
Structural Stability and Electronic Properties of Boron Phosphide Nanotubes: A Density Functional Theory Perspective

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
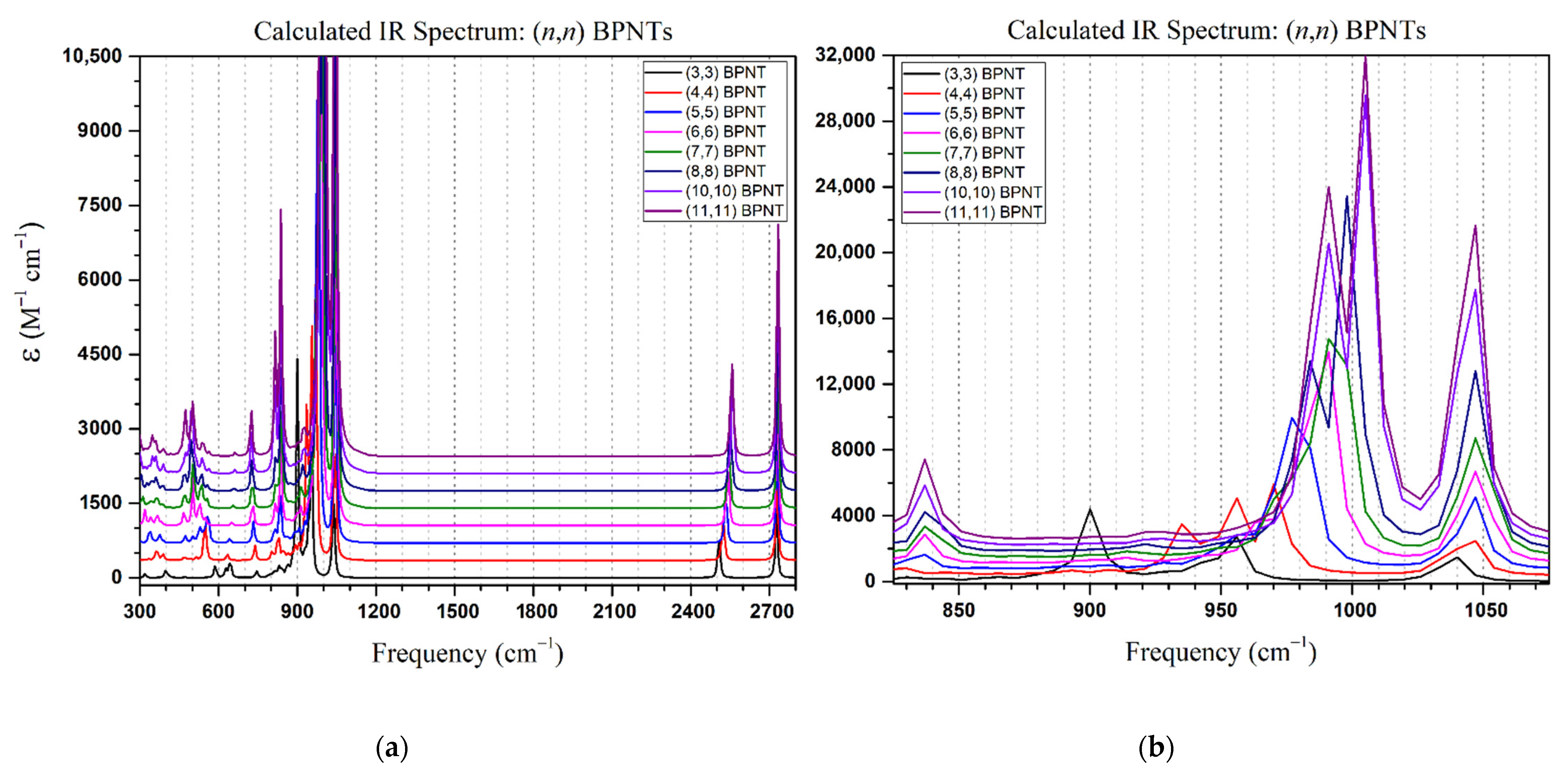
3. Results and Discussion
3.1. Structural Properties
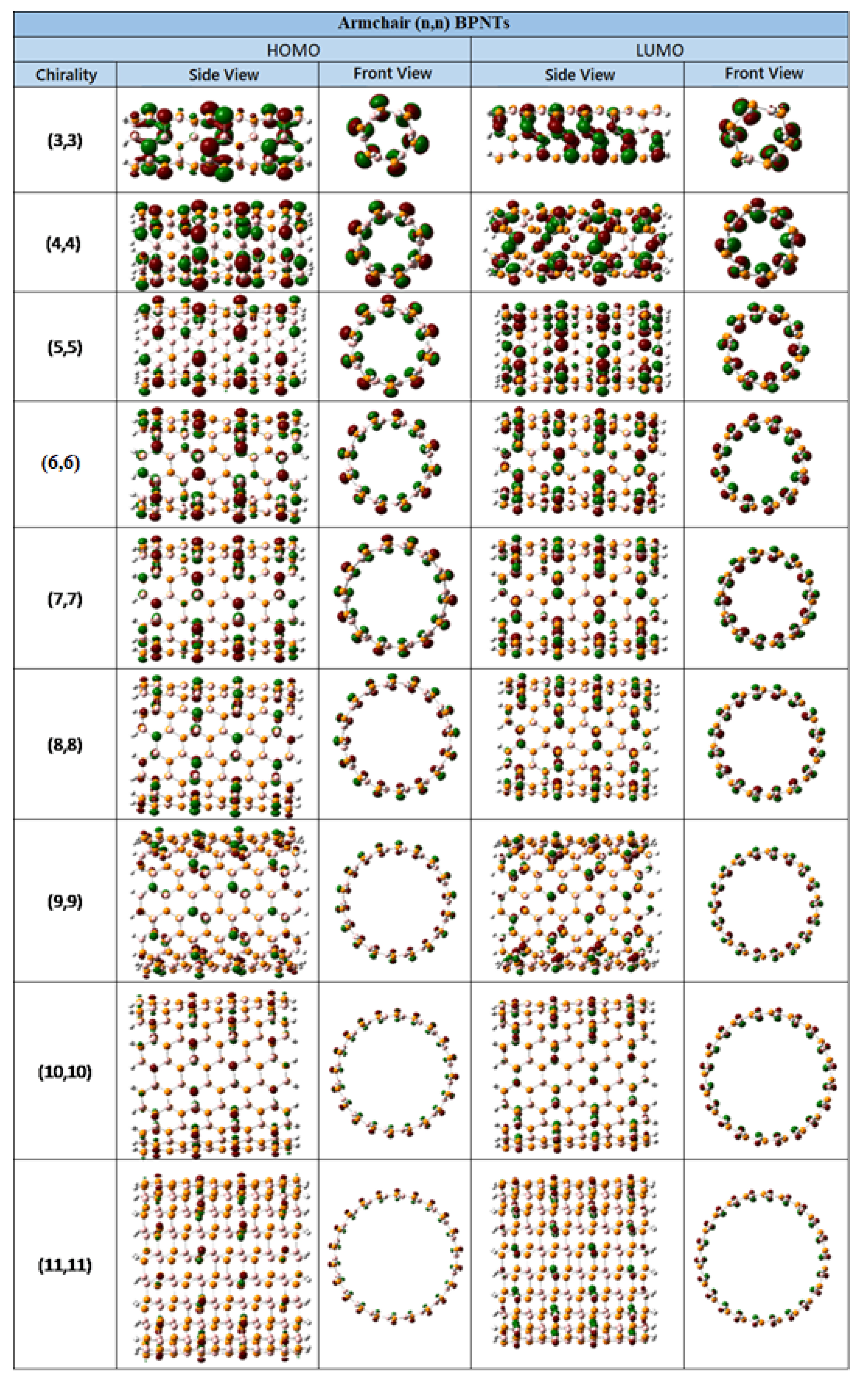
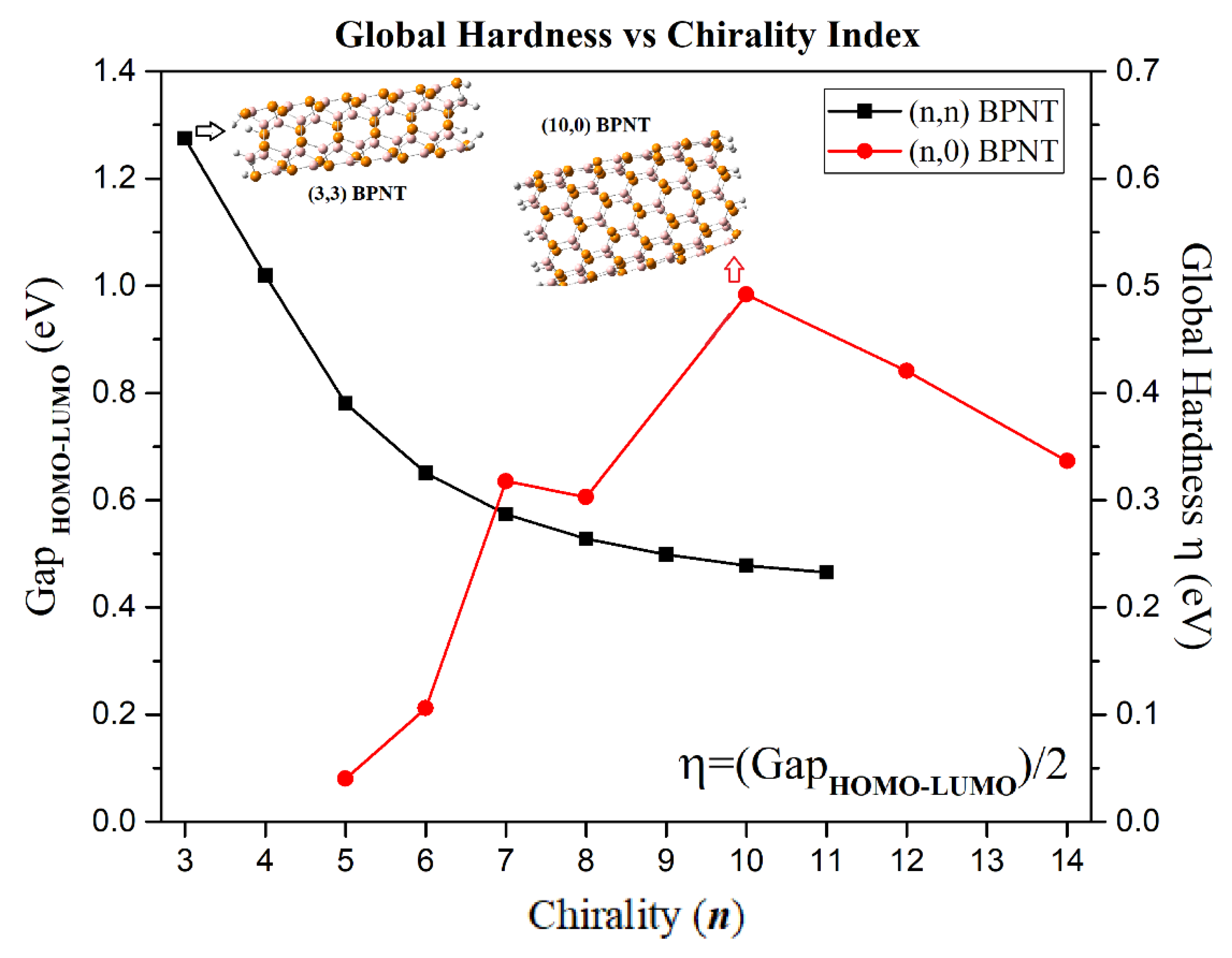
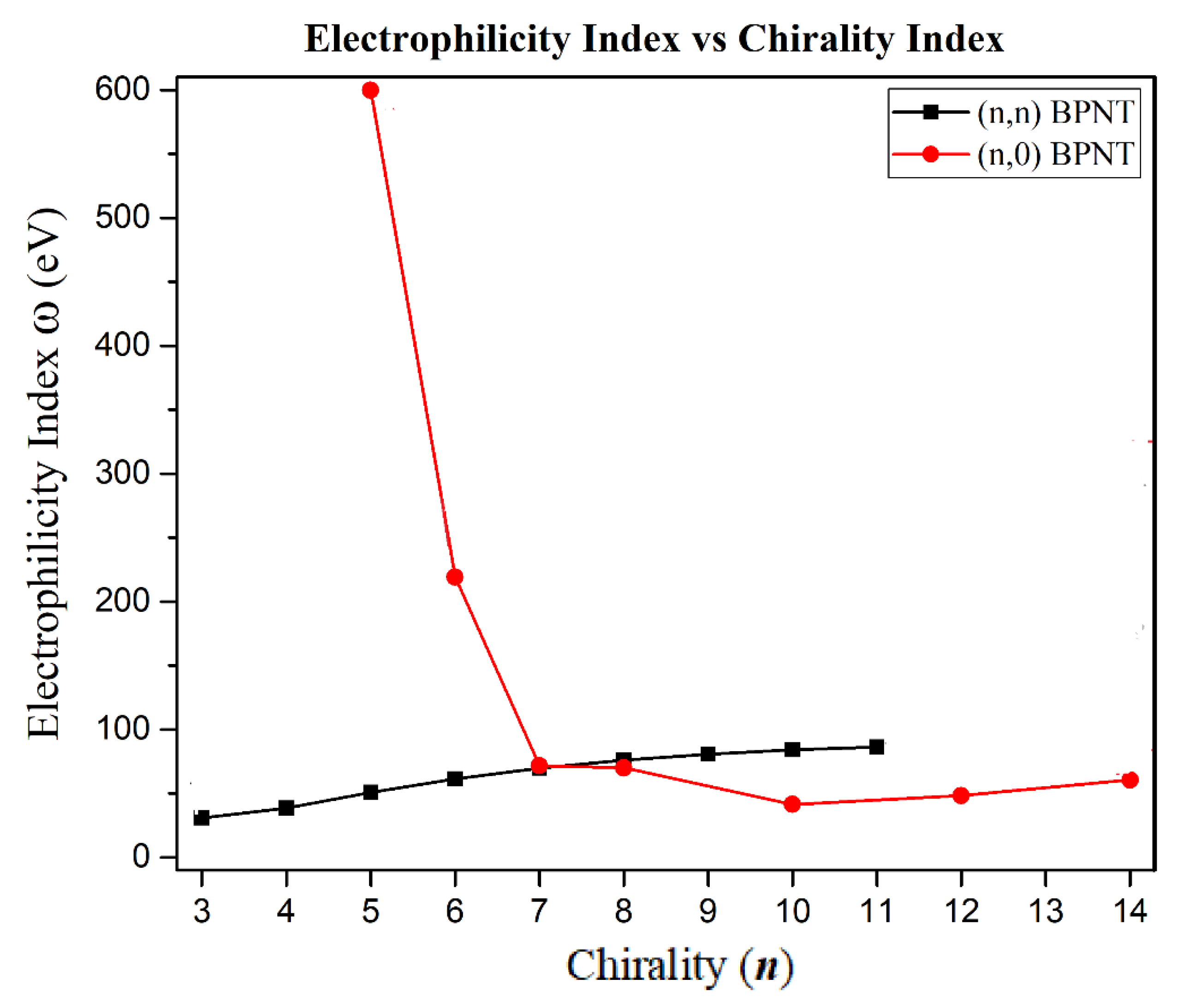
3.2. Electronic Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Rubio, A.; Corkill, J.L.; Cohen, M.L. Theory of graphitic boron nitride nanotubes. Phys. Rev. B Condens. Matter 1994, 49, 5081–5084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, N.G.; Luyken, R.J.; Cherrey, K.; Crespi, V.H.; Cohen, M.L.; Louie, S.G.; Zettl, A. Boron Nitride Nanotubes. Science 1995, 269, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Odom, T.W.; Huang, J.-L.; Kim, P.; Lieber, C.M. Structure and Electronic Properties of Carbon Nanotubes. J. Phys. Chem. B. 2000, 104, 2794–2809. [Google Scholar] [CrossRef]
- Sfeir, M.Y. Optical Spectroscopy of Individual Single-Walled Carbon Nanotubes of Defined Chiral Structure. Science 2006, 312, 554–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marana, N.L.; Albuquerque, A.R.; La Porta, F.A.; Longo, E.; Sambrano, J.R. Periodic density functional theory study of structural and electronic properties of single-walled zinc oxide and carbon nanotubes. J. Solid State Chem. 2016, 237, 36–47. [Google Scholar] [CrossRef]
- Dass, D.; Vaid, R. Chirality dependence of electronic band structure and density of states in single-walled carbon nanotubes. Afr. Rev. Phys. 2018, 12, 104–113. [Google Scholar]
- Blase, X.; Rubio, A.; Louie, S.G.; Cohen, M.L. Stability and Band Gap Constancy of Boron Nitride Nanotubes. Europhys. Lett. (EPL) 1994, 28, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Baei, M.T.; Moradi, A.V.; Torabi, P.; Moghimi, M. Adsorption properties of H2O2 trapped inside a boron phosphide nanotube. Monatsh. Chem. 2011, 143, 37–41. [Google Scholar] [CrossRef]
- Peyghan, A.A.; Baei, M.T.; Moghimi, M.; Hashemian, S. Theoretical Study of Phenol Adsorption on Pristine, Ga-Doped, and Pd-Decorated (6,0) Zigzag Single-Walled Boron Phosphide Nanotubes. J. Clust. Sci. 2012, 24, 49–60. [Google Scholar] [CrossRef]
- Sayyad-Alangi, S.Z.; Hashemian, S.; Baei, M.T. Covalent Functionalization of Pristine and Ga-Doped Boron Phosphide Nanotubes with Imidazole. Phosphorus Sulfur Silicon Relat. Elem. 2014, 189, 453–464. [Google Scholar] [CrossRef]
- Sayyad-Alangi, S.Z.; Baei, M.T.; Hashemian, S. Adsorption and electronic structure study of thiazole on the (6,0)zigzagsingle-walled boron phosphide nanotube. J. Sulphur Chem. 2013, 34, 407–420. [Google Scholar] [CrossRef]
- Beheshtian, J.; Baei, M.T.; Peyghan, A.A. Theoretical study of CO adsorption on the surface of BN, AlN, BP and AlP nanotubes. Surf. Sci. 2012, 606, 981–985. [Google Scholar] [CrossRef]
- Kanani, Y.; Baei, M.T.; Moradi, A.V.; Soltani, A. Adsorption mechanism of single OCN− and SCN− upon single-walled BP nanotubes. Phys. E Low Dimens. Syst. Nanostruct. 2014, 59, 66–74. [Google Scholar] [CrossRef]
- Soltani, A.; Taghartapeh, M.R.; Mighani, H.; Pahlevani, A.A.; Mashkoor, R. A first-principles study of the SCN− chemisorption on the surface of AlN, AlP, and BP nanotubes. Appl. Surf. Sci. 2012, 259, 637–642. [Google Scholar] [CrossRef]
- Wu, I.J.; Guo, G.Y. Optical properties of SiC nanotubes: An ab initio study. Phys. Rev. B Condens. Matter 2007, 76, 035343. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, M. Carbon doped boron phosphide nanotubes: A computational study. J. Mol. Model. 2010, 17, 89–96. [Google Scholar] [CrossRef]
- Esrafili, M.D. Carbon-Doped (6,0) Single-Walled Boron-Phosphide Nanotubes: A DFT Investigation of Electronic Structure, Surface Electrostatic Potential and QTAIM Analysis. Fuller. Nanotub. Carbon Nanostruct. 2014, 23, 142–147. [Google Scholar] [CrossRef]
- Baei, M.T.; Moghimi, M.; Torabi, P.; Moradi, A.V. The Ge-doped (6,0) zigzag single-walled boron phosphide nanotubes: A computational study. Comput. Theor. Chem. 2011, 972, 14–19. [Google Scholar] [CrossRef]
- Rezaei-Sameti, M. Gallium doped in armchair and zigzag models of boron phosphide nanotubes (BPNTs): A NMR study. Phys. B Condens. Matter 2012, 407, 3717–3721. [Google Scholar] [CrossRef]
- Baei, M.T.; Peyghan, A.A.; Moghimi, M. Electronic structure study of Si-doped (4,4) armchair single-walled boron phosphide nanotube as a semiconductor. Monatsh. Chem. 2012, 143, 1627–1635. [Google Scholar] [CrossRef]
- Baei, M.T.; Sayyad-Alangi, S.Z.; Moradi, A.V.; Torabi, P. NMR and NQR parameters of the SiC-doped on the (4,4) armchair single-walled BPNT: A computational study. J. Mol. Model. 2011, 18, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Baei, M.T. Ge-doped (4,4) armchair single-walles boron phosphide nanotube as a semiconductor: A computational study. Monatsh. Chem. 2012, 6, 881–889. [Google Scholar] [CrossRef]
- Srivastava, A.; Sharma, M.; Tyagi, N.; Kothari, S.L. Diameter Dependent Electronic Properties of Zigzag Single Wall BX (X = N, P, As) Nanotubes: Ab-Initio Study. J. Comput. Theor. Nanosci. 2012, 9, 1693–1699. [Google Scholar] [CrossRef]
- Salabat, K.; Azizi, K. A Computational Study on the Effects of Size and Chirality on the Electronic and Structural Properties of BP Nanotubes. In Proceedings of the 16th Iranian Physical Chemistry Conference, Mazandaran, Iran, 29–31 October 2013; Volume 16, pp. 867–869. Available online: https://www.sid.ir/FileServer/SE/247E20131694.pdf (accessed on 9 May 2021).
- Frish, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. G09W®; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hou, N.; Wu, Y.Y.; Wei, Q.; Liu, X.L.; Ma, X.J.; Zhang, M.; Wu, H.S. A comparative theoretical study on the electrical and nonlinear optical properties of Li atom adsorbed on AlN and BN single-walled nanotubes. J. Mol. Model. 2017, 23, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, N.; Head-Gordon, M. How Accurate Are the Minnesota Density Functionals for Noncovalent Interactions, Isomerization Energies, Thermochemistry, and Barrier Heights Involving Molecules Composed of Main-Group Elements? J. Chem. Theory Comput. 2016, 12, 4303–4325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar]
- Koopmans, T. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.v.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Juárez, A.R.; Anota, E.C.; Cocoletzi, H.H.; Ramírez, J.S.; Castro, M. Stability and electronic properties of armchair boron nitride/carbon nanotubes. Fuller. Nanotub. Carbon Nanostruct. 2017, 25, 716–725. [Google Scholar] [CrossRef]
- Beheshtian, J.; Soleymanabadi, H.; Kamfiroozi, M.; Ahmadi, A. The H2 dissociation on the BN, AlN, BP and AlP nanotubes: A comparative study. J. Mol. Model. 2011, 18, 2343–2348. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Giahi, M. Computational studies on boron nitride and boron phosphide nanotubes: Density functional calculations of boron-11 electric field gradient tensors. Phys. E Low Dimens. Syst. Nanostruc. 2010, 42, 1667–1669. [Google Scholar] [CrossRef]
- Arshadi, S.; Bekhradnia, A.R.; Alipour, F.; Abedini, S. Pure and carbon-doped boron phosphide (6,0) zigzag nanotube: A computational NMR study. Phys. B Condens. Matter 2015, 477, 1–7. [Google Scholar] [CrossRef]
- Rezaei-Sameti, M.; Yaghoobi, S. Theoretical study of adsorption of CO gas on pristine and AsGa-doped (4, 4) armchair models of BPNTs. Comput. Condens. Matter 2015, 3, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Parr, R.G. Density Functional Theory of Atoms and Molecules. Horiz. Quantum Chem. 1980, 3, 5–15. [Google Scholar]
- Chermette, H. Chemical reactivity indexes in density functional theory. J. Comput. Chem. 1999, 20, 129–154. [Google Scholar] [CrossRef]
- Roos, G.; Geerlings, P.; Messens, J. Enzymatic Catalysis: The Emerging Role of Conceptual Density Functional Theory. J. Phys. Chem. B 2009, 113, 13465–13475. [Google Scholar] [CrossRef]
- Hosseinzadeh, B.; Salimi Beni, A.; Eskandari, R.; Karami, M.; Khorram, M. Interaction of propylthiouracil, an anti-thyroid drug with boron nitride nanotube: A DFT study. Adsorption 2020, 26, 1385–1396. [Google Scholar] [CrossRef]
- Makiabadi, B.; Zakarianezhad, M.; Hosseini, S.S. Investigation and comparison of pristine/doped BN, AlN, and CN nanotubes as drug delivery systems for Tegafur drug: A theoretical study. Struct. Chem. 2021, 32, 1019–1037. [Google Scholar] [CrossRef]
- Chen, K.; Zeng, Q.; He, G.; Sarkar, A. Tyrosine amino acid as a sustainable material for chemical functionalization of single-wall BC2N nanotubes: Quantum chemical study. Struct. Chem. 2021, 32, 1197–1203. [Google Scholar] [CrossRef]
- Muz, İ.; Kurban, H.; Kurban, M. A DFT study on stability and electronic structure of AlN nanotubes. Mater. Today Commun. 2021, 26, 102118. [Google Scholar] [CrossRef]
- García-Toral, D.; Báez, R.M.; Sánchez, S.J.I.; Flores-Riveros, A.; Cocoletzi, G.H.; Rivas-Silva, J.F. Encapsulation of Pollutant Gaseous Molecules by Adsorption on Boron Nitride Nanotubes: A Quantum Chemistry Study. ACS Omega 2021, 6, 14824–14837. [Google Scholar] [CrossRef] [PubMed]
- Beheshtian, J.; Peyghan, A.A.; Bagheri, Z. Carbon nanotube functionalization with carboxylic derivatives: A DFT study. J. Mol. Model. 2013, 19, 391–396. [Google Scholar] [CrossRef]
- Chermahini, A.N.; Teimouri, A.; Farrokhpour, H. A DFT-D study on the interaction between lactic acid and single-wall carbon nanotubes. RSC Adv. 2015, 5, 97724–97733. [Google Scholar] [CrossRef]
- Mendoza-Wilson, A.M.; Glossman-Mitnik, D. CHIH-DFT study of the electronic properties and chemical reactivity of quercetin. J. Mol. Struct. THEOCHEM 2005, 716, 67–72. [Google Scholar] [CrossRef]
- Erkoç, Ş.; Malcioğlu, O.B.; Taşci, E. Structural and electronic properties of single-wall GaN nanotubes: Semi-empirical SCF-MO calculations. J. Mol. Struct. THEOCHEM 2004, 674, 1–5. [Google Scholar] [CrossRef]
- Kaur, J.; Singla, P.; Goel, N. Adsorption of oxazole and isoxazole on BNNT surface: A DFT study. Appl. Surf. Sci. 2015, 328, 632–640. [Google Scholar] [CrossRef]
- Sánchez, S.J.I.; Rivas-Silva, J.F.; García-Toral, D. Study of weak interactions of boron nitride nanotubes with anticancer drug by quantum chemical calculations. Theor. Chem. Acc. 2020, 139, 1–9. [Google Scholar] [CrossRef]
- Makiabadi, B.; Zakarianezhad, M.; Nasab, S.S. Theoretical study of interaction of NH2X (X = H, CH3, CH2OCH3, and CH2COOH) molecules with AlN and AlP nanotubes. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 81–87. [Google Scholar] [CrossRef]
- Mananghaya, M.R.; Santos, G.N.; Yu, D.N. Solubility of amide functionalized single wall carbon nanotubes: A quantum mechanical study. J. Mol. Liq. 2017, 242, 1208–1214. [Google Scholar] [CrossRef]
- Beheshtian, J.; Peyghan, A.A.; Tabar, M.B.; Bagheri, Z. DFT study on the functionalization of a BN nanotube with sulfamide. Appl. Surf. Sci. 2013, 266, 182–187. [Google Scholar] [CrossRef]
- Mananghaya, M. Modeling of single-walled carbon nanotubes functionalized with carboxylic and amide groups towards its solubilization in water. J. Mol. Liq. 2015, 212, 592–596. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chirality/Index | n = 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (n,0) | - | - | 5.56 | 6.46 | 7.43 | 8.44 | - | 10.42 | - | 12.55 | 14.40 |
| (n, n) | 5.19 | 6.85 | 8.15 | 9.99 | 12.11 | 13.91 | 15.55 | 17.38 | 19.31 | - | - |
| Nanotube | Bond Lengths (Ȧ) | Angle (°) | ||||||
|---|---|---|---|---|---|---|---|---|
| Armchair | Axial Length | B-P | P-H | B-H | P-B-P | B-P-B | H-P-B | H-B-P |
| (3,3) | 19.8894 | 1.9129 | 1.4259 | 1.1861 | 122.2794 | 109.5253 | 108.55 | 118.1869 |
| (4,4) | 19.9063 | 1.8924 | 1.4209 | 1.1859 | 119.2242 | 114.2072 | 109.4626 | 118.777 |
| (5,5) | 19.9237 | 1.8862 | 1.4187 | 1.1856 | 120.7065 | 115.1849 | 111.0487 | 119.6057 |
| (6,6) | 19.9376 | 1.8814 | 1.4166 | 1.1857 | 119.691 | 116.4117 | 111.9162 | 119.8233 |
| (7,7) | 19.9503 | 1.8798 | 1.4152 | 1.1856 | 119.6188 | 117.8321 | 112.3506 | 119.6171 |
| (8,8) | 19.9618 | 1.8771 | 1.4143 | 1.1856 | 119.4768 | 118.3511 | 112.997 | 119.7843 |
| (9,9) | 19.9708 | 1.8756 | 1.4134 | 1.1855 | 119.8691 | 118.3118 | 113.6048 | 119.9248 |
| (10,10) | 19.9805 | 1.8747 | 1.4127 | 1.1856 | 120.1455 | 118.9187 | 114.2501 | 120.085 |
| (11,11) | 19.9861 | 1.875 | 1.4122 | 1.1854 | 119.7613 | 119.2771 | 114.7068 | 120.1182 |
| Nanotube | Bond Lengths (Ȧ) | Angle (°) | ||||||
|---|---|---|---|---|---|---|---|---|
| Zigzag | Axial Length | B-P | P-H | B-H | P-B-P | B-P-B | H-P-B | H-B-P |
| (5,0) | 17.8614 | 1.9135 | 1.4351 | 1.1861 | 120.8479 | 105.0648 | 101.9944 | 117.7055 |
| (6,0) | 17.9346 | 1.8995 | 1.4316 | 1.1858 | 121.7117 | 110.9808 | 102.8412 | 117.5638 |
| (7,0) | 17.9806 | 1.8939 | 1.4294 | 1.1856 | 120.3019 | 111.5138 | 104.9868 | 118.5491 |
| (8,0) | 17.9966 | 1.8851 | 1.4279 | 1.1854 | 120.3811 | 114.968 | 105.9255 | 118.8265 |
| (10,0) | 18.0259 | 1.8819 | 1.4255 | 1.1852 | 119.7837 | 115.6483 | 107.5051 | 119.1812 |
| (12,0) | 18.0435 | 1.8778 | 1.424 | 1.1848 | 119.8862 | 116.4566 | 108.7472 | 119.3955 |
| (14,0) | 18.0537 | 1.8775 | 1.4228 | 1.185 | 120.2846 | 118.1968 | 109.7213 | 119.513 |
| Armchair BPNT | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Descriptors | (3,3) | (4,4) | (5,5) | (6,6) | (7,7) | (8,8) | (9,9) | (10,10) | (11,11) |
| −358,734.516 | −478,319.133 | −597,903.132 | −717,486.631 | −837,069.831 | −956,652.817 | −1,076,235.658 | −1,195,818.389 | −1,315,401.058 | |
| −6.8995 | −6.7725 | −6.6888 | −6.6428 | −6.6145 | −6.5971 | −6.5857 | −6.5764 | −6.5712 | |
| −5.6244 | −5.7531 | −5.9084 | −5.9922 | −6.0406 | −6.0694 | −6.0874 | −6.0988 | −6.1061 | |
| Eg Gap | 1.2751 | 1.0195 | 0.7804 | 0.6506 | 0.5739 | 0.5277 | 0.4983 | 0.4776 | 0.4651 |
| I = | 6.8996 | 6.7725 | 6.6888 | 6.6428 | 6.6145 | 6.5971 | 6.5857 | 6.5764 | 6.5712 |
| A = | 5.6244 | 5.7531 | 5.9084 | 5.9922 | 6.0406 | 6.0694 | 6.0874 | 6.0988 | 6.1061 |
| η = (I − A)/2 | 0.6376 | 0.5097 | 0.3902 | 0.3253 | 0.2870 | 0.2638 | 0.2492 | 0.2388 | 0.2326 |
| µ = −(I + A)/2 | −6.2620 | −6.2628 | −6.2986 | −6.3175 | −6.3275 | −6.3332 | −6.3365 | −6.3376 | −6.3387 |
| 30.7516 | 38.4741 | 50.8375 | 61.3418 | 69.7618 | 76.0120 | 80.5761 | 84.0923 | 86.3841 | |
| −2.8618 | −2.9193 | −2.9494 | −2.9665 | −2.9772 | −2.9843 | −2.9892 | −2.9927 | −2.9954 | |
| Zigzag BPNT | |||||||
|---|---|---|---|---|---|---|---|
| Descriptors | (5,0) | (6,0) | (7,0) | (8,0) | (10,0) | (12,0) | (14,0) |
| −348,742.4988 | −418,495.3783 | −488,247.8623 | −558,000.0492 | −697,503.8110 | −837,007.0566 | −976,509.9845 | |
| −6.9882 | −6.9210 | −7.0652 | −6.8106 | −6.8770 | −6.7910 | −6.7140 | |
| −6.9077 | −6.7092 | −6.4298 | −6.2049 | −5.8934 | −5.9500 | −6.0411 | |
| Eg Gap | 0.0805 | 0.2119 | 0.6354 | 0.6057 | 0.9836 | 0.8410 | 0.6729 |
| I = | 6.9882 | 6.9210 | 7.0652 | 6.8106 | 6.8770 | 6.7910 | 6.7140 |
| A = | 6.9077 | 6.7092 | 6.4298 | 6.2049 | 5.8934 | 5.9500 | 6.0411 |
| η = (I − A)/2 | 0.0403 | 0.1059 | 0.3177 | 0.3029 | 0.4918 | 0.4205 | 0.3365 |
| µ = −(I + A)/2 | −6.9480 | −6.8151 | −6.7475 | −6.5077 | −6.3852 | −6.3705 | −6.3776 |
| 599.5909 | 219.1985 | 71.6547 | 69.9151 | 41.4526 | 48.2548 | 60.4427 | |
| −2.8633 | −2.9090 | −2.9380 | −2.9575 | −2.9809 | −2.9939 | −3.0017 | |
| Summary of HOMO-LUMO Gap Values and Molecular Formula Reported in the Literature | ||||
|---|---|---|---|---|
| Chirality | Software + Method | HOMO-LUMO Gap | Molecular Formula | Reference |
| (5,0) | GAMESS/B3LYP/6-31G(d,p) | 1.62 | B27P27H10 | [34] |
| (5,0) | Gaussian16/M06-2X/6-31G(d) | 0.0805 | B35P35H10 | This work |
| (6,0) | Gaussian03/B3LYP/6-31G(d) | 2.27 | B24P24H12 | [12] |
| (6,0) | Gaussian03/B3LYP/6-31G(d) | 2.43 | B24P24H12 | [18] |
| (6,0) | GAMESS/B3LYP/6-31G(d) | 1.93 | B36P36H12 | [14] |
| (6,0) | Gaussian98/BLYP/6-31G(d) | 1.09 | B24P24H12 | [35] |
| (6,0) | GAMESS/B3LYP/6-31G(d) | 2.06 | B30P30H12 | [11] |
| (6,0) | Gaussian98/BLYP/6-31G(d) | 1.09 | B24P24H12 | [17] |
| (6,0) | Gaussian03/B3LYP/6-31G(d) | 2.06 | B30P30H12 | [10] |
| (6,0) | Gaussian98/B3LYP/6-311G(d,p) | 2.25 | B24P24H12 | [36] |
| (6,0) | Gaussian98/B3LYP/6-31G(d) | 2.27 | B24P24H12 | [15] |
| (6,0) | Gaussian16/M06-2X/6-31G(d) | 0.2118 | B42P42H12 | This work |
| (7,0) | GAMESS/B3LYP/6-31G(d) | 2.22 | B42P42H14 | [14] |
| (7,0) | Gaussian16/M06-2X/6-31G(d) | 0.6353 | B49P49H14 | This work |
| (8,0) | Gaussian03/B3LYP/6-31G(d) | 2.57 | B32P32H16 | [20] |
| (8,0) | Gaussian16/M06-2X/6-31G(d) | 0.6057 | B56P56H16 | This work |
| (4,4) | Gaussian98/BLYP/6-31G(d) | 1.77 | B28P28H16 | [17] |
| (4,4) | Gaussian03/B3LYP/6-31G(d) | 2.95 | B28P28H16 | [9] |
| (4,4) | Gaussian98/BLYP/6-31G(d) | 1.77 | B28P28H16 | [35] |
| (4,4) | GAMESS/B3LYP/6-31G(d) | 2.95 | B28P28H16 | [14] |
| (4,4) | Gaussian03/BLYP/6-31G(d) | 1.75 | B28P28H16 | [22] |
| (4,4) | Gaussian03/B3LYP/6-31G(d) | 2.95 | B28P28H16 | [22] |
| (4,4) | Gaussian03/B3LYP/6-31G(d) | 2.95 | B28P28H16 | [21] |
| (4,4) | Gaussian03/B3LYP/6-31G(d) | 2.95 | B32P32H16 | [37] |
| (4,4) | Gaussian16/M06-2X/6-31G(d) | 1.0194 | B48P48H16 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Toral, D.; Mendoza-Báez, R.; Chigo-Anota, E.; Flores-Riveros, A.; Vázquez-Báez, V.M.; Cocoletzi, G.H.; Rivas-Silva, J.F. Structural Stability and Electronic Properties of Boron Phosphide Nanotubes: A Density Functional Theory Perspective. Symmetry 2022, 14, 964. https://doi.org/10.3390/sym14050964
García-Toral D, Mendoza-Báez R, Chigo-Anota E, Flores-Riveros A, Vázquez-Báez VM, Cocoletzi GH, Rivas-Silva JF. Structural Stability and Electronic Properties of Boron Phosphide Nanotubes: A Density Functional Theory Perspective. Symmetry. 2022; 14(5):964. https://doi.org/10.3390/sym14050964
Chicago/Turabian StyleGarcía-Toral, Dolores, Raúl Mendoza-Báez, Ernesto Chigo-Anota, Antonio Flores-Riveros, Víctor M. Vázquez-Báez, Gregorio Hernández Cocoletzi, and Juan Francisco Rivas-Silva. 2022. "Structural Stability and Electronic Properties of Boron Phosphide Nanotubes: A Density Functional Theory Perspective" Symmetry 14, no. 5: 964. https://doi.org/10.3390/sym14050964
APA StyleGarcía-Toral, D., Mendoza-Báez, R., Chigo-Anota, E., Flores-Riveros, A., Vázquez-Báez, V. M., Cocoletzi, G. H., & Rivas-Silva, J. F. (2022). Structural Stability and Electronic Properties of Boron Phosphide Nanotubes: A Density Functional Theory Perspective. Symmetry, 14(5), 964. https://doi.org/10.3390/sym14050964