
Density Functional Theory Study of Substitution Effects on the Second-Order Nonlinear Optical Properties of Lindquist-Type Organo-Imido Polyoxometalates



Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Ground State Equilibrium Geometries
3.2. Natural Population Analysis (NPA) Charge Distributions
3.3. First Hyperpolarizabilities
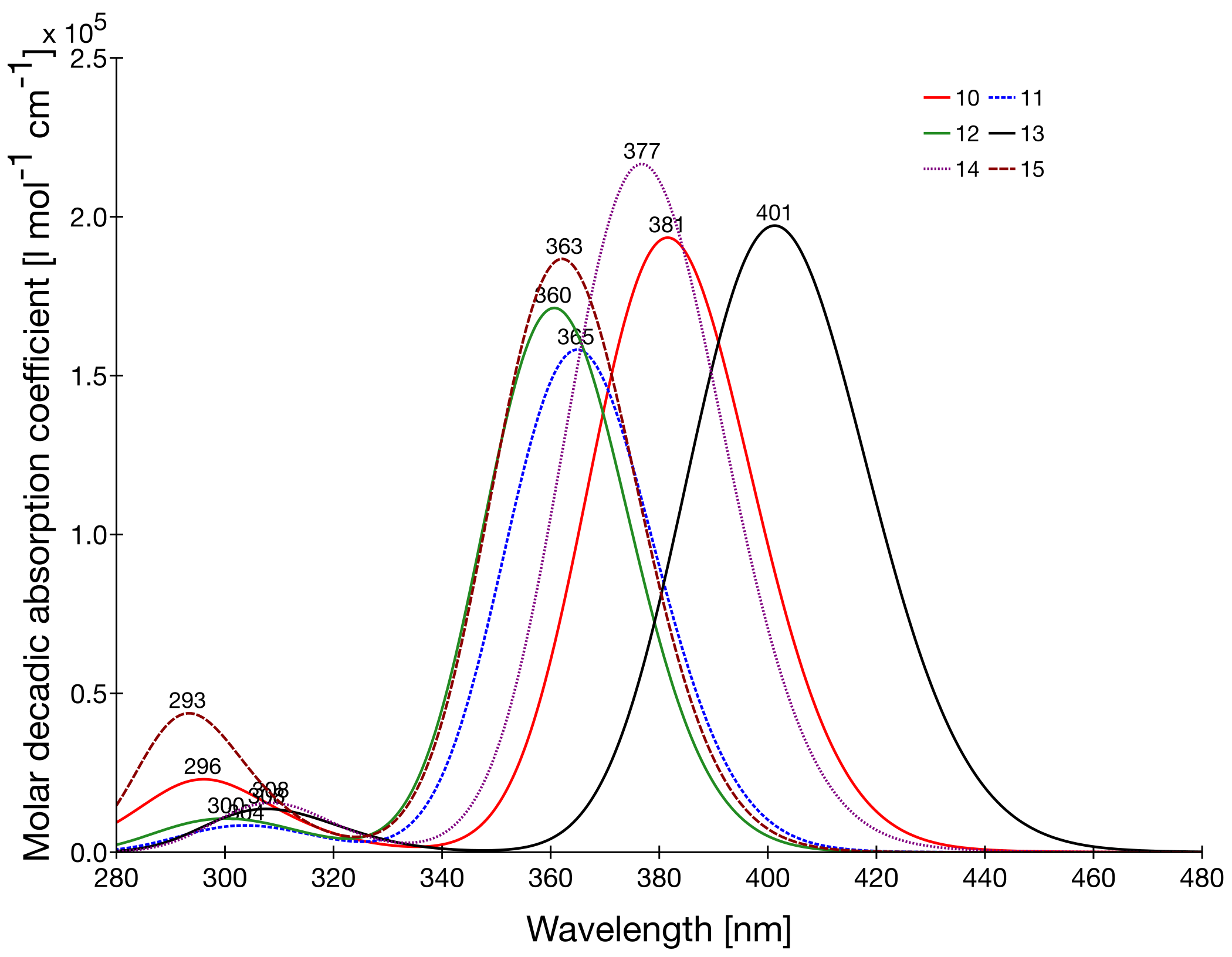
3.4. UV-Absorption Spectra and Interpretation of the β Values
4. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pope, M.T.; Müller, A. Polyoxometalate Chemistry: An Old Field with New Dimensions in Several Disciplines. Angew. Chem. Int. Ed. Engl. 1991, 30, 34–48. [Google Scholar] [CrossRef]
- Anyushin, A.V.; Kondinski, A.; Parac-Vogt, T.N. Hybrid Polyoxometalates as Post-functionalization Platforms: From Fundamentals to Emerging Applications. Chem. Soc. Rev. 2020, 49, 382–432. [Google Scholar] [CrossRef]
- Al-Yasari, A.; Van Steerteghem, N.; El Moll, H.; Clays, K.; Fielden, J. Donor—Acceptor Organo-Imido Polyoxometalates: High Transparency, High Activity Redox-Active NLO Chromophores. Dalton Trans. 2016, 45, 2818–2822. [Google Scholar] [CrossRef] [Green Version]
- Al-Yasari, A.; Van Steerteghem, N.; Kearns, H.; El Moll, H.; Faulds, K.; Wright, J.A.; Brunschwig, B.S.; Clays, K.; Fielden, J. Organoimido-Polyoxometalate Nonlinear Optical Chromophores: A Structural, Spectroscopic, and Computational Study. Inorg. Chem. 2017, 56, 10181–10194. [Google Scholar] [CrossRef] [Green Version]
- Al-Yasari, A.; Spence, P.; El Moll, H.; Van Steerteghem, N.; Horton, P.N.; Brunschwig, B.S.; Fielden, J. Fine-Tuning Polyoxometalate Non-Linear Optical Chromophores: A Molecular Electronic “Goldilocks” Effect. Dalton Trans. 2018, 47, 10415–10419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Yasari, A.; El Moll, H.; Purdy, R.; Vincent, B.K.; Spence, P.; Malval, J.P.; Fielden, J. Optical Third Order Non-Linear Optical And Electrochemical Properties Of Dipolar, Centrosymmetric and C2v Organoimido Polyoxometalate Derivatives. Phys. Chem. Chem. Phys. 2021, 23, 11807–11817. [Google Scholar] [CrossRef]
- Boulmier, A.; Vacher, A.; Zang, D.; Yang, S.; Saad, A.; Marrot, J.; Oms, O.; Mialane, P.; Ledoux, I.; Ruhlmann, L.; et al. Anderson-Type Polyoxometalates Functionalized by Tetrathiafulvalene Groups: Synthesis, Electrochemical Studies, and NLO Properties. Inorg. Chem. 2018, 57, 3742–3752. [Google Scholar] [CrossRef]
- Perez-Moreno, J.; Zhao, Y.; Clays, K.; Kuzyk, M.G.; Shen, Y.; Qiu, L.; Hao, J.; Guo, K. Modulated Conjugation as a Means of Improving the Intrinsic Hyperpolarizability. J. Am. Chem. Soc. 2009, 131, 5084–5093. [Google Scholar] [CrossRef]
- Castet, F.; Rodriguez, V.; Pozzo, J.L.; Ducasse, L.; Plaquet, A.; Champagne, B. Design and Characterization of Molecular Nonlinear Optical Switches. Acc. Chem. Res. 2013, 46, 2656–2665. [Google Scholar] [CrossRef] [PubMed]
- Karamanis, P.; Otero, N.; Pouchan, C. Unleashing the Quadratic Nonlinear Optical Responses of Graphene by Confining White-Graphene (h-BN) Sections in Its Framework. J. Am. Chem. Soc. 2014, 136, 7464–7473. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Park, J.; De Mey, K.; Hu, X.; Duncan, T.V.; Beratan, D.N.; Therien, M.J. Large Hyperpolarizabilities at Telecommunication-Relevant Wavelengths in Donor-Acceptor-Donor Nonlinear Optical Chromophores. ACS Cent. Sci. 2016, 2, 954–966. [Google Scholar] [CrossRef]
- Coe, B.J.; Rusanova, D.; Joshi, V.D.; Sanchez, S.; Vavra, J.; Khobragade, D.; Severa, L.; Cisarova, I.; Saman, D.; Pohl, R.; et al. Helquat Dyes: Helicene-like Push−Pull Systems with Large Second-Order Nonlinear Optical Responses. J. Org. Chem. 2016, 81, 1912–1920. [Google Scholar] [CrossRef]
- Lacroix, P.G.; Malfant, I.; Lepetit, C. Second-Order Nonlinear Optics in Coordination Chemistry: An Open Door towards Multi-Functional Materials and Molecular Switches. Coord. Chem. Rev. 2016, 308, 381–394. [Google Scholar] [CrossRef]
- Knoppe, S.; Hakkinen, H.; Verbiest, T.; Clays, K. Role of Donor and Acceptor Substituents on the Nonlinear Optical Properties of Gold Nanoclusters. J. Phys. Chem. C 2018, 122, 4019–4028. [Google Scholar] [CrossRef]
- Van Bezouw, S.; Koo, M.J.; Lee, S.C.; Lee, S.H.; Campo, J.; Kwon, O.P.; Wenseleers, W. Three-Stage pH-Switchable Organic Chromophores with Large Nonlinear Optical Responses and Switching Contrasts. Chem. Commun. 2018, 54, 7842–7845. [Google Scholar] [CrossRef] [Green Version]
- Tonnelé, C.; Champagne, B.; Muccioli, L.; Castet, F. Second-Order Nonlinear Optical Properties of Stenhouse Photoswitches: Insights from Density Functional Theory. Phys. Chem. Chem. Phys. 2018, 20, 27658–27667. [Google Scholar] [CrossRef] [PubMed]
- Lou, A.J.T.; Marks, T.J.A. Twist on Nonlinear Optics: Understanding the Unique Response of π-Twisted Chromophores. Acc. Chem. Res. 2019, 52, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, L.; Forni, A.; Cariati, E.; Malavasi, G.; Pasini, A. Solid-State Nonlinear Optical Properties of Mononuclear Copper (II) Complexes with Chiral Tridentate and Tetradentate Schiff Base Ligands. Materials 2019, 12, 3595. [Google Scholar] [CrossRef] [Green Version]
- Rothe, C.; Neusser, D.; Hoppe, N.; Dirnberger, K.; Vogel, W.; Gámez-Valenzuela, S.; Ludwigs, S. Push-Pull Chromophores for Electro-Optic Applications: From 1D Linear to β-branched Structures. Phys. Chem. Chem. Phys. 2020, 22, 2283–2294. [Google Scholar] [CrossRef]
- Qiu, S.; Morshedi, M.; Kodikara, M.S.; Du, J.; de Coene, Y.; Zhang, C.; Humphrey, M.G. Organometallic complexes for nonlinear optics. 66. Synthesis and quadratic nonlinear optical studies of trans-[Ru{C C {2, 5-C4H2S-(E)-CHCH} n-2, 5-C4H2S (NO2)} Cl (dppe) 2](n = 0–2). J. Organomet. Chem. 2020, 919, 121306. [Google Scholar] [CrossRef]
- Cesaretti, A.; Foggi, P.; Fortuna, C.G.; Elisei, F.; Spalletti, A.; Carlotti, B. Uncovering Structure—Property Relationships in Push–Pull Chromophores: A Promising Route to Large Hyperpolarizability and Two-Photon Absorption. J. Phys. Chem. 2020, 124, 15739–15748. [Google Scholar] [CrossRef]
- Ramos, T.N.; Canuto, S.; Champagne, B. Unraveling the Electric Field-Induced Second Harmonic Generation Responses of Stilbazolium Ion Pairs Complexes in Solution Using a Multiscale Simulation Method. J. Chem. Inf. Model. 2020, 60, 4817–4826. [Google Scholar] [CrossRef] [PubMed]
- Idney, B.; Tertius, L.F.; Leandro, R.F.; Herbert, C.G.; Marcos, A.C. Applicability of DFT Functionals for Evaluating the First Hyperpolarizability of Phenol Blue in Solution. J. Chem. Phys. 2021, 154, 094501. [Google Scholar] [CrossRef]
- Moris, M.; Van Den Eede, M.P.; Koeckelberghs, G.; Deshaume, O.; Bartic, C.; Clays, K.; Cleuvenbergen, S.; Verbiest, T. Solvent Role in the Self-Assembly of Poly (3-alkylthiophene): A Harmonic Light Scattering Study. Macromolecules 2021, 54, 2477–2484. [Google Scholar] [CrossRef]
- Castet, F.; Gillet, A.; Bureš, F.; Plaquet, A.; Rodriguez, V.; Champagne, B. Second-Order Nonlinear Optical Properties of Λ-Shaped Pyrazine Derivatives. Dye. Pigment. 2021, 184, 108850. [Google Scholar] [CrossRef]
- Verbiest, T.; Clays, K.; Rodriguez, V. Second-Order Nonlinear Optical Characterizations Techniques: An Introduction; CRC Press: New York, NY, USA, 2009. [Google Scholar]
- Yan, L.; Yang, G.; Guan, W.; Su, Z.; Wang, R. Density Functional Theory Study on the First Hyperpolarizabilities of Organoimido Derivatives of Hexamolybdates. J. Phys. Chem. B 2005, 109, 22332–22336. [Google Scholar] [CrossRef]
- Janjua, M.R.S.A.; Liu, C.G.; Guan, W.; Zhuang, J.; Muhammad, S.; Yan, L.K.; Su, Z.M. Prediction of Remarkably Large Second-Order Nonlinear Optical Properties of Organoimido-Substituted Hexamolybdates. J. Phys. Chem. 2009, 113, 3576–3587. [Google Scholar] [CrossRef]
- Rtibi, E.; Abderrabba, M.; Ayadi, S.; Champagne, B. Theoretical Assessment of the Second-Order Nonlinear Optical Responses of Lindqvist-Type Organoimido Polyoxometalates. Inorg. Chem. 2019, 58, 11210–11219. [Google Scholar] [CrossRef]
- Lescos, L.; Sitkiewicz, S.; Beaujean, P.; Blanchard-Desce, M.; Champagne, B.; Matito, R.E.; Castet, F. Performance of DFT Functionals for Calculating the Second-Order Nonlinear Optical Properties of Dipolar Merocyanines. Phys. Chem. Chem. Phys. 2020, 22, 16579–16594. [Google Scholar] [CrossRef]
- Champagne, B.; Perpète, E.A.; Jacquemin, D.; van Gisbergen, S.J.A.; Baerends, E.J.; Soubra-Ghaoui, C.; Kirtman, B. Assessment of Conventional Density Functional Schemes for Computing the Dipole Moment and (Hyper)polarizabilities of Push–Pull π-Conjugated Systems. J. Phys. Chem. 2000, 104, 4755–4763. [Google Scholar] [CrossRef]
- Bulat, F.A.; Toro-Labbé, A.; Champagne, B.; Kirtman, B.; Yang, W. Density-Functional Theory (hyper) polarizabilities of Push-Pull π-Conjugated Systems: Treatment of Exact Exchange and Role of Correlation. J. Chem. Phys. 2005, 123, 014319. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Autschbach, J. Influence of the Delocalization Error and Applicability of Optimal Functional Tuning in Density Functional Calculations of Nonlinear Optical Properties of Organic Donor-Acceptor Chromophores. ChemPhysChem 2013, 14, 2450–2461. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revis. C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Chai, J.D.; Head-Gordon, M. Long-range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. Density-Functional Thermochemistry. V. Systematic Optimization of Exchange-Correlation Functionals. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Van Gisbergen, S.J.A.; Snijders, J.G.; Baerends, E.J. Calculating Frequency-Dependent Hyperpolarizabilities Using Time-Dependent Density Functional Theory. J. Chem. Phys. 1998, 109, 10644–10656. [Google Scholar] [CrossRef] [Green Version]
- Helgaker, T.; Coriani, S.; Jørgensen, P.; Kristensen, K.; Olsen, J.; Ruud, K. Recent Advances in Wave Function-Based Methods of Molecular-Property Calculations. Chem. Rev. 2012, 112, 543–631. [Google Scholar] [CrossRef]
- Bersohn, R.; Pao, Y.H.; Frisch, H.L. Double-Quantum Light Scattering by Molecules. J. Chem. Phys. 1966, 45, 3184–3198. [Google Scholar] [CrossRef]
- Castet, F.; Bogdan, E.; Plaquet, A.; Ducasse, L.; Champagne, B.; Rodriguez, V. Reference Molecules for Nonlinear Optics: A Joint Experimental and Theoretical Investigation. J. Chem. Phys. 2012, 136, 024506. [Google Scholar] [CrossRef]
- Tuer, A.; Krouglov, S.; Cisek, R.; Tokarz, D.; Barzda, V. Three-Dimensional Visualization of the First Hyperpolarizability Tensor. J. Comput. Chem. 2011, 32, 1128–1134. [Google Scholar] [CrossRef]
- Orr, B.J.; Ward, J.F. Perturbation Theory of the Non-Linear Optical Polarization of an Isolated System. Mol. Phys. 1971, 20, 513–526. [Google Scholar] [CrossRef]
- Bishop, D.M. Explicit Non-divergent Formulas for Atomic and Molecular Dynamic Hyperpolarizabilities. J. Chem. Phys. 1994, 100, 6535–6542. [Google Scholar] [CrossRef]
- Oudar, J.L.; Chemla, D.S. Hyperpolarizabilities of the Nitroanilines and Their Relations to the Excited State Dipole Moment. J. Chem. Phys. 1977, 66, 2664–2668. [Google Scholar] [CrossRef]
- Zutterman, F.; Liégeois, V.; Champagne, B. Simulation of UV/visible Absorption Spectra of Fluorescent Protein Chromophore Models. ChemPhotoChem 2017, 1, 281–297. [Google Scholar] [CrossRef]
- Champagne, B.; Kirtman, B. Alternative Sum-Over-States Expressions for the First Hyperpolarizability of Push-Pull π-Conjugated Systems. J. Chem. Phys. 2006, 125, 024101. [Google Scholar] [CrossRef]
- Wei, Y.; Lu, M.; Cheung, C.F.-C.; Barnes, C.L.; Peng, Z. Functionalization of [MoW5O19]2- with Aromatic Amines: Synthesis of the First Arylimido Derivatives of Mixed-Metal Polyoxometalates. Inorg. Chem. 2001, 40, 5489–5490. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

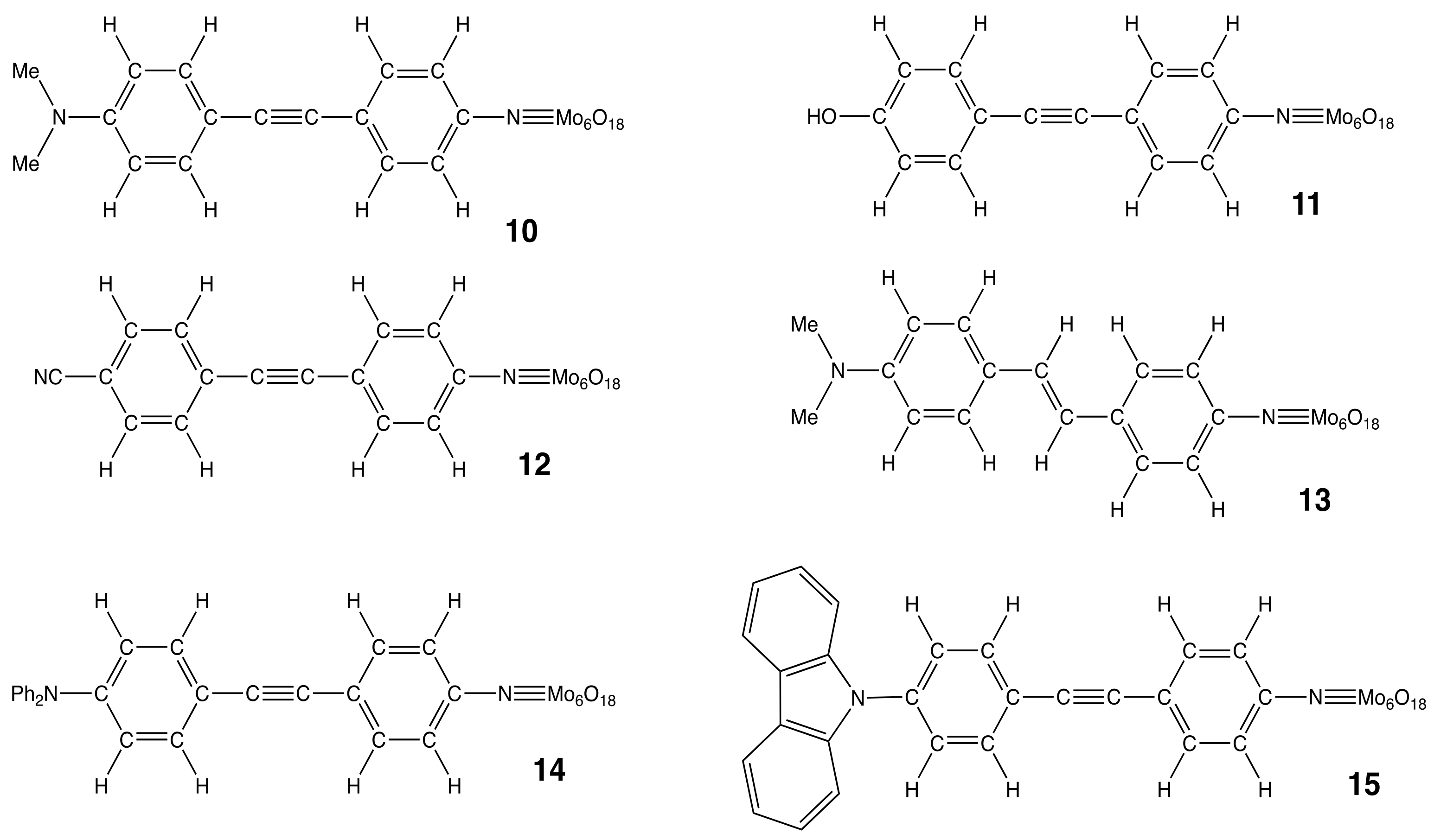
| Compounds | 10 | 11 | 12 | 13 | 14 | 15 | |||
|---|---|---|---|---|---|---|---|---|---|
| Method | DFT [29] | XRD [4] | DFT | DFT | DFT | DFT | XRD [5] | DFT 3 | XRD [5] |
| R-C1 | 1.367 | 1.368 | 1.352 | 1.432 | 1.371 | 1.403 | 1.385 | 1.413 | 1.447 |
| C1-C2 | 1.413 | 1.411 | 1.395 | 1.398 | 1.412 | 1.401 | 1.406 | 1.395 | 1.390 |
| C2-C3 | 1.382 | 1.385 | 1.386 | 1.383 | 1.382 | 1.383 | 1.388 | 1.385 | 1.392 |
| C3-C4 | 1.400 | 1.403 | 1.401 | 1.401 | 1.401 | 1.401 | 1.395 | 1.400 | 1.388 |
| C4-C5 | 1.424 | 1.430 | 1.427 | 1.427 | 1.462 | 1.426 | 1.448 | 1.428 | 1.474 |
| C5-C6 | 1.208 | 1.208 | 1.207 | 1.206 | 1.342 | 1.207 | 1.198 | 1.207 | 1.210 |
| C6-C7 | 1.425 | 1.433 | 1.426 | 1.426 | 1.465 | 1.426 | 1.438 | 1.427 | 1.489 |
| C7-C8 | 1.403 | 1.385 | 1.402 | 1.402 | 1.404 | 1.403 | 1.400 | 1.402 | 1.389 |
| C8-C9 | 1.384 | 1.384 | 1.382 | 1.383 | 1.382 | 1.383 | 1.362 | 1.383 | 1.391 |
| C9-C10 | 1.397 | 1.391 | 1.400 | 1.400 | 1.400 | 1.400 | 1.417 | 1.402 | 1.390 |
| C10-N | 1.373 | 1.395 | 1.375 | 1.374 | 1.374 | 1.374 | 1.382 | 1.378 | 1.373 |
| N-Mo | 1.727 | 1.737 | 1.729 | 1.730 | 1.727 | 1.728 | 1.7555 | 1.734 | 1.750 |
| Mo-O | 1.980 | 1.946 | 1.983 | 1.981 | 1.984 | 1.981 | 1.952 | 1.985 | 1.955 |
| Mo-Mo | 3.303 | 3.237 | 3.303 | 3.303 | 3.303 | 3.305 | 3.245 | 3.301 | 3.230 |
| C10-N-Mo | 169.6 | 168.3 | 170.0 | 169.9 | 174.9 | 176.7 | 172.3 | 168.3 | 160.6 |
| BLA 4 | 0.024 | 0.023 | 0.012 | 0.016 | 0.024 | 0.018 | 0.012 | 0.012 | −0.003 |

| Compounds | A | B | C | D | E | F |
|---|---|---|---|---|---|---|
| 10 (A = −NMe2, C = C≡C) 11 (A = −OH, C = C≡C) 12 (A = −CN, C = C≡C) 13 (A = NMe2, C = CH=CH) 14 (A = −NPh2, C = C≡C) 15 (A = −Carb., C = C≡C) | 0.01 −0.19 −0.04 0.00 −0.12 −0.21 | 0.04 0.22 0.01 0.04 0.14 0.21 | −0.01 −0.01 0.02 0.02 0.00 0.01 | 0.19 0.20 0.21 0.18 0.20 0.21 | −0.30 −0.30 −0.31 −0.29 −0.30 −0.31 | −1.93 −1.91 −1.89 −1.95 −1.92 −1.91 |
| Compounds | , Exp. | ||||
|---|---|---|---|---|---|
| λ = 1064 nm | λ = 1064 nm | λ = 1064 nm | |||
| 10 11 12 13 14 15 | 20.8 (4.75) 11.3 (4.75) 3.2 (4.55) 25.2 (4.77) 17.0 (4.49) 8.8 (4.37) | 42.4 (4.94) 19.2 (4.94) 5.4 (4.92) 58.5 (4.96) 36.6 (4.52) 16.6 (4.50) | 217 118 34 263 177 92 | 443 200 56 611 382 173 | 440 ± 55 / / / 590 ± 20 150 ±36 |
| POMs | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 10 11 12 13 14 15 | 3.25 (382) [2] 3.40 (365) [2] 3.44 (361) [2] 3.09 (401) [2] 3.29 (377) [2] 3.42 (362) [2] | 2.94 [5] - - - 2.98 [5] 3.20 [5] | 2.15 1.76 1.90 2.19 2.41 2.08 | 4.78 3.56 1.97 4.83 4.38 3.08 | 0.66 0.57 0.50 0.66 0.62 0.54 | 15.2 9.8 4.7 15.4 13.0 8.0 | 67.9 31.1 15.7 81.6 62.6 29.6 | 28.1 12.9 6.5 33.8 25.9 12.2 | 20.8 11.3 3.2 25.2 17.0 8.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rtibi, E.; Champagne, B. Density Functional Theory Study of Substitution Effects on the Second-Order Nonlinear Optical Properties of Lindquist-Type Organo-Imido Polyoxometalates. Symmetry 2021, 13, 1636. https://doi.org/10.3390/sym13091636
Rtibi E, Champagne B. Density Functional Theory Study of Substitution Effects on the Second-Order Nonlinear Optical Properties of Lindquist-Type Organo-Imido Polyoxometalates. Symmetry. 2021; 13(9):1636. https://doi.org/10.3390/sym13091636
Chicago/Turabian StyleRtibi, Emna, and Benoit Champagne. 2021. "Density Functional Theory Study of Substitution Effects on the Second-Order Nonlinear Optical Properties of Lindquist-Type Organo-Imido Polyoxometalates" Symmetry 13, no. 9: 1636. https://doi.org/10.3390/sym13091636
APA StyleRtibi, E., & Champagne, B. (2021). Density Functional Theory Study of Substitution Effects on the Second-Order Nonlinear Optical Properties of Lindquist-Type Organo-Imido Polyoxometalates. Symmetry, 13(9), 1636. https://doi.org/10.3390/sym13091636