

The Symmetric Active Site of Enantiospecific Enzymes




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
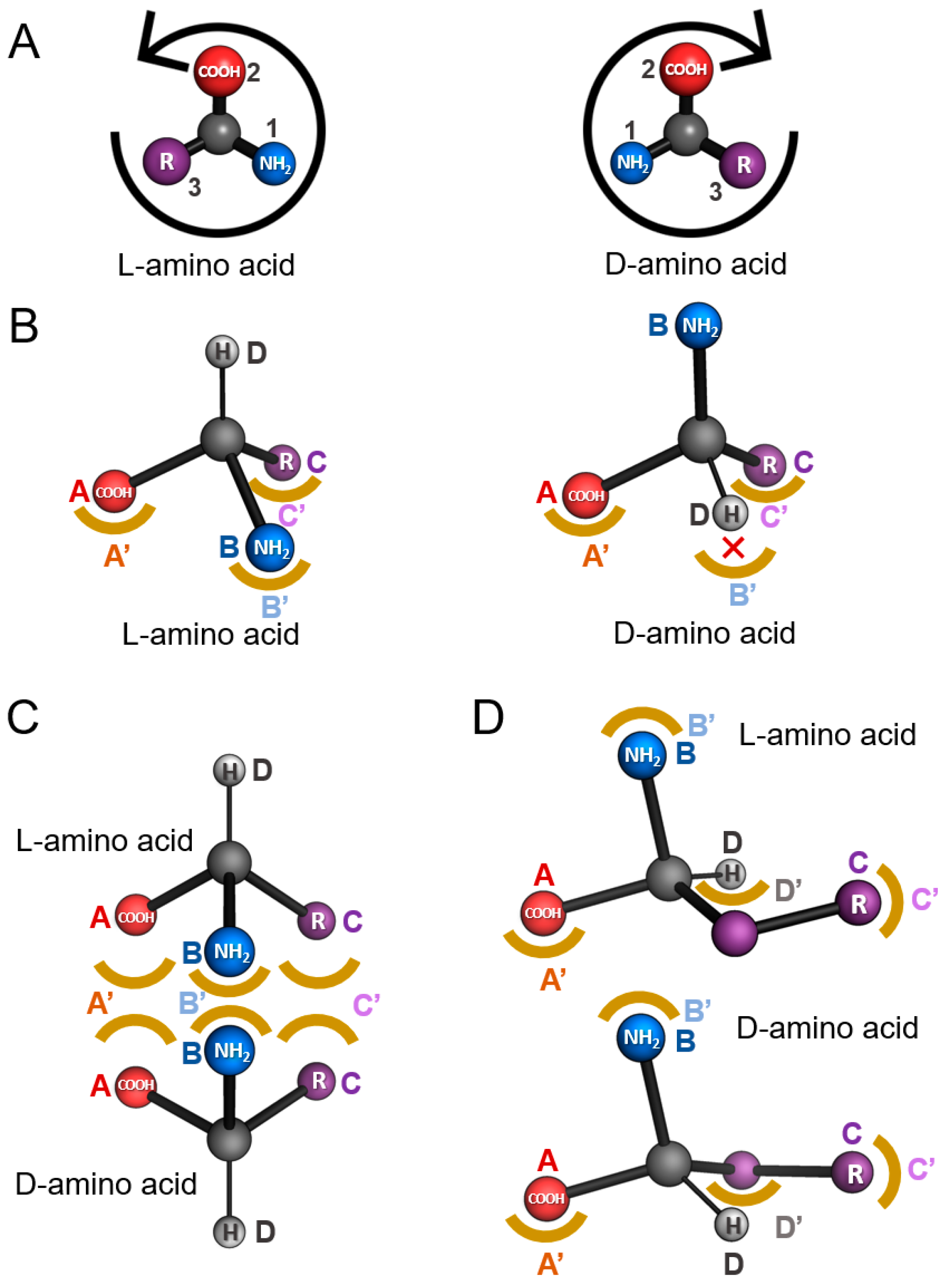
2. Models of Enantiospecificity in Protein-Ligand Interaction
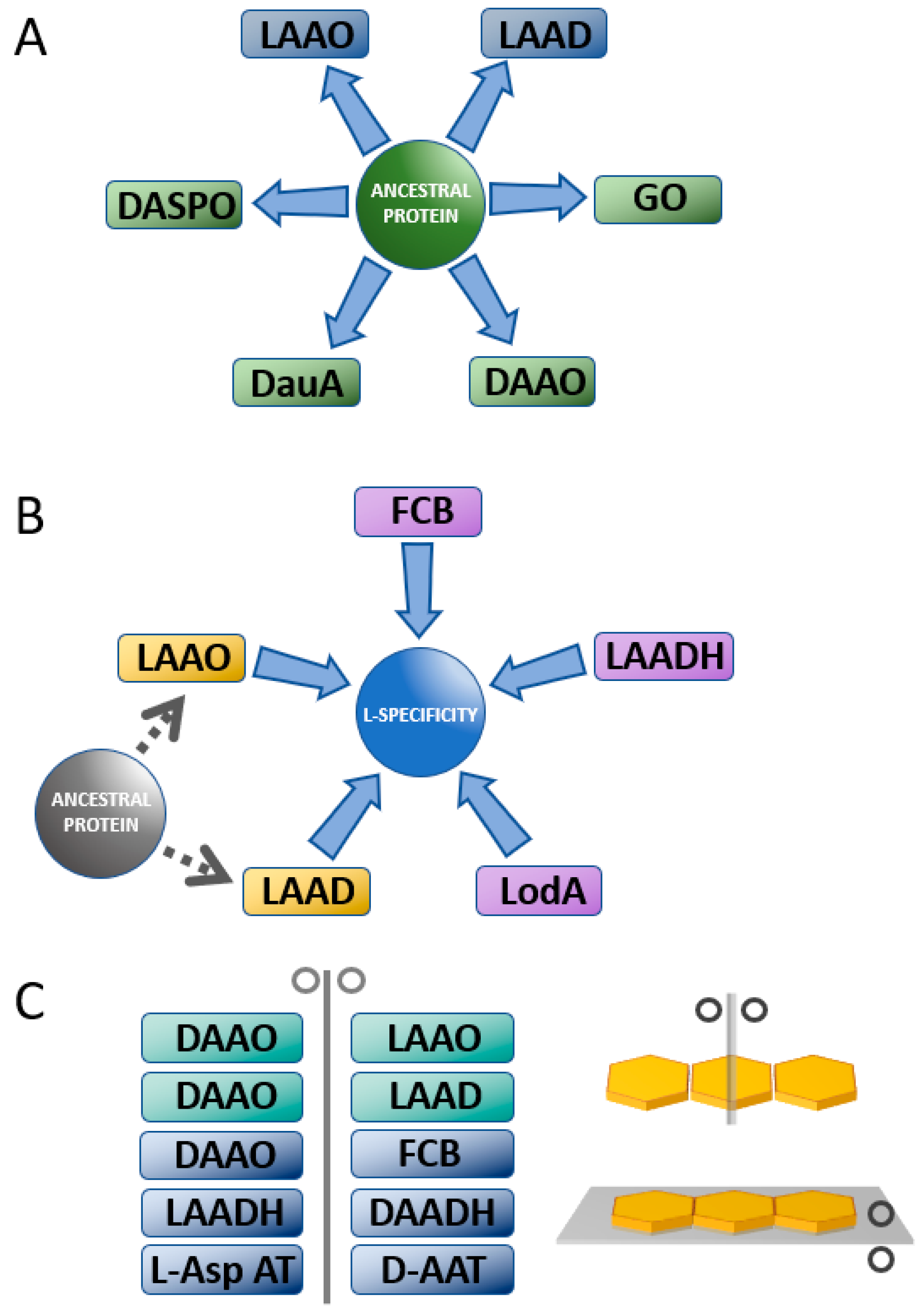
3. Enantiospecificity in Amino Acid Oxidases and Dehydrogenases
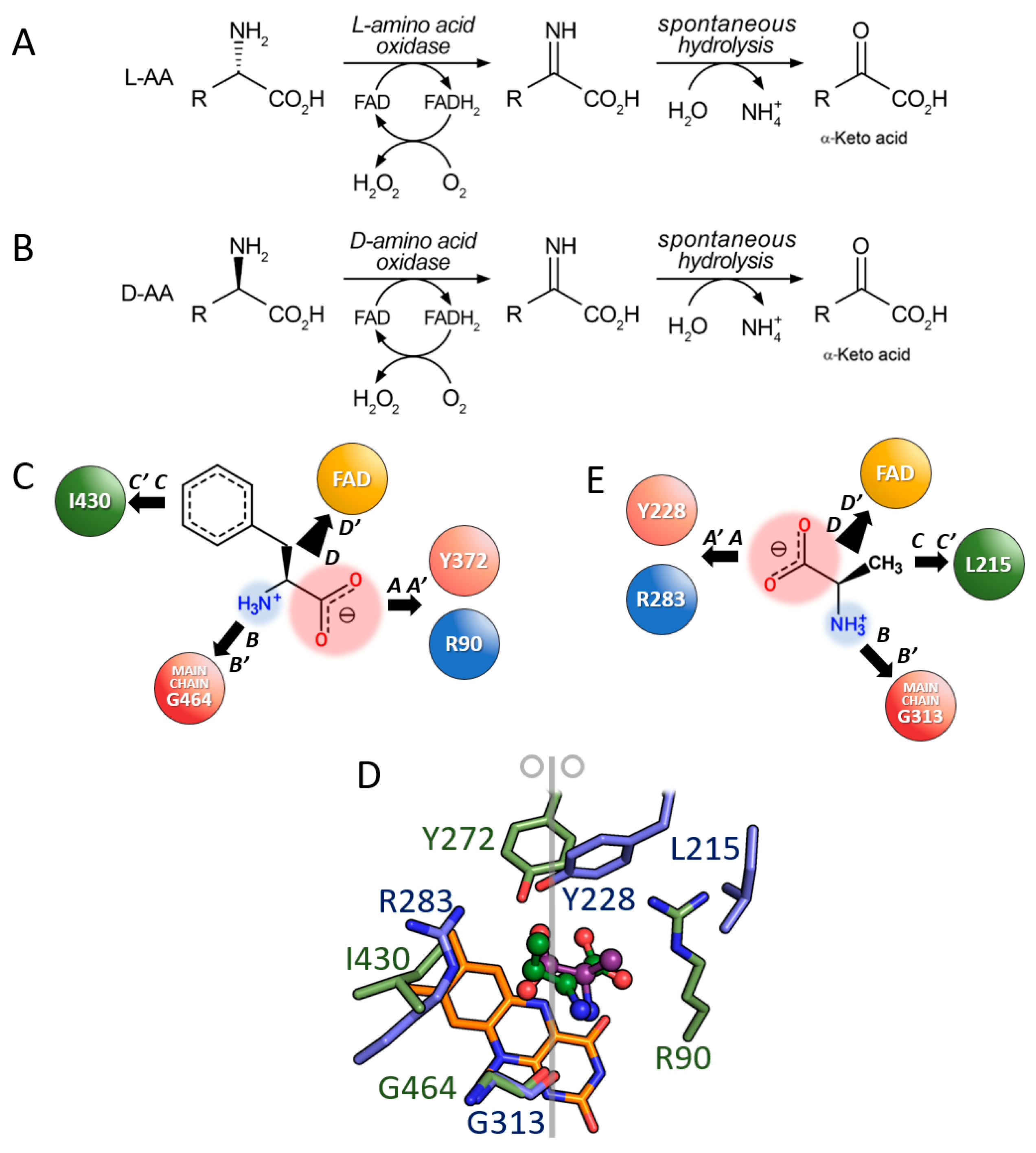
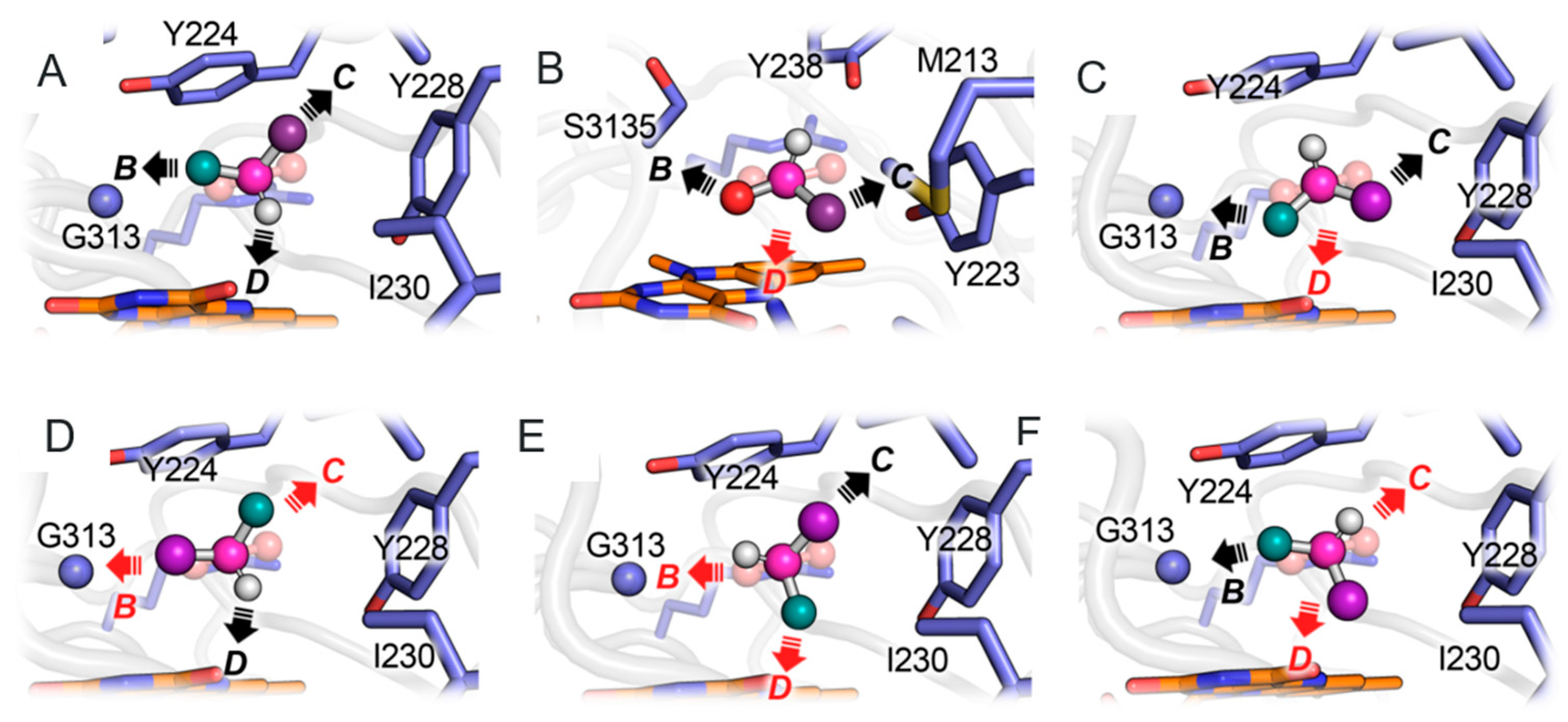
3.1. Comparison between D- and L-Amino Acid Oxidases
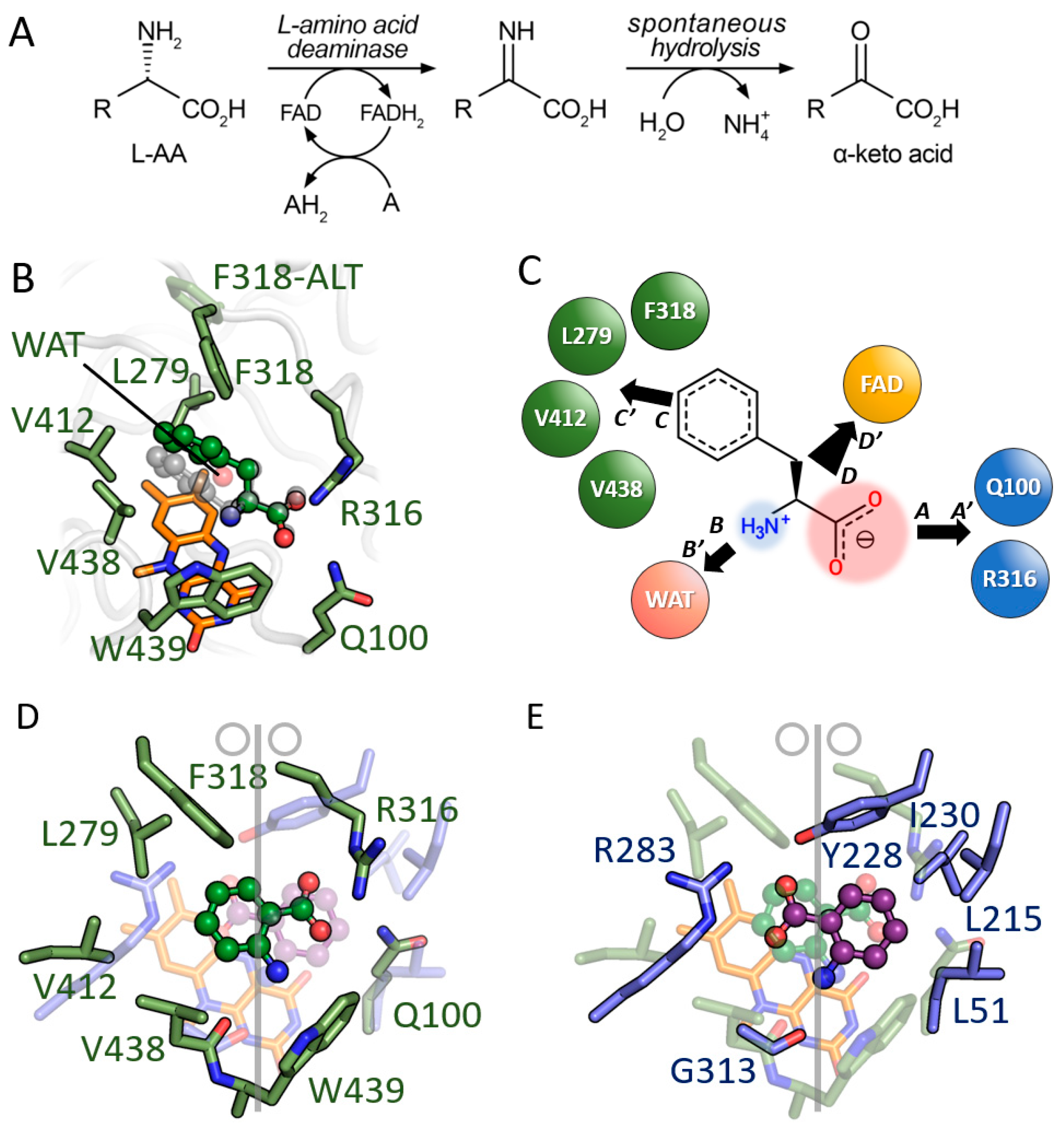
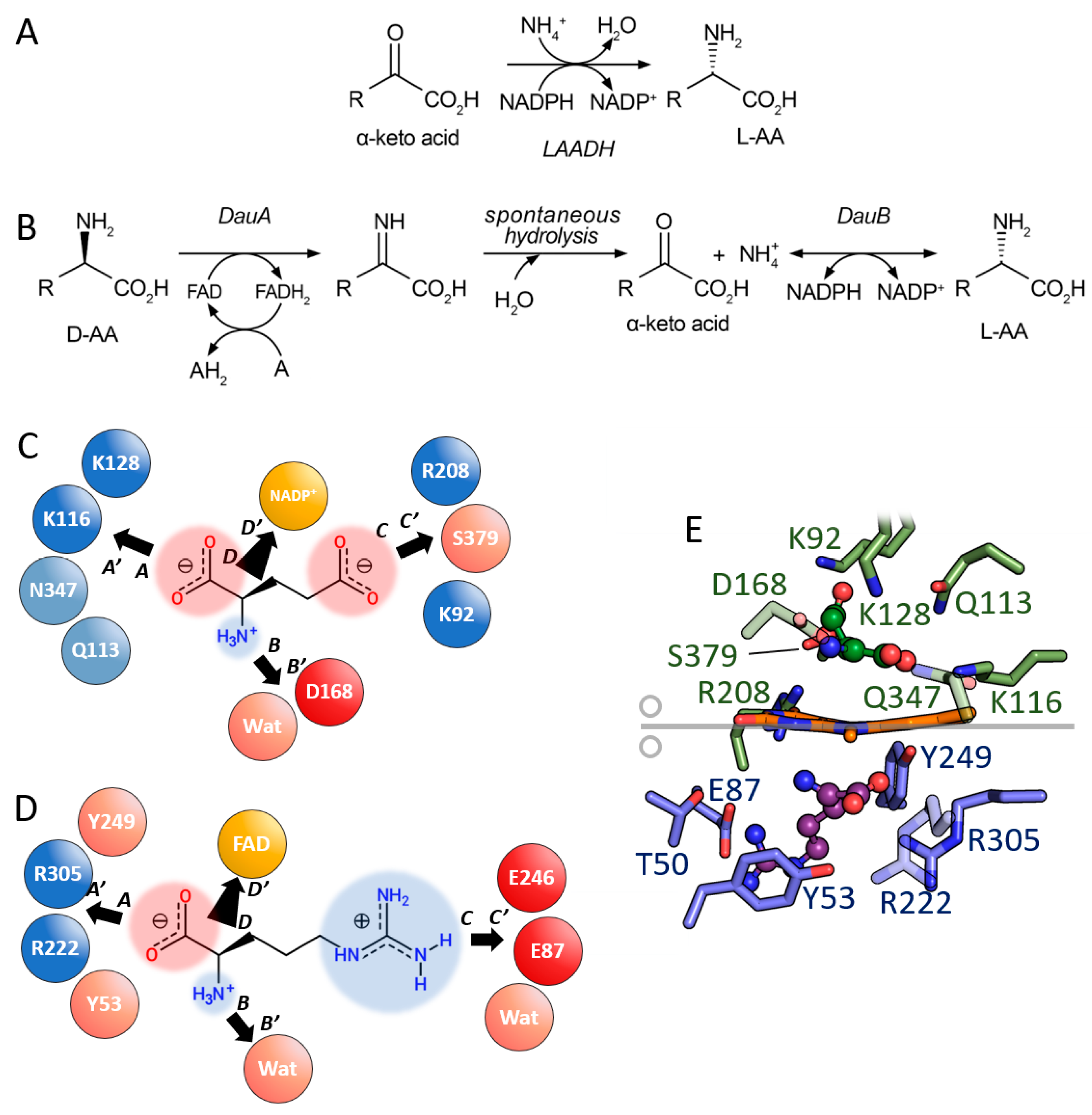
3.2. Comparison between D-Amino Acid Oxidases and L-Amino Acid Deaminases
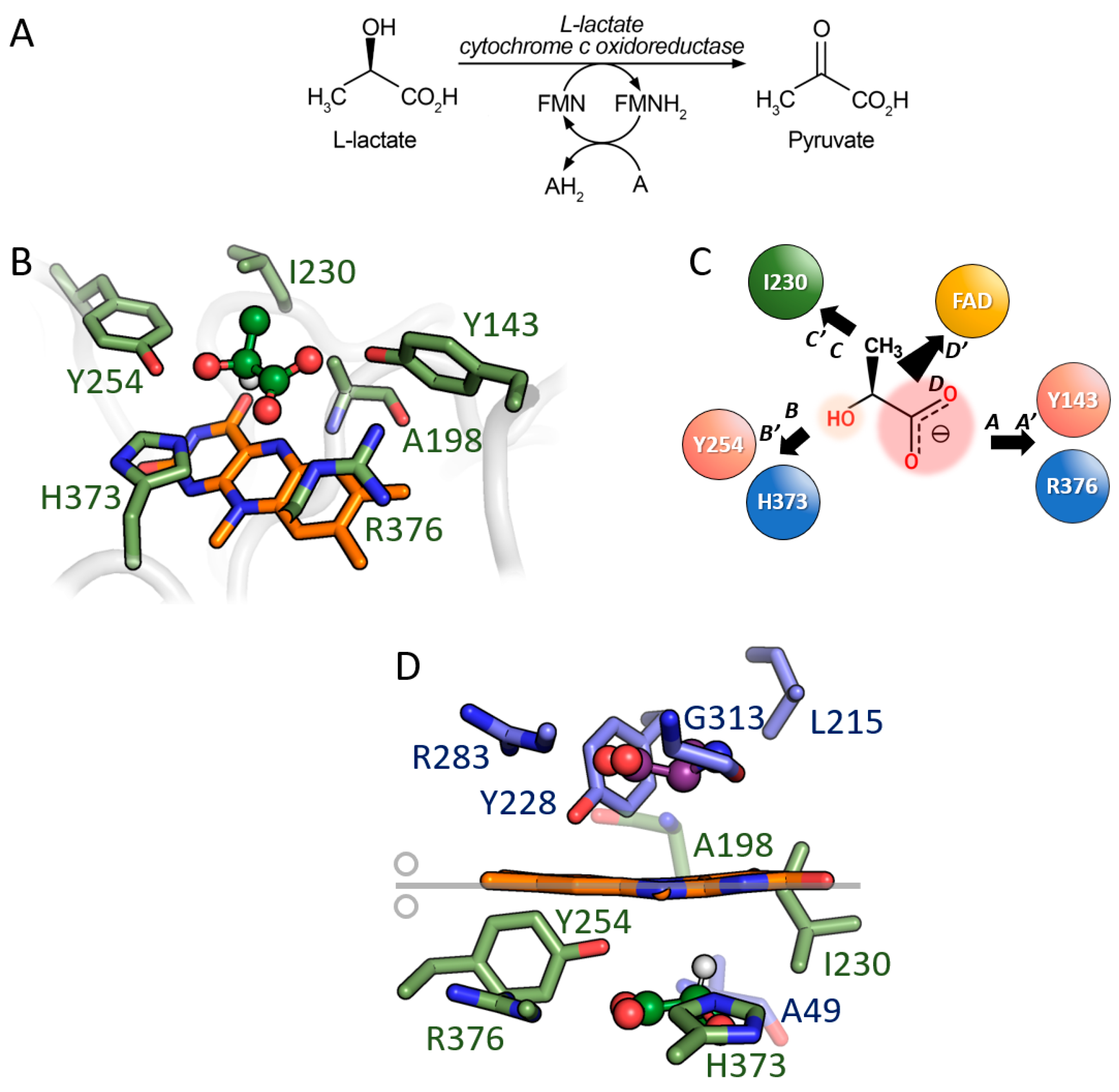
3.3. Comparison between D-Amino Acid Oxidases and L-Lactate Cytochrome c Oxidoreductase
3.4. Comparison between D- and L-Amino Acid Dehydrogenases
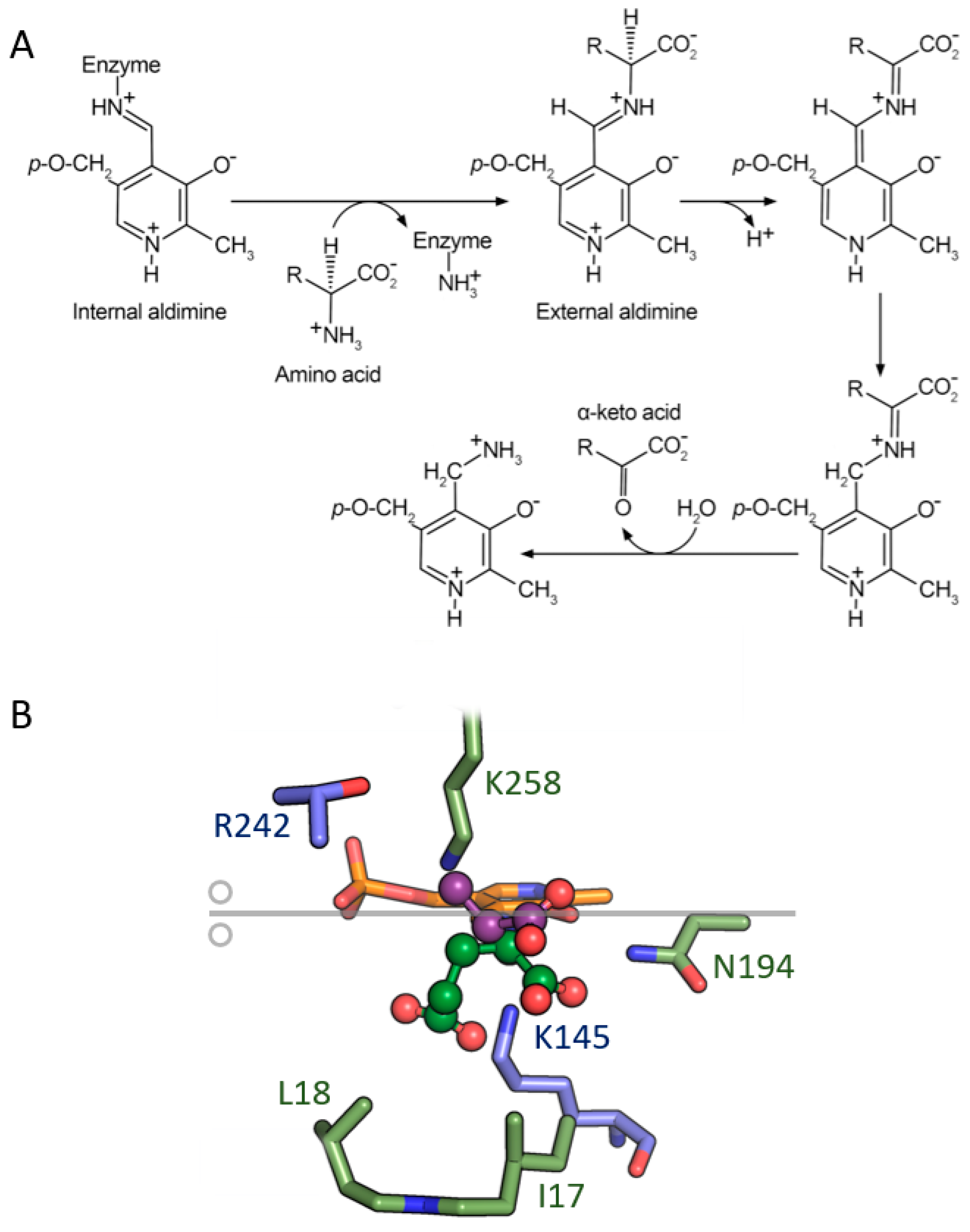
4. Aminotransferases
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gal, J. Pasteur and the Art of Chirality. Nat. Chem. 2017, 9, 604–605. [Google Scholar] [CrossRef]
- Pasteur, L. Recherches Sur Les Relations Qui Peuvent Exister Entre La Forme Cristalline, La Composition Chimique et Le Sens de La Polarisation Rotatoire. Ann. Chim. Phys. 1848, 24, 422–459. [Google Scholar]
- Fischer, E. Über Die Konfiguration Des Traubenzuckers Und Seiner Isomeren, I & II. Ber. Dtsch. Chem. Ges. 1891, 24, 2683–2687. [Google Scholar]
- Peretó, J.; López-García, P.; Moreira, D. Ancestral Lipid Biosynthesis and Early Membrane Evolution. Trends Biochem. Sci. 2004, 29, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Preciado, A.; Romero, H.; Peimbert, M. An Evolutionary Perspective on Amino Acids. Nat. Educ. 2010, 3, 29. [Google Scholar]
- Sanchez, Z.; Tani, A.; Kimbara, K. Extensive Reduction of Cell Viability and Enhanced Matrix Production in Pseudomonas Aeruginosa PAO1 Flow Biofilms Treated with a D-Amino Acid Mixture. Appl. Environ. Microbiol. 2013, 79, 1396–1399. [Google Scholar] [CrossRef] [PubMed]
- Hochbaum, A.I.; Kolodkin-Gal, I.; Foulston, L.; Kolter, R.; Aizenberg, J.; Losick, R. Inhibitory Effects of D-Amino Acids on Staphylococcus Aureus Biofilm Development. J. Bacteriol. 2011, 193, 5616–5622. [Google Scholar] [CrossRef] [PubMed]
- Leiman, S.A.; May, J.M.; Lebar, M.D.; Kahne, D.; Kolter, R.; Losick, R. D-Amino Acids Indirectly Inhibit Biofilm Formation in Bacillus Subtilis by Interfering with Protein Synthesis. J. Bacteriol. 2013, 195, 5391–5395. [Google Scholar] [CrossRef]
- Osborn, M.J. Structure and Biosynthesis of the Bacterial Cell Wall. Annu. Rev. Biochem. 1969, 38, 501–538. [Google Scholar] [CrossRef]
- Schell, M.J.; Brady, R.O., Jr.; Molliver, M.E.; Snyder, S.H. D-Serine as a Neuromodulator: Regional and Developmental Localizations in Rat Brain Glia Resemble NMDA Receptors. J. Neurosci. 1997, 17, 1604–1615. [Google Scholar] [CrossRef]
- Pollegioni, L.; Sacchi, S. Metabolism of the Neuromodulator D-serine. Cell Mol. Life Sci. 2010, 67, 2387–2404. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, M.M.; Santillo, A.; Chieffi Baccari, G. Current Knowledge of D-aspartate in Glandular Tissues. Amino Acids 2014, 46, 1805–1818. [Google Scholar] [CrossRef]
- Marcone, G.L.; Rosini, E.; Crespi, E.; Pollegioni, L. D-Amino Acids in Foods. Appl. Microbiol. Biotechnol. 2020, 104, 555–574. [Google Scholar] [CrossRef]
- Gordon, E.M.; Ondetti, M.A.; Pluscec, J.; Cimarusti, C.M.; Bonner, D.P.; Sykes, R.B. O-Sulfated. Beta.-Lactam Hydroxamic Acids (Monosulfactams). Novel Monocyclic. Beta.-Lactam Antibiotics of Synthetic Origin. J. Am. Chem. Soc. 1982, 104, 6053–6060. [Google Scholar] [CrossRef]
- Patel, R. Biocatalytic Synthesis of Chiral Alcohols and Amino Acids for Development of Pharmaceuticals. Biomolecules 2013, 3, 741–777. [Google Scholar] [CrossRef]
- Gustafsson, D.; Elg, M.; Lenfors, S.; Brjesson, I.; Teger-Nilsson, A.-C. Effects of Inogatran, a New Low-Molecular-Weight Thrombin Inhibitor, in Rat Models of Venous and Arterial Thrombosis, Thrombolysis and Bleeding Time. Blood Coagul. Fibrinolysis 1996, 7, 69–79. [Google Scholar] [CrossRef]
- Pollegioni, L.; Rosini, E.; Molla, G. Advances in Enzymatic Synthesis of D-Amino Acids. Int. J. Mol. Sci. 2020, 21, 3206. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.M.; Fernandes, C.; Remião, F.; Tiritan, M.E. Enantioselectivity in Drug Pharmacokinetics and Toxicity: Pharmacological Relevance and Analytical Methods. Molecules 2021, 26, 3113. [Google Scholar] [CrossRef]
- Shen, Z.; Lv, C.; Zeng, S. Significance and Challenges of Stereoselectivity Assessing Methods in Drug Metabolism. J. Pharm. Anal. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Leek, H.; Andersson, S. Preparative Scale Resolution of Enantiomers Enables Accelerated Drug Discovery and Development. Molecules 2017, 22, 158. [Google Scholar] [CrossRef]
- Easson, L.H.; Stedman, E. Studies on the Relationship between Chemical Constitution and Physiological Action. Biochem. J. 1933, 27, 1257–1266. [Google Scholar] [CrossRef]
- Mikhael, S.; Abrol, R. Chiral Graphs: Reduced Representations of Ligand Scaffolds for Stereoselective Biomolecular Recognition, Drug Design, and Enhanced Exploration of Chemical Structure Space. ChemMedChem 2019, 14, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Mesecar, A.D.; Koshland, D.E. A New Model for Protein Stereospecificity. Nature 2000, 403, 614–615. [Google Scholar] [CrossRef] [PubMed]
- Hanson, K.R. Phenylalanine Ammonia-Lyase: Mirror-Image Packing of D- and L-Phenylalanine and D- and L-Transition State Analogs into the Active Site. Arch. Biochem. Biophys. 1981, 211, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Bearne, S.L. Through the Looking Glass: Chiral Recognition of Substrates and Products at the Active Sites of Racemases and Epimerases. Chem. A Eur. J. 2020, 26, 10367–10390. [Google Scholar] [CrossRef]
- Bentley, R. Diastereoisomerism, Contact Points, and Chiral Selectivity: A Four-Site Saga. Arch. Biochem. Biophys. 2003, 414, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Kazemi, M.; Planas, F.; Himo, F. Modeling Enzymatic Enantioselectivity using Quantum Chemical Methodology. ACS Catal. 2020, 10, 6430–6449. [Google Scholar] [CrossRef]
- Thamim, M.; Thirumoorthy, K. Chiral Discrimination in a Mutated IDH Enzymatic Reaction in Cancer: A Computational Perspective. Eur. Biophys. J. 2020, 49, 549–559. [Google Scholar] [CrossRef]
- Wakamatsu, T.; Sakuraba, H.; Kitamura, M.; Hakumai, Y.; Fukui, K.; Ohnishi, K.; Ashiuchi, M.; Ohshima, T. Structural Insights into L-Tryptophan Dehydrogenase from a Photoautotrophic Cyanobacterium, Nostoc Punctiforme. Appl. Environ. Microbiol. 2017, 83, 16. [Google Scholar] [CrossRef]
- Li, C.; Lu, C.D. Arginine Racemization by Coupled Catabolic and Anabolic Dehydrogenases. Proc. Natl. Acad. Sci. USA 2009, 106, 906–911. [Google Scholar] [CrossRef]
- Pollegioni, L.; Motta, P.; Molla, G. L-Amino Acid Oxidase as Biocatalyst: A Dream Too Far? Appl. Microbiol. Biotechnol. 2013, 97, 9323–9341. [Google Scholar] [CrossRef] [PubMed]
- Lucas-Elío, P.; Gómez, D.; Solano, F.; Sanchez-Amat, A. The Antimicrobial Activity of Marinocine, Synthesized by Marinomonas Mediterranea, Is Due to Hydrogen Peroxide Generated by Its Lysine Oxidase Activity. J. Bacteriol. 2006, 188, 2493–2501. [Google Scholar] [CrossRef]
- Gómez, D.; Lucas-Elío, P.; Sanchez-Amat, A.; Solano, F. A Novel Type of Lysine Oxidase: L-Lysine-ε-Oxidase. Biochim. Et Biophys. Acta (BBA) Proteins Proteom. 2006, 1764, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Schriek, S.; Kahmann, U.; Staiger, D.; Pistorius, E.K.; Michel, K.-P. Detection of an L-Amino Acid Dehydrogenase Activity in Synechocystis Sp. PCC 6803. J. Exp. Bot. 2009, 60, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A. Metabolism of Amino-Acids. Biochem. J. 1935, 29, 1951–1969. [Google Scholar] [CrossRef]
- Mattevi, A.; Vanoni, M.A.; Todone, F.; Rizzi, M.; Teplyakov, A.; Coda, A.; Bolognesi, M.; Curti, B. Crystal Structure of D-Amino Acid Oxidase: A Case of Active Site Mirror-Image Convergent Evolution with Flavocytochrome B2. Proc. Natl. Acad. Sci. USA 1996, 93, 7496–7501. [Google Scholar] [CrossRef]
- Pilone, M.S.; Pollegioni, L. D-Amino Acid Oxidase as an Industrial Biocatalyst. Biocatal. Biotransformation 2002, 20, 145–159. [Google Scholar] [CrossRef]
- Job, V.; Molla, G.; Pilone, M.S.; Pollegioni, L. Overexpression of a Recombinant Wild-Type and His-Tagged Bacillus Subtilis Glycine Oxidase in Escherichia Coli. Eur. J. Biochem. 2002, 269, 1456–1463. [Google Scholar] [CrossRef]
- Job, V.; Marcone, G.L.; Pilone, M.S.; Pollegioni, L. Glycine Oxidase from Bacillus Subtilis. Characterization of a New Flavoprotein. J. Biol. Chem. 2002, 277, 6985–6993. [Google Scholar] [CrossRef]
- Mörtl, M.; Diederichs, K.; Welte, W.; Molla, G.; Motteran, L.; Andriolo, G.; Pilone, M.S.; Pollegioni, L. Structure-Function Correlation in Glycine Oxidase from Bacillus Subtilis. J. Biol. Chem. 2004, 279, 29718–29727. [Google Scholar] [CrossRef]
- Settembre, E.; Begley, T.P.; Ealick, S.E. Structural Biology of Enzymes of the Thiamin Biosynthesis Pathway. Curr. Opin. Struct. Biol. 2003, 13, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Umhau, S.; Pollegioni, L.; Molla, G.; Diederichs, K.; Welte, W.; Pilone, M.S.; Ghisla, S. The X-Ray Structure of D-Amino Acid Oxidase at Very High Resolution Identifies the Chemical Mechanism of Flavin-Dependent Substrate Dehydrogenation. Proc. Natl. Acad. Sci. USA 2000, 97, 12463–12468. [Google Scholar] [CrossRef] [PubMed]
- Pollegioni, L.; Piubelli, L.; Sacchi, S.; Pilone, M.S.; Molla, G. Physiological Functions of D-Amino Acid Oxidases: From Yeast to Humans. Cell. Mol. Life Sci. 2007, 64, 1373–1394. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, P.D.; Cheah, J.; Coulombe, R.; Macheroux, P.; Ghisla, S.; Vrielink, A. The Structure of L-Amino Acid Oxidase Reveals the Substrate Trajectory into an Enantiomerically Conserved Active Site. EMBO J. 2000, 19, 4204–4215. [Google Scholar] [CrossRef]
- Yu, Z.; Qiao, H. Advances in Non-Snake Venom l-Amino Acid Oxidase. Appl. Biochem. Biotechnol. 2012, 167, 1–13. [Google Scholar] [CrossRef]
- Lukasheva, E.V.; Efremova, A.A.; Treshalina, E.M.; Arinbasarova, A.Y.; Medentzev, A.G.; Berezov, T.T. L-Amino Acid Oxidases: Properties and Molecular Mechanisms of Action. Biochem. Mosc. Suppl. B Biomed. Chem. 2011, 5, 337–345. [Google Scholar] [CrossRef]
- Izidoro, L.F.M.; Sobrinho, J.C.; Mendes, M.M.; Costa, T.R.; Grabner, A.N.; Rodrigues, V.M.; da Silva, S.L.; Zanchi, F.B.; Zuliani, J.P.; Fernandes, C.F.C.; et al. Snake Venom L-Amino Acid Oxidases: Trends in Pharmacology and Biochemistry. Biomed. Res. Int. 2014, 2014, 196754. [Google Scholar] [CrossRef]
- Nielsen, V.G. Characterization of L-Amino Acid Oxidase Derived from Crotalus Adamanteus Venom: Procoagulant and Anticoagulant Activities. Int. J. Mol. Sci. 2019, 20, 4853. [Google Scholar] [CrossRef]
- Yang, C.A.; Cheng, C.H.; Lo, C.T.; Liu, S.Y.; Lee, J.W.; Peng, K.C. A Novel L-Amino Acid Oxidase from Trichoderma Harzianum ETS 323 Associated with Antagonism of Rhizoctonia Solani. J. Agric. Food Chem. 2011, 59, 4519–4526. [Google Scholar] [CrossRef]
- Mai-Prochnow, A.; Lucas-Elio, P.; Egan, S.; Thomas, T.; Webb, J.S.; Sanchez-Amat, A.; Kjelleberg, S. Hydrogen Peroxide Linked to Lysine Oxidase Activity Facilitates Biofilm Differentiation and Dispersal in Several Gram-Negative Bacteria. J. Bacteriol. 2008, 190, 5493–5501. [Google Scholar] [CrossRef]
- Kitani, Y.; Kikuchi, N.; Zhang, G.; Ishizaki, S.; Shimakura, K.; Shiomi, K.; Nagashima, Y. Antibacterial Action of L-Amino Acid Oxidase from the Skin Mucus of Rockfish Sebastes Schlegelii. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 149, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Kasai, K.; Ishikawa, T.; Nakamura, T.; Miura, T. Antibacterial Properties of L-Amino Acid Oxidase: Mechanisms of Action and Perspectives for Therapeutic Applications. Appl. Microbiol. Biotechnol. 2015, 99, 7847–7857. [Google Scholar] [CrossRef] [PubMed]
- Puiffe, M.-L.; Lachaise, I.; Molinier-Frenkel, V.; Castellano, F. Antibacterial Properties of the Mammalian L-Amino Acid Oxidase IL4I1. PLoS ONE 2013, 8, e54589. [Google Scholar] [CrossRef] [PubMed]
- Saam, J.; Rosini, E.; Molla, G.; Schulten, K.; Pollegioni, L.; Ghisla, S. O2 Reactivity of Flavoproteins: Dynamic Access of Dioxygen to the Active Site and Role of a H+ Relay System in D-Amino Acid Oxidase. J. Biol. Chem. 2010, 285, 24439–24446. [Google Scholar] [CrossRef] [PubMed]
- Ball, J.; Gannavaram, S.; Gadda, G. Structural Determinants for Substrate Specificity of Flavoenzymes Oxidizing D-Amino Acids. Arch. Biochem. Biophys. 2018, 660, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Molla, G.; Porrini, D.; Job, V.; Motteran, L.; Vegezzi, C.; Campaner, S.; Pilone, M.S.; Pollegioni, L. Role of Arginine 285 in the Active Site of Rhodotorula gracilis D-amino Acid Oxidase. A Site-Directed Mutagenesis Study. J. Biol. Chem. 2000, 275, 24715–24721. [Google Scholar] [CrossRef]
- Kawazoe, T.; Park, H.K.; Iwana, S.; Tsuge, H.; Fukui, K. Human D-Amino Acid Oxidase: An Update and Review. Chem. Rec. 2007, 7, 305–315. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, L.; Zhang, Z.; Du, B.; Xiao, Y.; Yang, K.; Gong, L.; Wu, L.; Li, X.; He, Y. pH-Dependent Enantioselectivity of D-Amino Acid Oxidase in Aqueous Solution. Sci. Rep. 2017, 7, 2994. [Google Scholar] [CrossRef]
- Pollegioni, L.; Diederichs, K.; Molla, G.; Umhau, S.; Welte, W.; Ghisla, S.; Pilone, M.S. Yeast D-Amino Acid Oxidase: Structural Basis of Its Catalytic Properties. J. Mol. Biol. 2002, 324, 535–546. [Google Scholar] [CrossRef]
- Takahashi, S.; Takahashi, T.; Kera, Y.; Matsunaga, R.; Shibuya, H.; Yamada, R.H. Cloning and Expression in Escherichia Coli of the D-Aspartate Oxidase Gene from the Yeast Cryptococcus Humicola and Characterization of the Recombinant Enzyme. J. Biochem. 2004, 135, 533–540. [Google Scholar] [CrossRef]
- Negri, A.; Ceciliani, F.; Tedeschi, G.; Simonic, T.; Ronchi, S. The Primary Structure of the Flavoprotein D-Aspartate Oxidase from Beef Kidney. J. Biol. Chem. 1992, 267, 11865–11871. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Kakuichi, T.; Fujii, K.; Kera, Y.; Yamada, R. Physiological Role Of D-Aspartate Oxidase in the Assimilation and Detoxification Of D-Aspartate in the Yeast Cryptococcus Humicola. Yeast 2005, 22, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, Y.; Katane, M.; Miyamoto, T.; Sekine, M.; Sakamoto, T.; Imai, H.; Homma, H. Secreted D-Aspartate Oxidase Functions in C. Elegans Reproduction and Development. FEBS J. 2019, 286, 124–138. [Google Scholar] [CrossRef]
- Molla, G.; Chaves-Sanjuan, A.; Savinelli, A.; Nardini, M.; Pollegioni, L. Structure and Kinetic Properties of Human D-Aspartate Oxidase, the Enzyme-Controlling d-Aspartate Levels in Brain. FASEB J. 2020, 34, 1182–1197. [Google Scholar] [CrossRef] [PubMed]
- Katane, M.; Kawata, T.; Nakayama, K.; Saitoh, Y.; Kaneko, Y.; Matsuda, S.; Saitoh, Y.; Miyamoto, T.; Sekine, M.; Homma, H. Characterization of the Enzymatic and Structural Properties of Human D-Aspartate Oxidase and Comparison with Those of the Rat and Mouse Enzymes. Biol. Pharm. Bull. 2015, 38, 298–305. [Google Scholar] [CrossRef]
- Takahashi, S. D-Aspartate Oxidase: Distribution, Functions, Properties, and Biotechnological Applications. Appl. Microbiol. Biotechnol. 2020, 104, 2883–2895. [Google Scholar] [CrossRef]
- Tedeschi, G.; Negri, A.; Mortarino, M.; Ceciliani, F.; Simonic, T.; Faotto, L.; Ronchi, S. L-Aspartate Oxidase from Escherichia Coli. II. Interaction with C4 Dicarboxylic Acids and Identification of a Novel L-Aspartate:Fumarate Oxidoreductase Activity. Eur. J. Biochem. 1996, 239, 427–433. [Google Scholar] [CrossRef]
- Motta, P.; Molla, G.; Pollegioni, L.; Nardini, M. Structure-Function Relationships in L-Amino Acid Deaminase, a Flavoprotein Belonging to a Novel Class of Biotechnologically Relevant Enzymes. J. Biol. Chem. 2016, 291, 10457–10475. [Google Scholar] [CrossRef]
- Takahashi, E.; Ito, K.; Yoshimoto, T. Cloning of L-Amino Acid Deaminase Gene from Proteus Vulgaris. Biosci. Biotechnol. Biochem. 1999, 63, 2244–2247. [Google Scholar] [CrossRef]
- Molla, G.; Melis, R.; Pollegioni, L. Breaking the Mirror: L-Amino Acid Deaminase, a Novel Stereoselective Biocatalyst. Biotechnol. Adv. 2017, 35, 657–668. [Google Scholar] [CrossRef]
- Stefan, R.I.; Bokretsion, R.G.; van Staden, J.F.; Aboul-Enein, H.Y. Determination of L- And D-Enantiomers of Methotrexate Using Amperometric Biosensors. Talanta 2003, 60, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Inaba, Y.; Mizukami, K.; Hamada-Sato, N.; Kobayashi, T.; Imada, C.; Watanabe, E. Development of a L-Alanine Sensor for the Monitoring of a Fermentation Using the Improved Selectivity by the Combination of D-Amino Acid Oxidase and Pyruvate Oxidase. Biosens. Bioelectron. 2003, 19, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, R.; Serra, B.; Reviejo, A.J.; Pingarrón, J.M. Chiral Analysis of Amino Acids Using Electrochemical Composite Bienzyme Biosensors. Anal. Biochem. 2001, 298, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Rosini, E.; Molla, G.; Rossetti, C.; Pilone, M.S.; Pollegioni, L.; Sacchi, S. A Biosensor for All D-Amino Acids Using Evolved D-Amino Acid Oxidase. J. Biotechnol. 2008, 135, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Pernot, P.; Mothet, J.P.; Schuvailo, O.; Soldatkin, A.; Pollegioni, L.; Pilone, M.; Adeline, M.T.; Cespuglio, R.; Marinesco, S. Characterization of a Yeast D-Amino Acid Oxidase Microbiosensor for D-Serine Detection in the Central Nervous System. Anal. Chem. 2008, 80, 1589–1597. [Google Scholar] [CrossRef]
- Pollegioni, L.; Molla, G. New Biotech Applications from Evolved D-Amino Acid Oxidases. Trends Biotechnol. 2011, 29, 276–283. [Google Scholar] [CrossRef]
- Melis, R.; Rosini, E.; Pirillo, V.; Pollegioni, L.; Molla, G. In Vitro Evolution of an L-Amino Acid Deaminase Active on L-1-Naphthylalanine. Catal. Sci. Technol. 2018, 8, 5359–5367. [Google Scholar] [CrossRef]
- Moynihan, K.; Elion, G.B.; Pegram, C.; Reist, C.J.; Wellner, D.; Bigner, D.D.; Griffith, O.W.; Friedman, H.S. L -Amino Acid Oxidase (LOX) Modulation of Melphalan Activity against Intracranial Glioma. Cancer Chemother. Pharmacol. 1996, 39, 179–186. [Google Scholar] [CrossRef]
- Lukasheva, E.V.; Berezov, T.T. L-Lysine Alpha-Oxidase: Physicochemical and Biological Properties. Biochemistry 2002, 67, 1152–1158. [Google Scholar] [CrossRef]
- Sukhacheva, M.V.; Zhuravleva, N.I. Properties and Prospects of Practical Use of Extracellular L-Glutamate Oxidase from Streptomyces Sp. Z-11-6. Prikl. Biokhim. Mikrobiol. 2004, 40, 173–177. [Google Scholar]
- Gherardini, P.F.; Wass, M.N.; Helmer-Citterich, M.; Sternberg, M.J.E. Convergent Evolution of Enzyme Active Sites Is Not a Rare Phenomenon. J. Mol. Biol. 2007, 372, 817–845. [Google Scholar] [CrossRef] [PubMed]
- Ogura, R.; Wakamatsu, T.; Mutaguchi, Y.; Doi, K.; Ohshima, T. Biochemical Characterization of an L-Tryptophan Dehydrogenase from the Photoautotrophic Cyanobacterium Nostoc Punctiforme. Enzyme Microb. Technol. 2014, 60, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.J.; Waugh, M.L.; Wang, X.-G.; Stillman, T.J.; Turnbull, A.P.; Engel, P.C.; Rice, D.W. Determinants of Substrate Specificity in the Superfamily of Amino Acid Dehydrogenases. Biochemistry 1997, 36, 16109–16115. [Google Scholar] [CrossRef]
- Britton, K.L.; Baker, P.J.; Engel, P.C.; Rice, D.W.; Stillman, T.J. Evolution of Substrate Diversity in the Superfamily of Amino Acid Dehydrogenases. J. Mol. Biol. 1993, 234, 938–945. [Google Scholar] [CrossRef]
- Heydari, M.; Ohshima, T.; Nunoura-Kominato, N.; Sakuraba, H. Highly Stable L-Lysine 6-Dehydrogenase from the Thermophile Geobacillus Stearothermophilus Isolated from a Japanese Hot Spring: Characterization, Gene Cloning and Sequencing, and Expression. Appl. Environ. Microbiol. 2004, 70, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, Q.; Wang, H. Chiral Resolution of DL-glutamic Acid by a Chiral Additive. J. Chem. Technol. Biotechnol. 2022, 97, 1240–1246. [Google Scholar] [CrossRef]
- Son, H.F.; Kim, I.K.; Kim, K.J. Structural Insights into Domain Movement and Cofactor Specificity of Glutamate Dehydrogenase from Corynebacterium Glutamicum. Biochem. Biophys. Res. Commun. 2015, 459, 387–392. [Google Scholar] [CrossRef]
- Tomita, T.; Yin, L.; Nakamura, S.; Kosono, S.; Kuzuyama, T.; Nishiyama, M. Crystal Structure of the 2-Iminoglutarate-Bound Complex of Glutamate Dehydrogenase from Corynebacterium Glutamicum. FEBS Lett. 2017, 591, 1611–1622. [Google Scholar] [CrossRef]
- Jones, H.; Venables, W.A. Effects of Solubilisation on Some Properties of the Membrane-Bound Respiratory Enzyme D-Amino Acid Dehydrogenase of Escherichia Coli. FEBS Lett. 1983, 151, 189–192. [Google Scholar] [CrossRef]
- Olsiewski, P.J.; Kaczorowski, G.J.; Walsh, C. Purification and Properties of D-Amino Acid Dehydrogenase, an Inducible Membrane-Bound Iron-Sulfur Flavoenzyme from Escherichia Coli B. J. Biol. Chem. 1980, 255, 4487–4494. [Google Scholar] [CrossRef]
- Xu, J.; Bai, Y.; Fan, T.; Zheng, X.; Cai, Y. Expression, Purification, and Characterization of a Membrane-Bound D-Amino Acid Dehydrogenase from Proteus Mirabilis JN458. Biotechnol. Lett. 2017, 39, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Todone, F.; Vanoni, M.A.; Mozzarelli, A.; Bolognesi, M.; Coda, A.; Curti, B.; Mattevi, A. Active Site Plasticity in D-Amino Acid Oxidase: A Crystallographic Analysis. Biochemistry 1997, 36, 5853–5860. [Google Scholar] [CrossRef] [PubMed]
- Ouedraogo, D.; Souffrant, M.; Vasquez, S.; Hamelberg, D.; Gadda, G. Importance of Loop L1 Dynamics for Substrate Capture and Catalysis in Pseudomonas Aeruginosa D-Arginine Dehydrogenase. Biochemistry 2017, 56, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.; Huber, R.; Laber, B.; Pohlenz, H.-D.; Messerschmidt, A. Crystal Structure of the Pyridoxal-5′-Phosphate Dependent Cystathionine β-Lyase FromEscherichia Coliat 1.83 Å. J. Mol. Biol. 1996, 262, 202–224. [Google Scholar] [CrossRef]
- Percudani, R.; Peracchi, A. The B6 Database: A Tool for the Description and Classification of Vitamin B6-Dependent Enzymatic Activities and of the Corresponding Protein Families. BMC Bioinform. 2009, 10, 273. [Google Scholar] [CrossRef]
- Liang, J.; Han, Q.; Tan, Y.; Ding, H.; Li, J. Current Advances on Structure-Function Relationships of Pyridoxal 5′-Phosphate-Dependent Enzymes. Front. Mol. Biosci. 2019, 6, 4. [Google Scholar] [CrossRef]
- Grishin, N.V.; Phillips, M.A.; Goldsmith, E.J. Modeling of the Spatial Structure of Eukaryotic Ornithine Decarboxylases. Protein Sci. 1995, 4, 1291–1304. [Google Scholar] [CrossRef]
- Schneider, G.; Käck, H.; Lindqvist, Y. The Manifold of Vitamin B6 Dependent Enzymes. Structure 2000, 8, R1–R6. [Google Scholar] [CrossRef]
- Yoshimura, T.; Goto, M. D-Amino Acids in the Brain: Structure and Function of Pyridoxal Phosphate-Dependent Amino Acid Racemases. FEBS J. 2008, 275, 3527–3537. [Google Scholar] [CrossRef]
- Toney, M.D.; Kirsch, J.F. The K258R Mutant of Aspartate Aminotransferase Stabilizes the Quinonoid Intermediate. J. Biol. Chem. 1991, 266, 23900–23903. [Google Scholar] [CrossRef]
- Toney, M.D. Controlling Reaction Specificity in Pyridoxal Phosphate Enzymes. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2011, 1814, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Bezsudnova, E.Y.; Popov, V.O.; Boyko, K.M. Structural Insight into the Substrate Specificity of PLP Fold Type IV Transaminases. Appl. Microbiol. Biotechnol. 2020, 104, 2343–2357. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, A.; D’Arrigo, P.; Gefflaut, T.; Molla, G.; Pollegioni, L.; Rosini, E.; Rossi, C.; Servi, S. Multistep Enzyme Catalysed Deracemisation of 2-Naphthyl Alanine. Biocatal. Biotransformation 2006, 24, 409–413. [Google Scholar] [CrossRef]
- Steffen-Munsberg, F.; Vickers, C.; Kohls, H.; Land, H.; Mallin, H.; Nobili, A.; Skalden, L.; van den Bergh, T.; Joosten, H.J.; Berglund, P.; et al. Bioinformatic Analysis of a PLP-Dependent Enzyme Superfamily Suitable for Biocatalytic Applications. Biotechnol. Adv. 2015, 33, 566–604. [Google Scholar] [CrossRef]
- Yu, C.; Li, X.; Zhang, N.; Wen, D.; Liu, C.; Li, Q. Inhibition of Biofilm Formation by D-Tyrosine: Effect of Bacterial Type and D-Tyrosine Concentration. Water Res. 2016, 92, 173–179. [Google Scholar] [CrossRef]
- Kolodkin-Gal, I.; Romero, D.; Cao, S.; Clardy, J.; Kolter, R.; Losick, R. D-Amino Acids Trigger Biofilm Disassembly. Science 2010, 328, 627–629. [Google Scholar] [CrossRef]
- Miyamoto, T.; Homma, H. D-Amino Acid Metabolism in Bacteria. J. Biochem. 2021, 170, 5–13. [Google Scholar] [CrossRef]
- Miyamoto, T.; Katane, M.; Saitoh, Y.; Sekine, M.; Homma, H. Involvement of Penicillin-Binding Proteins in the Metabolism of a Bacterial Peptidoglycan Containing a Non-Canonical D-Amino Acid. Amino Acids 2020, 52, 487–497. [Google Scholar] [CrossRef]
- Li, E.; Wu, J.; Wang, P.; Zhang, D. D-Phenylalanine Inhibits Biofilm Development of a Marine Microbe, Pseudoalteromonas Sp. SC2014. FEMS Microbiol. Lett. 2016, 363, fnw198. [Google Scholar] [CrossRef]
- Warraich, A.A.; Mohammed, A.R.; Perrie, Y.; Hussain, M.; Gibson, H.; Rahman, A. Evaluation of Anti-Biofilm Activity of Acidic Amino Acids and Synergy with Ciprofloxacin on Staphylococcus Aureus Biofilms. Sci. Rep. 2020, 10, 9021. [Google Scholar] [CrossRef]
- Nakade, Y.; Iwata, Y.; Furuichi, K.; Mita, M.; Hamase, K.; Konno, R.; Miyake, T.; Sakai, N.; Kitajima, S.; Toyama, T.; et al. Gut Microbiota–Derived D-Serine Protects against Acute Kidney Injury. JCI Insight 2018, 3, 97957. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.J.; Hariri, B.M.; McMahon, D.B.; Chen, B.; Doghramji, L.; Adappa, N.D.; Palmer, J.N.; Kennedy, D.W.; Jiang, P.; Margolskee, R.F.; et al. Bacterial D-Amino Acids Suppress Sinonasal Innate Immunity through Sweet Taste Receptors in Solitary Chemosensory Cells. Sci. Signal 2017, 10, 7703. [Google Scholar] [CrossRef] [PubMed]
- Kepert, I.; Fonseca, J.; Müller, C.; Milger, K.; Hochwind, K.; Kostric, M.; Fedoseeva, M.; Ohnmacht, C.; Dehmel, S.; Nathan, P.; et al. D-Tryptophan from Probiotic Bacteria Influences the Gut Microbiome and Allergic Airway Disease. J. Allergy Clin. Immunol. 2017, 139, 1525–1535. [Google Scholar] [CrossRef]
- Miyamoto, T.; Moriya, T.; Katane, M.; Saitoh, Y.; Sekine, M.; Sakai-Kato, K.; Oshima, T.; Homma, H. Identification of a Novel D-amino Acid Aminotransferase Involved in D-glutamate Biosynthetic Pathways in the Hyperthermophile Thermotoga Maritima. FEBS J. 2022, 289, 5933–5946. [Google Scholar] [CrossRef]
- Sugio, S.; Petsko, G.A.; Manning, J.M.; Soda, K.; Ringe, D. Crystal Structure of a D-Amino Acid Aminotransferase: How the Protein Controls Stereoselectivity. Biochemistry 1995, 34, 9661–9669. [Google Scholar] [CrossRef]
- Dunathan, H.C. Stereochemical Aspects of Pyridoxal Phosphate Catalysis. Adv. Enzymol. Relat. Areas Mol. Biol. 2006, 35, 79–134. [Google Scholar]
- Soda, K.; Yoshimura, T.; Esaki, N. Stereospecificity for the Hydrogen Transfer of Pyridoxal Enzyme Reactions. Chem. Rec. 2001, 1, 373–384. [Google Scholar] [CrossRef]
- Almo, S.C.; Smith, D.L.; Danishefsky, A.T.; Ringe, D. The Structural Basis for the Altered Substrate Specificity of the R292D Active Site Mutant of Aspartate Aminotransferase from E. coli. Protein Eng. Des. Sel. 1994, 7, 405–412. [Google Scholar] [CrossRef]
- Bezsudnova, E.Y.; Boyko, K.M.; Popov, V.O. Properties of Bacterial and Archaeal Branched-Chain Amino Acid Aminotransferases. Biochemistry 2017, 82, 1572–1591. [Google Scholar] [CrossRef]
- Goto, M.; Miyahara, I.; Hayashi, H.; Kagamiyama, H.; Hirotsu, K. Crystal Structures of Branched-Chain Amino Acid Aminotransferase Complexed with Glutamate and Glutarate: True Reaction Intermediate and Double Substrate Recognition of the Enzyme. Biochemistry 2003, 42, 3725–3733. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosini, E.; Pollegioni, L.; Molla, G. The Symmetric Active Site of Enantiospecific Enzymes. Symmetry 2023, 15, 1017. https://doi.org/10.3390/sym15051017
Rosini E, Pollegioni L, Molla G. The Symmetric Active Site of Enantiospecific Enzymes. Symmetry. 2023; 15(5):1017. https://doi.org/10.3390/sym15051017
Chicago/Turabian StyleRosini, Elena, Loredano Pollegioni, and Gianluca Molla. 2023. "The Symmetric Active Site of Enantiospecific Enzymes" Symmetry 15, no. 5: 1017. https://doi.org/10.3390/sym15051017
APA StyleRosini, E., Pollegioni, L., & Molla, G. (2023). The Symmetric Active Site of Enantiospecific Enzymes. Symmetry, 15(5), 1017. https://doi.org/10.3390/sym15051017