
Sub-Hz Differential Rotational Spectroscopy of Enantiomers


Abstract
:1. Introduction
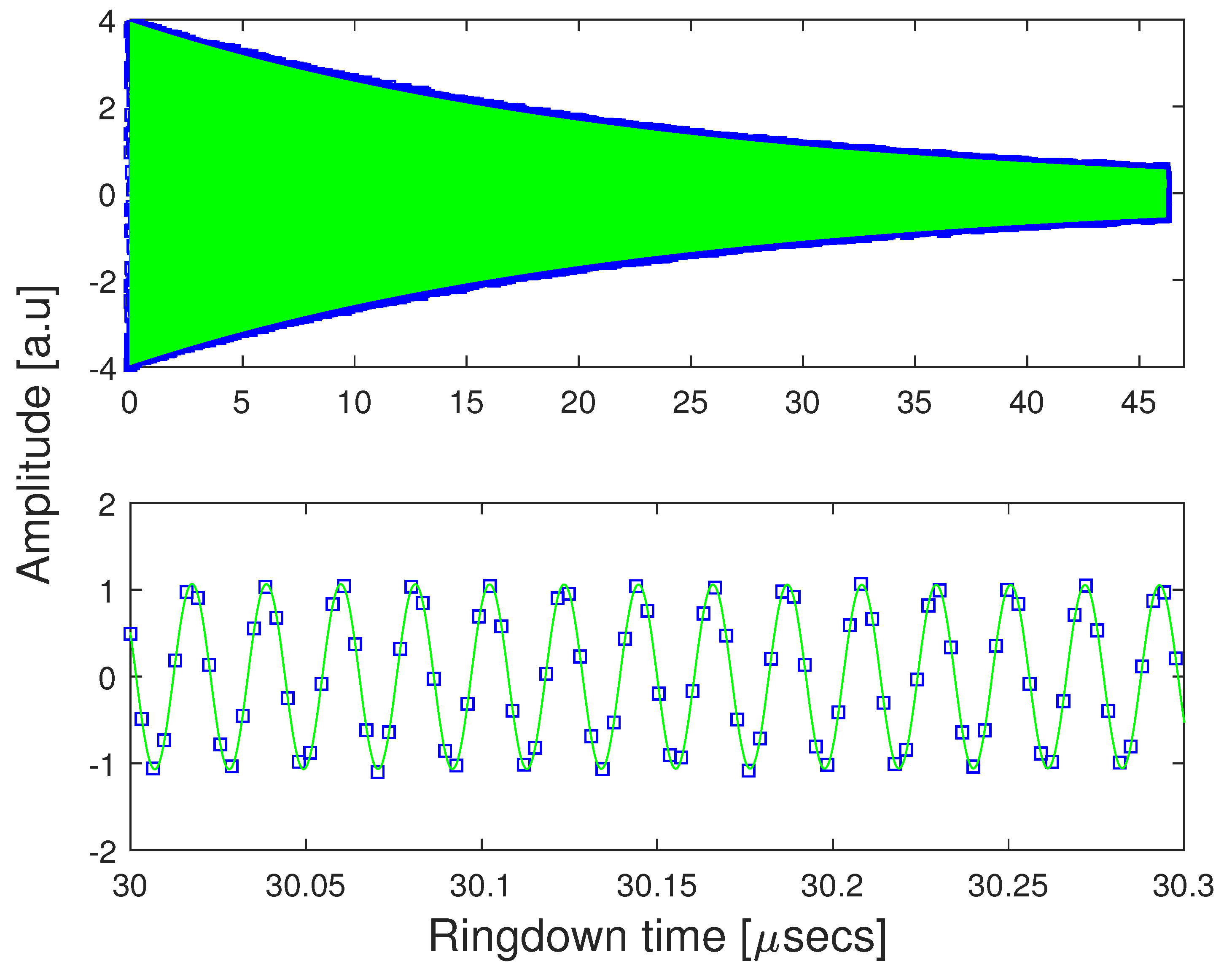
2. Materials and Methods
3. Results
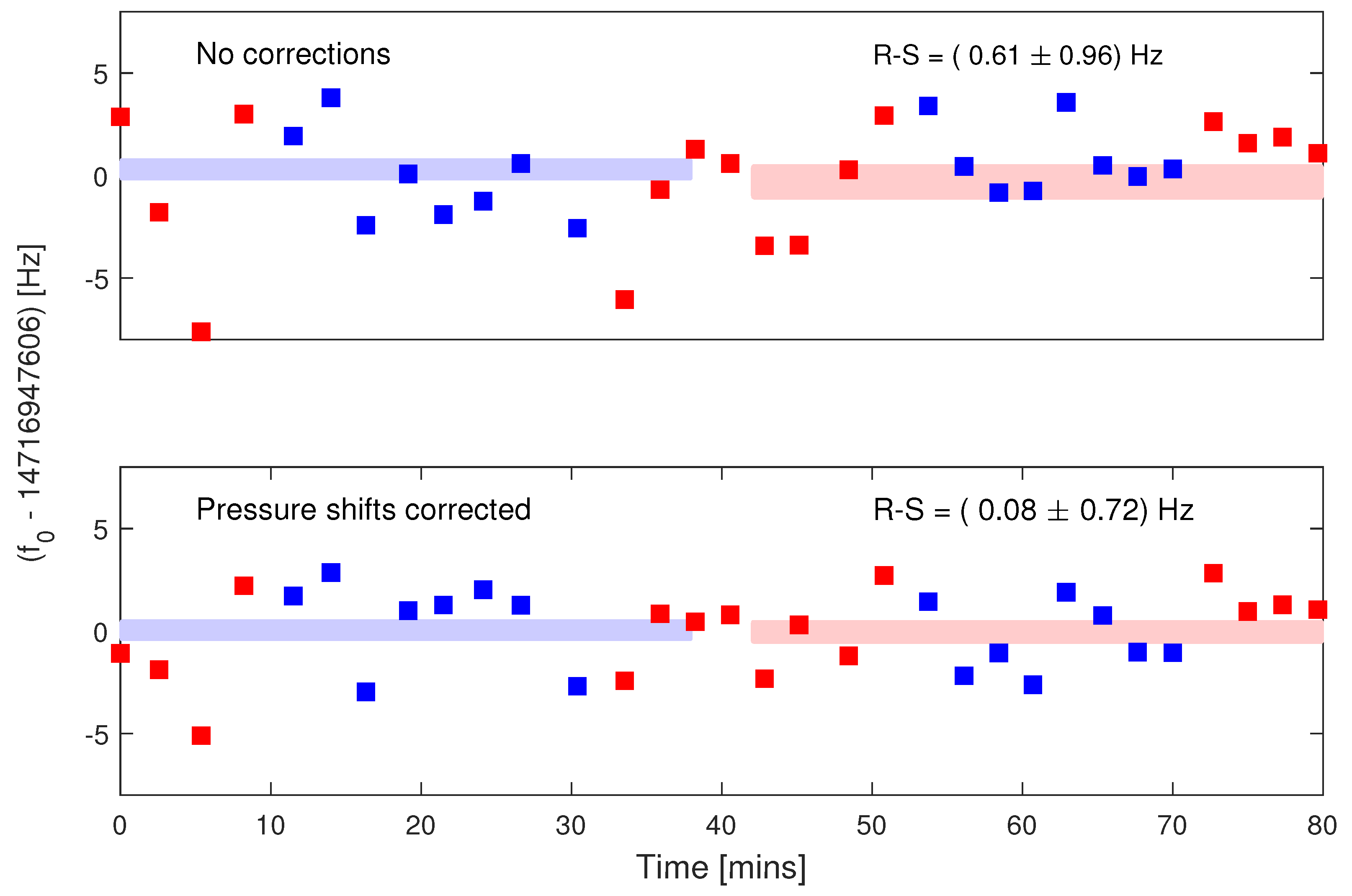
3.1. Analysis of Systematics
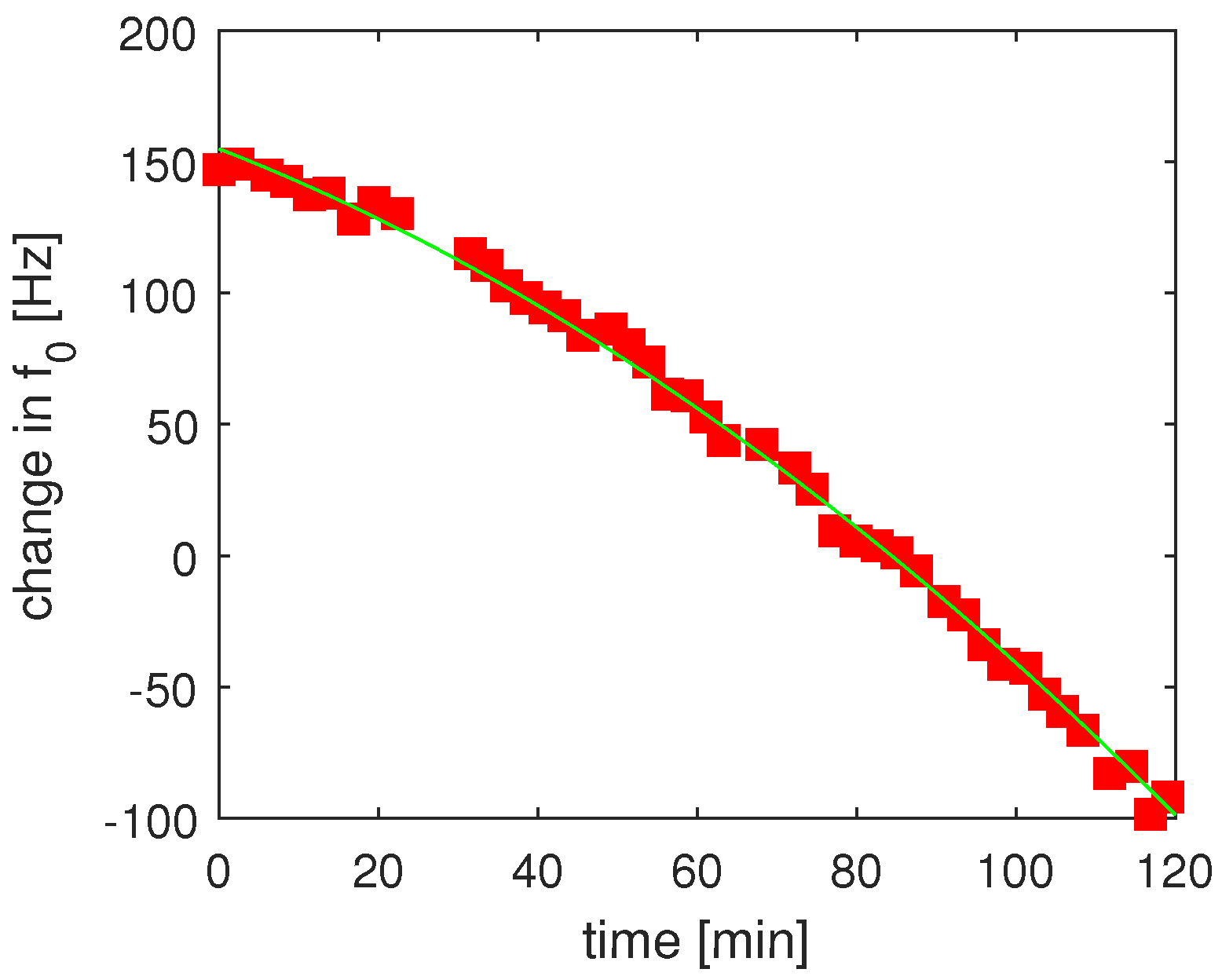
3.1.1. DC Stark Shifts
3.1.2. Pressure Shifts
3.1.3. Systematic Shifts between Samples
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fukuyama, T. Searching for new physics beyond the standard model in electric dipole moment. Int. J. Mod. Phys. A 2012, 27, 1230015. [Google Scholar] [CrossRef]
- Hudson, J.; Sauer, B.; Tarbutt, M.; Hinds, E. Measurement of the electron electric dipole moment using YbF molecules. Phys. Rev. Lett. 2002, 89, 023003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreev, V.; Hutzler, N. Improved limit on the electric dipole moment of the electron. Nature 2018, 562, 355–360. [Google Scholar]
- Tarbutt, M.R.; Hudson, J.J.; Sauer, B.E.; Hinds, E.A. Preparation and manipulation of molecules for fundamental physics tests. arXiv 2008, arXiv:0803.0967. [Google Scholar]
- DeMille, D.; Cahn, S.B.; Murphree, D.; Rahmlow, D.A.; Kozlov, M.G. Using molecules to measure nuclear spin-dependent parity violation. Phys. Rev. Lett. 2008, 100, 023003. [Google Scholar] [CrossRef] [Green Version]
- Wall, T. Preparation of cold molecules for high-precision measurements. J. Phys. B At. Mol. Opt. Phys. 2016, 49, 243001. [Google Scholar] [CrossRef]
- Tarbutt, M.; Sauer, B.; Hudson, J.; Hinds, E. Design for a fountain of YbF molecules to measure the electron’s electric dipole moment. New J. Phys. 2013, 15, 053034. [Google Scholar] [CrossRef]
- Safronova, M.; Budker, D.; DeMille, D.; Kimball, D.F.J.; Derevianko, A.; Clark, C.W. Search for new physics with atoms and molecules. Rev. Mod. Phys. 2018, 90, 025008. [Google Scholar] [CrossRef] [Green Version]
- Lemeshko, M.; Krems, R.V.; Doyle, J.M.; Kais, S. Manipulation of molecules with electromagnetic fields. Mol. Phys. 2013, 111, 1648–1682. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.; Friedrich, B.; Krems, R.; Masnou-Seeuws, F. Quo vadis, cold molecules? Eur. Phys. J. D-At. Mol. Opt. Plasma Phys. 2004, 31, 149–164. [Google Scholar]
- Wu, C.S.; Ambler, E.; Hayward, R.W.; Hoppes, D.D.; Hudson, R.P. Experimental test of parity conservation in beta decay. Phys. Rev. 1957, 105, 1413. [Google Scholar] [CrossRef]
- Edwards, N.; Phipp, S.; Baird, P.; Nakayama, S. Precise measurement of parity nonconserving optical rotation in atomic thallium. Phys. Rev. Lett. 1995, 74, 2654. [Google Scholar] [CrossRef] [PubMed]
- Meekhof, D.; Vetter, P.; Majumder, P.; Lamoreaux, S.; Fortson, E. Optical-rotation technique used for a high-precision measurement of parity nonconservation in atomic lead. Phys. Rev. A 1995, 52, 1895. [Google Scholar] [CrossRef]
- Macpherson, M.; Zetie, K.; Warrington, R.; Stacey, D.; Hoare, J. Precise measurement of parity nonconserving optical rotation at 876 nm in atomic bismuth. Phys. Rev. Lett. 1991, 67, 2784. [Google Scholar] [CrossRef]
- Tsigutkin, K.; Dounas-Frazer, D.; Family, A.; Stalnaker, J.E.; Yashchuk, V.V.; Budker, D. Observation of a large atomic parity violation effect in ytterbium. Phys. Rev. Lett. 2009, 103, 071601. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, S. Molecular homochirality and the parity-violating energy difference. A critique with new proposals. Chirality 2008, 20, 84–95. [Google Scholar] [CrossRef]
- Stevenson, C.D.; Davis, J.P. Magnetars and Magnetic Separation of Chiral Radicals in Interstellar Space: Homochirality. J. Phys. Chem. A 2019, 123, 9587–9593. [Google Scholar] [CrossRef]
- Bonner, W.A. Parity violation and the evolution of biomolecular homochirality. Chirality 2000, 12, 114–126. [Google Scholar] [CrossRef]
- Darquié, B.; Stoeffler, C.; Shelkovnikov, A.; Daussy, C.; Amy-Klein, A.; Chardonnet, C.; Zrig, S.; Guy, L.; Crassous, J.; Soulard, P.; et al. Progress toward the first observation of parity violation in chiral molecules by high-resolution laser spectroscopy. Chirality 2010, 22, 870–884. [Google Scholar] [CrossRef] [Green Version]
- Quack, M.; Stohner, J.; Willeke, M. High-resolution spectroscopic studies and theory of parity violation in chiral molecules. Annu. Rev. Phys. Chem. 2008, 59, 741–769. [Google Scholar] [CrossRef]
- Laerdahl, J.K.; Schwerdtfeger, P. Fully relativistic ab initio calculations of the energies of chiral molecules including parity-violating weak interactions. Phys. Rev. A 1999, 60, 4439. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Gierlich, J.; Bollwein, T. Large Parity-Violation Effects in Heavy-Metal-Containing Chiral Compounds. Angew. Chem. Int. Ed. 2003, 42, 1293–1296. [Google Scholar] [CrossRef] [PubMed]
- Darquié, B.; Saleh, N.; Tokunaga, S.K.; Srebro-Hooper, M.; Ponzi, A.; Autschbach, J.; Decleva, P.; Garcia, G.A.; Crassous, J.; Nahon, L. Valence-shell photoelectron circular dichroism of ruthenium (III)-tris-(acetylacetonato) gas-phase enantiomers. Phys. Chem. Chem. Phys. 2021, 23, 24140–24153. [Google Scholar] [CrossRef]
- Schnell, M.; Küpper, J. Tailored molecular samples for precision spectroscopy experiments. Faraday Discuss. 2011, 150, 33–49. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Kühn, A.; Bast, R.; Laerdahl, J.K.; Faglioni, F.; Lazzeretti, P. The vibrational spectrum of camphor from ab initio and density functional theory and parity violation in the C–C*–CO bending mode. Chem. Phys. Lett. 2004, 383, 496–501. [Google Scholar] [CrossRef]
- Lahamer, A.; Mahurin, S.; Compton, R.; House, D.; Laerdahl, J.; Lein, M.; Schwerdtfeger, P. Search for a parity-violating energy difference between enantiomers of a chiral iron complex. Phys. Rev. Lett. 2000, 85, 4470. [Google Scholar] [CrossRef]
- Tokunaga, S.K.; Stoeffler, C.; Auguste, F.; Shelkovnikov, A.; Daussy, C.; Amy-Klein, A.; Chardonnet, C.; Darquié, B. Probing weak force-induced parity violation by high-resolution mid-infrared molecular spectroscopy. Mol. Phys. 2013, 111, 2363–2373. [Google Scholar] [CrossRef] [Green Version]
- Cournol, A.; Manceau, M.; Pierens, M.; Lecordier, L.; Tran, D.; Santagata, R.; Argence, B.; Goncharov, A.; Lopez, O.; Abgrall, M.; et al. A new experiment to test parity symmetry in cold chiral molecules using vibrational spectroscopy. Quantum Electron. 2019, 49, 288. [Google Scholar] [CrossRef] [Green Version]
- Quack, M.; Stohner, J. Influence of parity violating weak nuclear potentials on vibrational and rotational frequencies in chiral molecules. Phys. Rev. Lett. 2000, 84, 3807. [Google Scholar] [CrossRef]
- Blanchard, J.W.; King, J.P.; Sjolander, T.F.; Kozlov, M.G.; Budker, D. Molecular parity nonconservation in nuclear spin couplings. Phys. Rev. Res. 2020, 2, 023258. [Google Scholar] [CrossRef]
- Eills, J.; Blanchard, J.W.; Bougas, L.; Kozlov, M.G.; Pines, A.; Budker, D. Measuring molecular parity nonconservation using nuclear-magnetic-resonance spectroscopy. Phys. Rev. A 2017, 96, 042119. [Google Scholar] [CrossRef] [Green Version]
- Senami, M.; Ito, K. Asymmetry of electron chirality between enantiomeric pair molecules and the origin of homochirality in nature. Phys. Rev. A 2019, 99, 012509. [Google Scholar] [CrossRef]
- Arimondo, E.; Glorieux, P.; Oka, T. Observation of inverted infrared lamb dips in separated optical isomers. Opt. Commun. 1977, 23, 369–372. [Google Scholar] [CrossRef]
- Ziskind, M.; Daussy, C.; Marrel, T.; Chardonnet, C. Improved sensitivity in the search for a parity-violating energy difference in the vibrational spectrum of the enantiomers of CHFClBr. Eur. Phys. J. D-At. Mol. Opt. Plasma Phys. 2002, 20, 219–225. [Google Scholar] [CrossRef]
- Balle, T.; Flygare, W. Fabry–Perot cavity pulsed Fourier transform microwave spectrometer with a pulsed nozzle particle source. Rev. Sci. Instrum. 1981, 52, 33–45. [Google Scholar] [CrossRef]
- Grabow, J.U.; Palmer, E.S.; McCarthy, M.C.; Thaddeus, P. Supersonic-jet cryogenic-resonator coaxially oriented beam-resonator arrangement Fourier transform microwave spectrometer. Rev. Sci. Instrum. 2005, 76, 093106. [Google Scholar] [CrossRef]
- Schnell, M.; Banser, D.; Grabow, J.U. Coaxially aligned electrodes for Stark-effect applied in resonators using a supersonic jet Fourier transform microwave spectrometer. Rev. Sci. Instrum. 2004, 75, 2111–2115. [Google Scholar] [CrossRef]
- Richard, J.P.; Hamilton, J. Cryogenic monocrystalline silicon Fabry–Perot cavity for the stabilization of laser frequency. Rev. Sci. Instrum. 1991, 62, 2375–2378. [Google Scholar] [CrossRef]
- Kuvshinskii, M.V.; Oreshkin, S.I.; Popov, S.M.; Rudenko, V.N.; Yudin, I.S.; Azarova, V.V.; Blagov, S.V. Tests of cryogenic Fabry–Perot cavity with mirrors on different substrates. Appl. Sci. 2019, 9, 230. [Google Scholar] [CrossRef] [Green Version]
- Porterfield, J.P.; Satterthwaite, L.; Eibenberger, S.; Patterson, D.; McCarthy, M.C. High sensitivity microwave spectroscopy in a cryogenic buffer gas cell. Rev. Sci. Instrum. 2019, 90, 053104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, G. The thermal and electrical conductivity of copper at low temperatures. Aust. J. Phys. 1953, 6, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Bize, S.; Sortais, Y.; Mandache, C.; Clairon, A.; Salomon, C. Cavity frequency pulling in cold atom fountains. IEEE Trans. Instrum. Meas. 2001, 50, 503–506. [Google Scholar] [CrossRef]
- Viennet, J.; Audoin, C.; Desaintfuscien, M. Cavity pulling in passive frequency standards. IEEE Trans. Instrum. Meas. 1972, 21, 204–209. [Google Scholar] [CrossRef]
- Godone, A.; Micalizio, S.; Levi, F.; Calosso, C. Microwave cavities for vapor cell frequency standards. Rev. Sci. Instrum. 2011, 82, 074703. [Google Scholar] [CrossRef]
- Patterson, D.; Doyle, J.M. Sensitive chiral analysis via microwave three-wave mixing. Phys. Rev. Lett. 2013, 111, 023008. [Google Scholar] [CrossRef] [Green Version]
- Field, D.; Plekan, O.; Cassidy, A.; Balog, R.; Jones, N.; Dunger, J. Spontaneous electric fields in solid films: Spontelectrics. Int. Rev. Phys. Chem. 2013, 32, 345–392. [Google Scholar] [CrossRef]
- Balog, R.; Cicman, P.; Jones, N.; Field, D. Spontaneous Dipole Alignment in Films of N2O. Phys. Rev. Lett. 2009, 102, 073003. [Google Scholar] [CrossRef] [Green Version]
- Plekan, O.; Cassidy, A.; Balog, R.; Jones, N.C.; Field, D. A new form of spontaneously polarized material. Phys. Chem. Chem. Phys. 2011, 13, 21035–21044. [Google Scholar] [CrossRef] [PubMed]
- Plekan, O.; Rosu-Finsen, A.; Cassidy, A.M.; Lasne, J.; McCoustra, M.R.; Field, D. A review of recent progress in understanding the spontelectric state of matter. Eur. Phys. J. D 2017, 71, 1–8. [Google Scholar] [CrossRef]
- Western, C.M. PGOPHER: A program for simulating rotational, vibrational and electronic spectra. J. Quant. Spectrosc. Radiat. Transf. 2017, 186, 221–242. [Google Scholar] [CrossRef] [Green Version]
- Reed, Z.; Long, D.; Fleurbaey, H.; Hodges, J. SI-traceable molecular transition frequency measurements at the 10–12 relative uncertainty level. Optica 2020, 7, 1209–1220. [Google Scholar] [CrossRef]
- Patterson, D.; Doyle, J.M. Bright, guided molecular beam with hydrodynamic enhancement. J. Chem. Phys. 2007, 126, 154307. [Google Scholar] [CrossRef] [Green Version]
- Hutzler, N.R.; Lu, H.I.; Doyle, J.M. The buffer gas beam: An intense, cold, and slow source for atoms and molecules. Chem. Rev. 2012, 112, 4803–4827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, D.; Rasmussen, J.; Doyle, J.M. Intense atomic and molecular beams via neon buffer-gas cooling. New J. Phys. 2009, 11, 055018. [Google Scholar] [CrossRef]
- Xiao, D.; Lancaster, D.M.; Allen, C.H.; Taylor, M.J.; Lancaster, T.A.; Shaw, G.; Hutzler, N.R.; Weinstein, J.D. Shaped nozzles for cryogenic buffer-gas beam sources. Phys. Rev. A 2019, 99, 013603. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Uncertainty | Fractional Precision | Technique | Author |
|---|---|---|---|---|
| camphor | 300 kHz | 1.25 × 10−8 | ro-vibrational | Arimondo et al [33] |
| CHClFBr | 8 Hz | 2.5 × 10−13 | vibrational | M. Ziskind et al [34] |
| iron complex | 45 kHz | 1.2 × 10−14 | Mössbauer | A.S. Lahamer et al [26] |
| undetermined, heavy | 0.1 Hz | 1 × 10−15 (proposed) | IR Ramsey interfer. | A. Cournol et al. [28] |
| 1,2-propanediol | 0.72 Hz | 9.7 × 10−11 | rotational | present work |
| Systematic Effect | Error Budget (Hz) |
|---|---|
| uncorrected | (0.61 ± 0.93) |
| DC stark shift | <0.5 (see discussion) |
| pressure shift | (0.53 ± 0.24) |
| amplitude shift | <0.1 (see discussion) |
| statistical error | (0.08 ± 0.72) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Satterthwaite, L.; Koumarianou, G.; Sorensen, D.; Patterson, D. Sub-Hz Differential Rotational Spectroscopy of Enantiomers. Symmetry 2022, 14, 28. https://doi.org/10.3390/sym14010028
Satterthwaite L, Koumarianou G, Sorensen D, Patterson D. Sub-Hz Differential Rotational Spectroscopy of Enantiomers. Symmetry. 2022; 14(1):28. https://doi.org/10.3390/sym14010028
Chicago/Turabian StyleSatterthwaite, Lincoln, Greta Koumarianou, Daniel Sorensen, and David Patterson. 2022. "Sub-Hz Differential Rotational Spectroscopy of Enantiomers" Symmetry 14, no. 1: 28. https://doi.org/10.3390/sym14010028
APA StyleSatterthwaite, L., Koumarianou, G., Sorensen, D., & Patterson, D. (2022). Sub-Hz Differential Rotational Spectroscopy of Enantiomers. Symmetry, 14(1), 28. https://doi.org/10.3390/sym14010028