez, J.; Díaz-Cabiale, Z.; Moratalla, R.; de la Calle, A. Expression of D4 dopamine receptors in striatonigral and striatopallidal neurons in the rat striatum. Brain Res. 2003, 989, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Kliem, M.A.; Pare, J.F.; Khan, Z.U.; Wichmann, T.; Smith, Y. Ultrastructural localization and function of dopamine D1-like receptors in the substantia nigra pars reticulata and the internal segment of the globus pallidus of parkinsonian monkeys. Eur. J. Neurosci. 2010, 31, 836–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.W.; Jin, Y.; Matta, S.G.; Xu, M.; Zhou, F.M. An ultra-short dopamine pathway regulates basal ganglia output. J. Neurosci. 2009, 29, 10424–10435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wichmann, T.; Soares, J. Neuronal firing before and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. J. Neurophysiol. 2006, 95, 2120–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cáceres-Chávez, V.A.; Hernández-Martínez, R.; Pérez-Ortega, J.; Herrera-Valdez, M.A.; Aceves, J.J.; Galarraga, E.; Bargas, J. Acute dopamine receptor blockade in substantia nigra pars reticulata: A possible model for drug-induced Parkinsonism. J. Neurophysiol. 2018, 120, 2922–2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgkvist, A.; Avegno, E.M.; Wong, M.Y.; Kheirbek, M.A.; Sonders, M.S.; Hen, R.; Sulzer, D. Loss of Striatonigral GABAergic Presynaptic Inhibition Enables Motor Sensitization in Parkinsonian Mice. Neuron 2015, 87, 976–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta-García, J.; Hernández-Chan, N.; Paz-Bermúdez, F.; Sierra, A.; Erlij, D.; Aceves, J.; Florán, B. D4 and D1 dopamine receptors modulate [3H] GABA release in the substantia nigra pars reticulata of the rat. Neuropharmacology 2009, 57, 725–730. [Google Scholar] [CrossRef]
- Erlij, D.; Acosta-García, J.; Rojas-Márquez, M.; González-Hernández, B.; Escartín-Perez, E.; Aceves, J.; Florán, B. Dopamine D4 receptor stimulation in GABAergic projections of the globus pallidus to the reticular thalamic nucleus and the substantia nigra reticulata of the rat decreases locomotor activity. Neuropharmacology 2012, 62, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Mathai, A.; Smith, Y. The corticostriatal and corticosubthalamic pathways: Two entries, one target. So what? Front. Syst. Neurosci. 2011, 5, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fremeau, R.T., Jr.; Troyer, M.D.; Pahner, I.; Nygaard, G.O.; Tran, C.H.; Reimer, R.J.; Bellocchio, E.E.; Fortin, D.; Storm-Mathisen, J.; Edwards, R.H. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 2001, 31, 247–260. [Google Scholar] [CrossRef] [Green Version]
- Nisenbaum, E.S.; Wilson, C.J. Potassium currents responsible for inward and outward rectification in rat neostriatal spiny projection neurons. J. Neurosci. 1995, 15, 4449–4463. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, J.L.; Day, M.; Surmeier, D.J. Synaptically driven state transitions in distal dendrites of striatal spiny neurons. Nat. Neurosci. 2011, 14, 881–888. [Google Scholar] [CrossRef]
- Perrin, E.; Venance, L. Bridging the gap between striatal plasticity and learning. Curr. Opin. Neurobiol. 2019, 54, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Flajolet, M.; Greengard, P.; Surmeier, D.J. Dichotomous dopaminergic control of striatal synaptic plasticity. Science 2008, 321, 848–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Plotkin, J.L.; Francardo, V.; Ko, W.K.; Xie, Z.; Li, Q.; Fieblinger, T.; Wess, J.; Neubig, R.R.; Lindsley, C.W.; et al. M4 Muscarinic Receptor Signaling Ameliorates Striatal Plasticity Deficits in Models of L-DOPA-Induced Dyskinesia. Neuron 2015, 88, 762–773. [Google Scholar] [CrossRef] [Green Version]
- Baufreton, J.; Zhu, Z.T.; Garret, M.; Bioulac, B.; Johnson, S.W.; Taupignon, A.I. Dopamine receptors set the pattern of activity generated in subthalamic neurons. FASEB J. 2005, 19, 1771–1777. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J.; Mercer, J.N.; Chan, C.S. Autonomous pacemakers in the basal ganglia: Who needs excitatory synapses anyway? Curr. Opin. Neurobiol. 2005, 15, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.; Huang, K.W.; Sabatini, B.L. Globus Pallidus Externus Neurons Expressing parvalbumin Interconnect the Subthalamic Nucleus and Striatal Interneurons. PLoS ONE 2016, 11, e0149798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouty-Colomer, L.A.; Michel, F.J.; Baude, A.; Lopez-Pauchet, C.; Dufour, A.; Cossart, R.; Hammond, C. Mouse subthalamic nucleus neurons with local axon collaterals. J. Comp. Neurol. 2018, 526, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Bazzari, A.H.; Parri, H.R. Neuromodulators and Long-Term Synaptic Plasticity in Learning and Memory: A Steered-Glutamatergic Perspective. Brain Sci. 2019, 9, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, V.; Di Maio, A.; Marino, G.; Cardinale, A.; Natale, G.; De Rosa, A.; Campanelli, F.; Mancini, M.; Napolitano, F.; Avallone, L.; et al. Rapamycin, by Inhibiting mTORC1 Signaling, Prevents the Loss of Striatal Bidirectional Synaptic Plasticity in a Rat Model of L-DOPA-Induced Dyskinesia. Front. Aging. Neurosci. 2020, 12, 230. [Google Scholar] [CrossRef] [PubMed]
- Parker, P.R.; Lalive, A.L.; Kreitzer, A.C. Pathway-Specific Remodeling of Thalamostriatal Synapses in Parkinsonian Mice. Neuron 2016, 89, 734–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, S.L.; Chen, B.; Lo, C.; Gertler, T.S.; Warre, R.; Surmeier, J.D.; Brotchie, J.M.; Nash, J.E. Selective loss of bi-directional synaptic plasticity in the direct and indirect striatal output pathways accompanies generation of parkinsonism and l-DOPA induced dyskinesia in mouse models. Neurobiol. Dis. 2014, 71, 334–344. [Google Scholar] [CrossRef] [Green Version]
- Froux, L.; Le Bon-Jego, M.; Miguelez, C.; Normand, E.; Morin, S.; Fioramonti, S.; Barresi, M.; Frick, A.; Baufreton, J.; Taupignon, A. D5 dopamine receptors control glutamatergic AMPA transmission between the motor cortex and subthalamic nucleus. Sci. Rep. 2018, 8, 8858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Singh, S.; Shukla, S. Physiological and Functional Basis of Dopamine Receptors and Their Role in Neurogenesis: Possible Implication for Parkinson’s disease. J. Exp. Neurosci. 2018, 12, 1179069518779829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, S.; Tkatch, T.; Atherton, J.F.; Wilson, C.J.; Bevan, M.D. D2-like dopamine receptors modulate SKCa channel function in subthalamic nucleus neurons through inhibition of Cav2.2 channels. J. Neurophysiol. 2008, 99, 442–459. [Google Scholar] [CrossRef] [Green Version]
- Beurrier, C.; Congar, P.; Bioulac, B.; Hammond, C. Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J. Neurosci. 1999, 19, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Baufreton, J.; Garret, M.; Rivera, A.; de la Calle, A.; Gonon, F.; Dufy, B.; Bioulac, B.; Taupignon, A. D5 (not D1) dopamine receptors potentiate burst-firing in neurons of the subthalamic nucleus by modulating an L-type calcium conductance. J. Neurosci. 2003, 23, 816–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loucif, K.C.; Wilson, C.L.; Baig, R.; Lacey, M.G.; Stanford, I.M. Functional interconnectivity between the globus pallidus and the subthalamic nucleus in the mouse brain slice. J. Physiol. 2005, 567, 977–987. [Google Scholar] [CrossRef]
- McIver, E.L.; Atherton, J.F.; Chu, H.Y.; Cosgrove, K.E.; Kondapalli, J.; Wokosin, D.; Surmeier, D.J.; Bevan, M.D. Maladaptive Downregulation of Autonomous Subthalamic Nucleus Activity following the Loss of Midbrain Dopamine Neurons. Cell Rep. 2019, 28, 992–1002.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathai, A.; Ma, Y.; Paré, J.F.; Villalba, R.M.; Wichmann, T.; Smith, Y. Reduced cortical innervation of the subthalamic nucleus in MPTP-treated parkinsonian monkeys. Brain 2015, 138, 946–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, H.Y.; McIver, E.L.; Kovaleski, R.F.; Atherton, J.F.; Bevan, M.D. Loss of Hyperdirect Pathway Cortico-Subthalamic Inputs Following Degeneration of Midbrain Dopamine Neurons. Neuron 2017, 95, 1306–1318.e5. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Wang, Y.; Jiang, H.F.; Liu, J.H.; Jia, J.; Wang, K.; Zhao, F.; Luo, M.H.; Luo, M.M.; Wang, X.M. Impaired glutamatergic projection from the motor cortex to the subthalamic nucleus in 6-hydroxydopamine-lesioned hemi-parkinsonian rats. Exp. Neurol. 2018, 300, 135–148. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, J.R.; Chen, L.; Ge, S.N.; Wang, J.L.; Yan, Z.Q.; Jia, D.; Zhu, J.L.; Gao, G.D. Decreased HCN2 expression in STN contributes to abnormal high-voltage spindles in the cortex and globus pallidus of freely moving rats. Brain Res. 2015, 1618, 17–28. [Google Scholar] [CrossRef]
- Levy, R.; Hutchison, W.D.; Lozano, A.M.; Dostrovsky, J.O. High-frequency synchronization of neuronal activity in the subthalamic nucleus of parkinsonian patients with limb tremor. J. Neurosci. 2000, 20, 7766–7775. [Google Scholar] [CrossRef] [Green Version]
- Mallet, N.; Pogosyan, A.; Sharott, A.; Csicsvari, J.; Bolam, J.P.; Brown, P.; Magill, P.J. Disrupted dopamine transmission and the emergence of exaggerated beta oscillations in subthalamic nucleus and cerebral cortex. J. Neurosci. 2008, 28, 4795–4806. [Google Scholar] [CrossRef] [PubMed]
- Baufreton, J.; Atherton, J.F.; Surmeier, D.J.; Bevan, M.D. Enhancement of excitatory synaptic integration by GABAergic inhibition in the subthalamic nucleus. J. Neurosci. 2005, 25, 8505–8517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibañez-Sandoval, O.; Hernández, A.; Florán, B.; Galarraga, E.; Tapia, D.; Valdiosera, R.; Erlij, D.; Aceves, J.; Bargas, J. Control of the subthalamic innervation of substantia nigra pars reticulata by D1 and D2 dopamine receptors. J. Neurophysiol. 2006, 95, 1800–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, J.P.; Feyder, M.; Miguelez, C.; Garcia, L.; Morin, S.; Choquet, D.; Hosy, E.; Bezard, E.; Fisone, G.; Bioulac, B.H.; et al. Dopamine-dependent long-term depression at subthalamo-nigral synapses is lost in experimental parkinsonism. J. Neurosci. 2013, 33, 14331–14341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faynveitz, A.; Lavian, H.; Jacob, A.; Korngreen, A. Proliferation of Inhibitory Input to the Substantia Nigra in Experimental Parkinsonism. Front. Cell Neurosci. 2019, 13, 417. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romaus-Sanjurjo, D.; Custodia, A.; Aramburu-Núñez, M.; Posado-Fernández, A.; Vázquez-Vázquez, L.; Camino-Castiñeiras, J.; Leira, Y.; Pías-Peleteiro, J.M.; Aldrey, J.M.; Ouro, A.; et al. Symmetric and Asymmetric Synapses Driving Neurodegenerative Disorders. Symmetry 2021, 13, 2333. https://doi.org/10.3390/sym13122333
Romaus-Sanjurjo D, Custodia A, Aramburu-Núñez M, Posado-Fernández A, Vázquez-Vázquez L, Camino-Castiñeiras J, Leira Y, Pías-Peleteiro JM, Aldrey JM, Ouro A, et al. Symmetric and Asymmetric Synapses Driving Neurodegenerative Disorders. Symmetry. 2021; 13(12):2333. https://doi.org/10.3390/sym13122333
Chicago/Turabian StyleRomaus-Sanjurjo, Daniel, Antía Custodia, Marta Aramburu-Núñez, Adrián Posado-Fernández, Laura Vázquez-Vázquez, Javier Camino-Castiñeiras, Yago Leira, Juan Manuel Pías-Peleteiro, José Manuel Aldrey, Alberto Ouro, and et al. 2021. "Symmetric and Asymmetric Synapses Driving Neurodegenerative Disorders" Symmetry 13, no. 12: 2333. https://doi.org/10.3390/sym13122333
APA StyleRomaus-Sanjurjo, D., Custodia, A., Aramburu-Núñez, M., Posado-Fernández, A., Vázquez-Vázquez, L., Camino-Castiñeiras, J., Leira, Y., Pías-Peleteiro, J. M., Aldrey, J. M., Ouro, A., & Sobrino, T. (2021). Symmetric and Asymmetric Synapses Driving Neurodegenerative Disorders. Symmetry, 13(12), 2333. https://doi.org/10.3390/sym13122333