Symmetry Breaking in NMR Spectroscopy: The Elucidation of Hidden Molecular Rearrangement Processes

Abstract
: Variable-temperature NMR spectroscopy is probably the most convenient and sensitive technique to monitor changes in molecular structure in solution. Rearrangements that are rapid on the NMR time-scale exhibit simplified spectra, whereby non-equivalent nuclear environments yield time-averaged resonances. At lower temperatures, when the rate of exchange is sufficiently reduced, these degeneracies are split and the underlying “static” molecular symmetry, as seen by X-ray crystallography, becomes apparent. Frequently, however, such rearrangement processes are hidden, even when they become slow on the NMR time-scale, because the molecular point group remains unchanged. Judicious symmetry breaking, such as by substitution of a molecular fragment by a similar, but not identical moiety, or by the incorporation of potentially diastereotopic (chemically non-equivalent) nuclei, allows the elucidation of the kinetics and energetics of such processes. Examples are chosen that include a wide range of rotations, migrations and other rearrangements in organic, inorganic and organometallic chemistry.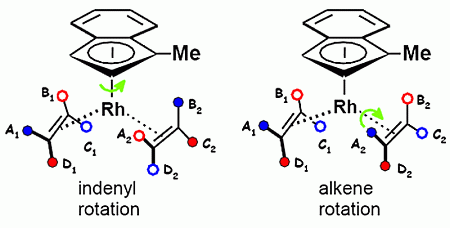
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Symmetry-breaking plays a crucial role in many aspects of nuclear magnetic resonance spectroscopy. For example, theoreticians calculating the simplest spin-spin coupling constant, 1JHH in dihydrogen, need an experimental measurement to validate their predictions [1]. Evidently, this is not obtainable from H2 itself since the two nuclei are equivalent and the observed gas phase spectrum is a singlet. However, isotopic substitution, as in HD, yields duplicate values of 1JHD as 43.3 Hz, not only from the 1:1:1 triplet in the proton spectrum, but also from the 1:1 doublet in the deuterium spectrum (the nuclear spin values, I, for 1H and 2D are ½ and 1, respectively). The unobservable 1JHH is now readily calculated since 1JHH/1JHD = γH/γD = 6.51, where γ is the magnetogyric ratio for the relevant nucleus; the experimental value for 1JHH is therefore 282 Hz.
Variable-temperature NMR spectroscopy is probably the most convenient and widely-used technique to study molecular rearrangement processes in solution. It frequently allows elucidation not only of the mechanism of rearrangement, but also the activation energies and entropies of the process or processes involved. It is commonly the case that molecular rearrangements occur very rapidly at room temperature, thus equilibrating nuclear environments that are in fact non-equivalent in the static system, as seen for example by X-ray crystallography. Lowering the temperature slows the exchange processes on the NMR time-scale, thus revealing the underlying “instantaneous” structure and breaking the time-averaged symmetry.
Typically, bullvalene, 1, a C10H10 isomer (see Scheme 1), exhibits a single resonance in both the 1H and 13C NMR regimes at room temperature, but at low temperatures each is split into a 3:3:3:1 peak ratio [2], in accord with the solid state structure revealed by X-ray crystallography [3]. In this case, a series of rapid Cope rearrangements—[3,3] sigmatropic shifts in Woodward-Hoffmann orbital symmetry terminology [4]—in which each carbon can occupy any position, become slow on the NMR time-scale and so reveal the underlying C3v molecular geometry. However, there is no need to introduce additional labels to break the three-fold symmetry since it is immediately exposed merely by lowering the temperature.
2. Symmetry Breaking as a Probe for Barriers to Undetectable Molecular Rotations
2.1. Alkyl and Tripodal Rotations in (C6R6)M(CO)3 Systems
In contrast to the dynamic behaviour of bullvalene, the situation is different in hexaethylbenzene (HEB) whereby alternate alkyl groups lie above and below the plane of the arene, giving rise to D3d symmetry that renders all six ethyl groups NMR equivalent. Attachment of a π-bonded metal tricarbonyl fragment, as in (η6-C6Et6)Cr(CO)3, 2, breaks the symmetry between the faces of the arene ring and lowers the point group to “time-averaged” C6v when both ethyl and tripodal rotation are fast on the NMR time-scale (Scheme 2).
As the temperature is lowered, ethyl rotation slows to the point such that the alkyl substitutents are “frozen out” into proximal and distal sets relative to the π-bonded Cr(CO)3 fragment [5,6]. However, it is not possible to tell whether or not tripodal rotation has slowed since the system retains its C3v symmetry in either case. As shown in Scheme 3, replacement one of the carbon monoxide ligands by the structurally similar, cylindrical thiocarbonyl moiety lowers the symmetry to CS in (η6-C6Et6)Cr(CO)2CS, 3, and splits the ethyl groups into a 2:1:2:1 pattern when tripodal rotation is slow [7]. Even more striking, introduction of a third different ligand, a nitrosyl group, as in [(η6-C6Et6)Cr(CO)(CS)NO]+, 4, completely breaks the degeneracy of the six ethyl substituents and results in an eighteen peak 13C NMR spectrum since, in the point group C1, each of the six methyl, methylene and ring carbon environments is now different, as shown in Figure 1 [8].
Of course, it is important that substitution to lower the molecular symmetry does not incur any significant electronic or steric effect that would unduly perturb the system. To this end, in the hexaethylbenzene-chromium system, where carbon monoxide units were sequentially replaced by thiocarbonyl and nitrosyl ligands, the compounds 2, 3 and 4 were each characterized by X-ray crystallography to ensure consistency of the molecular geometry.
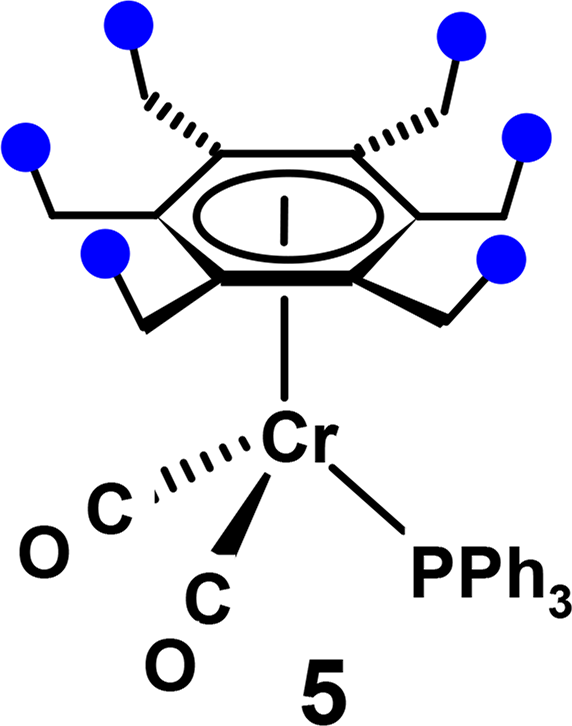
In contrast, however, an attempt to enhance the hindrance to rotation by incorporation of a bulky triphenylphosphine group into the tripod, as in (η6-C6Et6)Cr(CO)2PPh3, 5, led instead to a different rotamer whereby the six ethyls are always distal, and the NMR spectrum is unchanged upon lowering the temperature [6].

The barrier to tripodal rotation in molecules of the type (arene)M(CO)3 is of significance since it has been proposed that the orientation of the tripod directs the site of attack at the arene ring by an incoming nucleophile. Consequently, approaches towards controlling the orientation of the tripod, either sterically or electronically, have been widely investigated [9].
The question of slowed rotation of a π-bonded chromium tricarbonyl fragment has also been investigated by breaking the symmetry of the hexa-substituted arene rather than by rendering chiral the tripodal moiety. Replacement of one ethyl group by an acetyl substituent, as in pentaethylacetophenone-tricarbonylchromium, 6, leaves the steric environment of the Cr(CO)3 group essentially unchanged, but slowed tripodal rotation splits the three-fold degeneracy such that the 13CO NMR resonances now exhibit a 2:1 pattern [10]. As with the HEB complexes 2 through 4, the tripodal rotational barrier in 6 was again 9.5 kcal·mol−1.

Another approach to measuring the barriers to alkyl and tripodal rotations in (η6-C6R6)Cr(CO)3 systems was reported in an elegant parallel investigation by Kilway and Siegel [11]. In this case, the arene chosen was [1,4-bis(4,4-dimethyl-3-oxopentyl)-2,3,5,6-tetraethylbenzene]tricarbonylchromium, 7. In this molecule, with its alternating up-down pattern of substituents, the two ketones are situated on opposite faces of the central ring, one distal the other proximal. When alkyl and tripodal rotations are slowed on the NMR time-scale, the molecule has only mirror symmetry (CS) thus splitting the Cr(13CO)3 resonance into a 2:1 pattern. As the temperature is gradually raised, the chromium carbonyl resonances coalesce into a singlet, but the two ketonic environments remain non-equivalent until the onset of alkyl rotations, at which point the molecule has effective C2v symmetry. The barriers to ethyl and tripodal rotation were found to be 11.8 and 9.5 kcal·mol−1, respectively, in excellent agreement with the earlier work [8,10].
2.2. Rotations of Peripheral Ring Substituents in (C5Ph5)- and (C5R5)-Metal Complexes
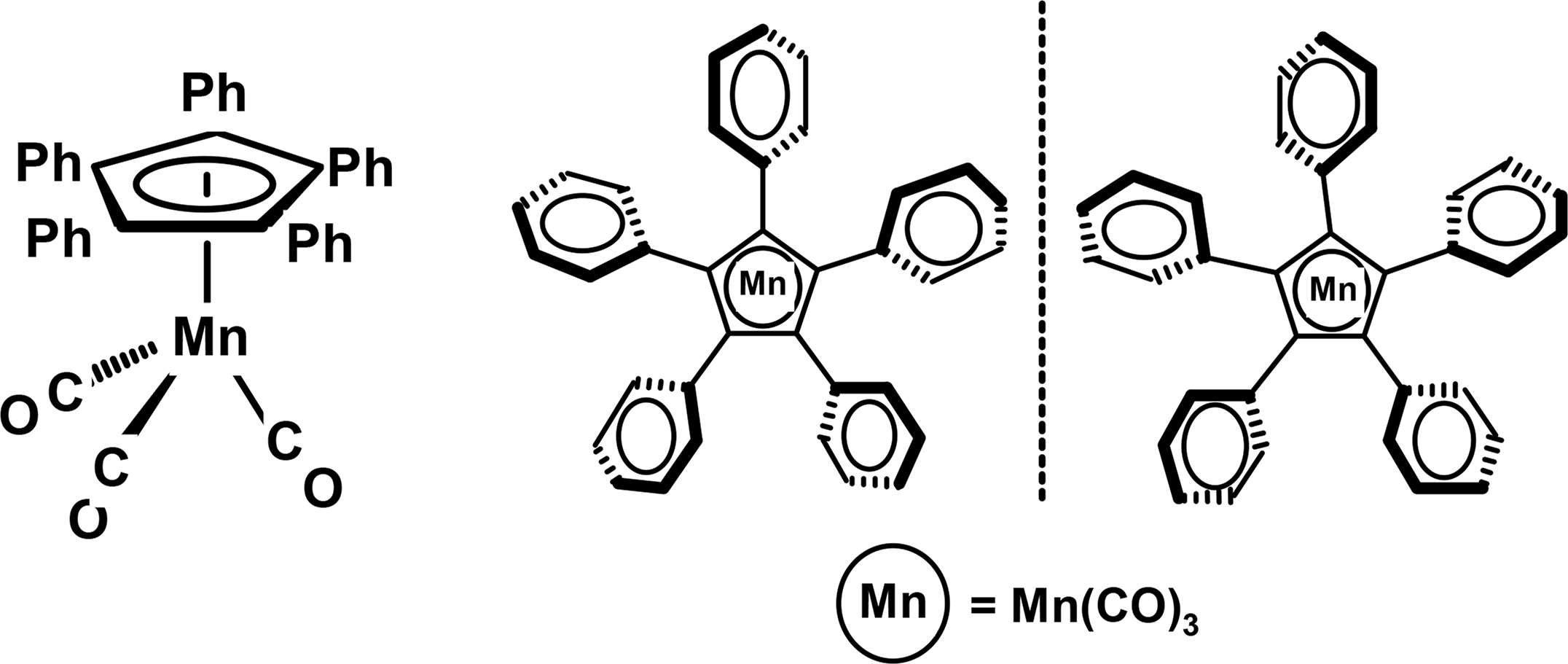
Perphenylated ring systems, CnPhn, where n ranges from 3 through 7, adopt propeller conformations whereby the twist angles made by the peripheral rings with respect to the central ring reflect the compromise between electronically favoured conjugative coplanarity and the ugly reality of steric hindrance. As the ring size increases, the angle ω, subtended by adjacent moieties at the centre of the internal ring, decreases: C3Ph3 120°, C4Ph4 90°, C5Ph5 72°, C6Ph6 60°, C7Ph7 51.4°. Although increasing the ring size lengthens the radial distance of the external groups from the ring centre, this is more than compensated for by the diminishing value of ω. As a result, the peripheral rings find themselves in an increasingly crowded locale such that the twist angles for [C5Ph5]−, C6Ph6 and [C7Ph7]+ are approximately 50°, 70° and 80°, respectively [12,13]. These systems have been heavily investigated as molecular analogues of propellers or gears, although only those with an even number of blades would be capable of correlated conrotatory motion [14,15]. They adopt Dn symmetry, but peripheral phenyl rotations cannot be monitored unless the symmetry is lowered either by incorporation of ortho or meta substituents into these rings or by π-complexation to an organometallic moiety. In the case of a non-chiral tripod, typified by (η5-C5Ph5)Mn(CO)3 [16], rapid rotation of phenyls and of the tripod generates time-averaged C5v symmetry, but separation into clockwise and counter-clockwise orientations of the peripheral phenyls, as in Figure 2, gives rise to enantiomers of C5 symmetry [17].
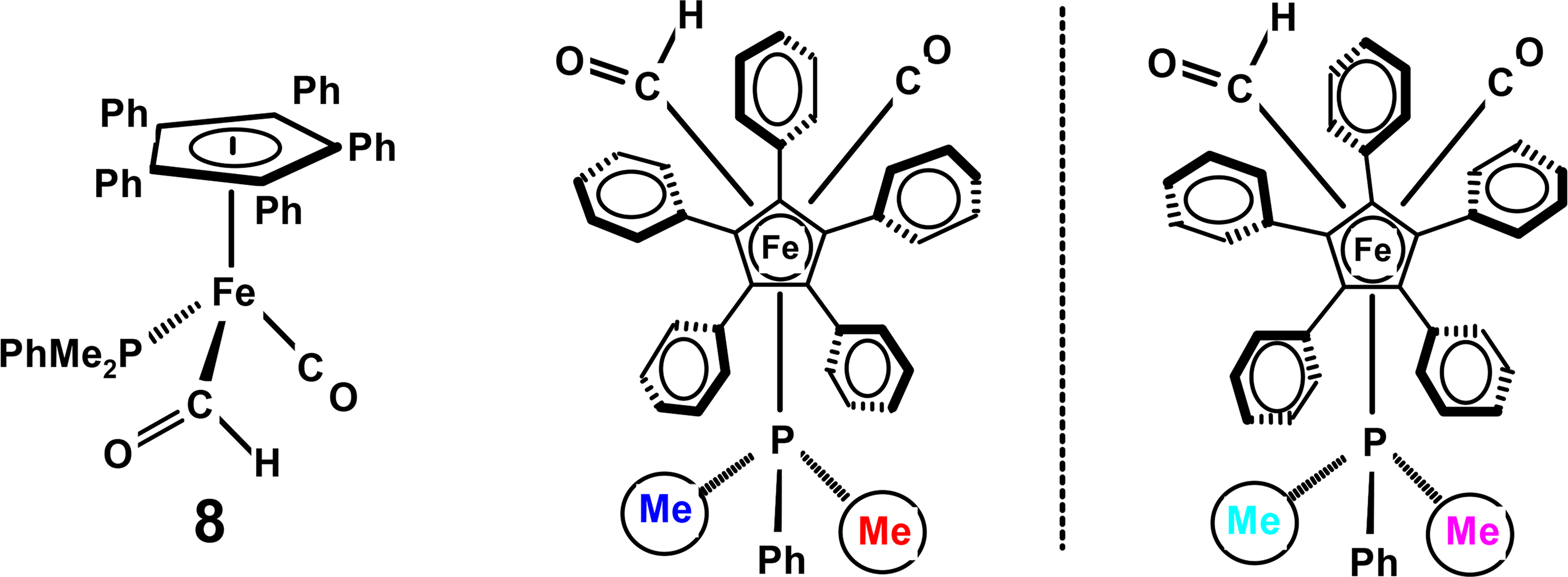
However, when phenyl rotation (or even oscillation) becomes slow on the NMR time-scale, the presence of a chiral tripod, as in (η5-C5Ph5)Fe(CO)(CHO)PMe2Ph, 8, provides another stereocentre and generates C1-symmetric diastereomers (Figure 3). This situation renders the phosphorus nuclei diastereotopic, thus giving rise to two 31P NMR resonances whose decoalescence behaviour allows the evaluation of the barrier to interconversion of the C5Ph5 helices as 11.7 ± 0.3 kcal·mol−1. Moreover, within each molecule, the two methyl groups on the phosphine ligand are also diastereotopic, giving rise to a pair of signals; hence, when interconversion of the C5Ph5 five-bladed propellers becomes slow on the NMR time-scale, we see two sets of diastereotopic methyl pairs [18].
On a 500 MHz spectrometer at 173 K, slowed tripodal rotation splits the five-fold degeneracy of the C5Ph5 moiety in 8 and yields a barrier of 8.7 ± 0.3 kcal·mol−1 [18]. It is evident that the marked difference between the activation energies of these two processes shows that they are not correlated; indeed, the tripod continues to spin rapidly even when rotation of the peripheral phenyls has become slow on the NMR time-scale. The net result is that sequentially enhanced symmetry breaking allows complete determination of the stereodynamic behaviour of the system.
One should emphasise that when monitoring fluxional processes in chiral molecules containing diastereotopic groups there is no requirement for enantiomeric or optical purity; indeed, such systems are normally racemic. Each individual molecule generates separate resonances for the diastereotopic nuclei, and each one is indistinguishable by NMR from its enantiomer in an achiral solvent [19].
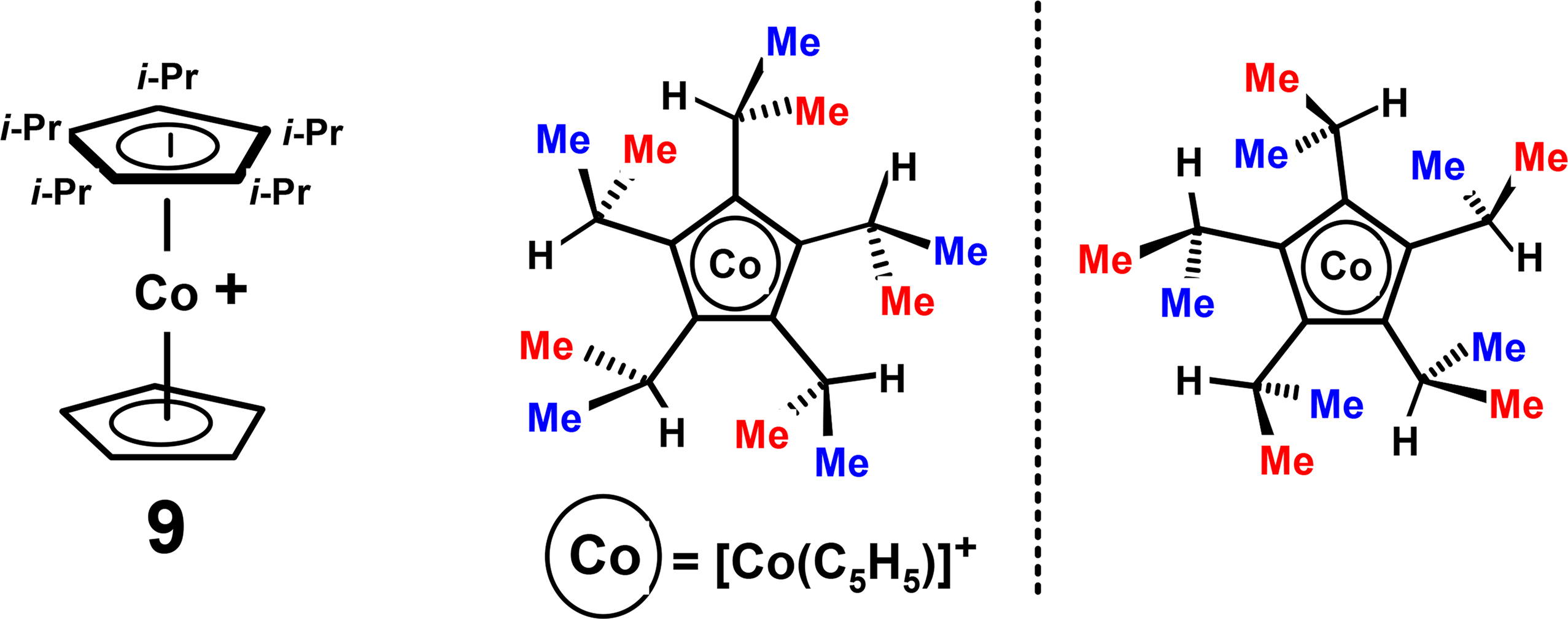
Another interesting molecule is the very crowded cobalt sandwich cation [(C5iPr5)Co(C5H5)]+, 9, reported by Gloaguen and Astruc [20], in which the isopropyl substituents are arranged such that they all point in the same direction, thus giving rise to C5-symmetric enantiomers (Figure 4). At room temperature the 1H and 13C NMR spectra reveal the presence of two methyl environments, exo and endo, that only coalesce at 100 °C indicating a substantial barrier to racemization of 17 kcal·mol−1. The methyl protons are rendered diastereotopic by the helicity of the pentaisopropylcyclopentadienyl ring that senses the clockwise and counter-clockwise orientations of the gear-meshed substituents. Once again, it is incorporation of a π-complexed moiety, Co(C5H5), that breaks the C5h symmetry of the C5iPr5 ring and allows detection and measurement of the barrier to alkyl rotation.
2.3. Incorporation of Diastereotopic Nuclei to Detect Molecular Rearrangements
2.3.1. Metal Cluster Cations
The remarkable ease of formation and stability of primary carbocations capping a triangle of organo-transition metal fragments, as in [(OC)9Co3CCH2]+, 10, prompted numerous experimental and theoretical studies [21]. Molecular orbital calculations by Schilling and Hoffmann [22] indicated that in the ground state the carbon-carbon linkage of the vinylidene fragment is not aligned with the three-fold axis of the metal triangle, but rather interacts directly with a single cobalt vertex so as to accept electron density from a filled metal d orbital (Scheme 4). Moreover, it was suggested that the capping fragment should migrate between the cluster vertices in an antarafacial fashion.
This proposal was brilliantly explored in an ingenious experiment from the Mislow group that verified not only the ground state geometry, but also the details of the fluxional process [23]. The cation 11 contains an isopropyl substituent as a 13C NMR probe for chiral conformations (Scheme 5). In the calculated ground state the cation has C1 symmetry which would render diastereotopic the methyls of the isopropyl group; gratifyingly, at low temperature, two methyl resonances are indeed observed. Moreover, as the temperature is raised, these two peaks coalesce as would be required for the antarafacial migration pathway via a transition state that possesses a molecular mirror plane; this process equilibrates the two methyl environments, and the measured activation energy was found to be 10.5 kcal·mol−1. In this case, the initial broken symmetry verifies the tilted ground state, whereas the recovery of mirror symmetry validates the antarafacial nature of the migration process.
The non-linear nature of other metal cluster stabilized cations, such as the ketenylidene cluster [(OC)9Co3C–C=O]+, 12, originally synthesized by Seyferth [24], has also been verified by 13C NMR which revealed a 6:3 splitting of the metal carbonyl resonances at low temperature [25] (Scheme 6).
2.3.2. Mixed-Metal Square-Pyramidal Clusters
With the goal of combining the reactivity of metal surfaces with the control achievable in homogeneous systems, especially with regard to asymmetric syntheses, the area of mixed metal clusters has been intensely studied for many years [26]. Clearly, such an undertaking would presuppose that the chirality of the cluster not be lost during the reaction sequence. Typical of such systems are the square-based pyramidal M3C2 clusters, in particular those in which the three metal vertices are different and the C2 unit is derived from an alkyne. As exemplars, we have selected the clusters [(C5H5)Ni·Ni(C5H5)·Fe(CO)3][PhC≡CCO2iPr], 13, and [(C5H5)Ni·Co(CO)3·Fe(CO)3][PhC≡CCO2iPr], 14, in which the isopropyl group acts as a probe for the chiral nature of the cluster.
The unsymmetrical nature of the alkyne moiety in 13 renders the cluster chiral; the (cyclopentadienyl)nickel groups are non-equivalent, as are the diastereotopic methyls of the isopropyl substituent. One can now test for two different processes. One could envisage rotation of the nickel-nickel vector relative to the iron-carbon-carbon triangle that would interconvert the two nickel environments, but not racemize the molecule; that is, the diastereotopic character of the Me2CH group would be unaffected. In contrast, a formal rotation of the alkyne moiety relative to the Ni2Fe triangle not only racemizes the molecular cluster, but also interconverts the two [(C5H5)Ni environments (Scheme 7). Were this to be the only rearrangement process, the activation energies for the nickel exchange and coalescence of the methyl peaks must be identical. Gratifyingly, the observed barrier in each case is 15 ± 0.5 kcal·mol−1 [27].
One must, however, consider yet another possible occurrence. Dissociation of the cluster into non-complexed alkyne and an unsaturated metal triangle (perhaps weakly stabilized by solvent) would simultaneously equilibrate not only the methyls but also the cyclopentadienyl-nickel environments. It is necessary, therefore, to ensure that the process is indeed intramolecular. This was accomplished by making all three metal vertices different, as in the iron-cobalt-nickel cluster, 14. As illustrated in Scheme 8, acetylene rotation does not interconvert enantiomers but merely diastereomers. Thus, when alkyne rotation is slow on the NMR time-scale, each diastereomer will show in the methyl region four peaks—two magnetically non-equivalent methyls, each doublet split by its adjacent isopropyl hydrogen.
Indeed, at room temperature one sees two major diastereomers, each with its four peak pattern in the methyl region, but at elevated temperatures there still appears a four-line spectrum indicating that the stereochemical integrity of the chiral cluster is conserved. The experimentally observable barrier of 16.5 ± 0.5 kcal·mol−1 in the Fe–Co–Ni cluster, 14, is similar to that observed in the Ni2Fe compound, 13, indicating that the same mechanistic pathway is involved [27]. Molecular orbital calculations [28] revealed that the rearrangement proceeds via a “modified windscreen-wiper” motion (Scheme 9), analogous to that previously proposed for the isolobal C5H5+ nido cluster [29,30]. Note that simple circumambulation of the alkyne round the periphery of the metal triangle (Scheme 10) does not racemize the cluster.
2.3.3. Diphos and Arphos Complexes of Cobalt Clusters
The potential use of chiral metal clusters as catalysts for asymmetric syntheses inevitably demands that they maintain their stereochemical integrity, and not suffer racemization under reaction conditions. Tetrahedral carbynyl-tricobalt clusters react with chelating ligands, such as diphosphines, to form complexes of the type RCCo3(CO)7(L-L) having CS symmetry. Use of 1-diphenylarsino-3-diphenylphosphinoethane (arphos), Ph2As–CH2CH2–PPh2, breaks the mirror symmetry and renders the cluster chiral. However, attempts to separate the enantiomers consistently failed, implying that racemization was occurring. To monitor this situation, it was necessary to incorporate a diastereotopic probe, and this was accomplished by introducing an isopropyl ester as the substituent on the capping carbyne. At room temperature, the 1H NMR spectrum of (arphos)(CO)7Co3C–CO2CHMe2, 15, in the methyl region appears as a doublet (due to splitting by the adjacent hydrogen); evidently, the system racemizes at this temperature. However, when cooled to 223 K, this resonance undergoes decoalescence to form two sets of doublets as the diastereotopicity of the isopropyl methyls becomes apparent. Simulation of the variable-temperature spectra yields a relatively modest barrier of 13 kcal·mol−1, which accounts for the facile racemization seen at room temperature. The mechanism apparently involves fragmentation of the arsenic-cobalt linkage to leave a dangling diphenylarsino group that can either return to its original position or migrate onto the neighbouring cobalt vertex (Scheme 11), thus interconverting R and S cluster isomers [31]. (The absolute configuration of chiral tetrahedral clusters, such as 15, can be conveniently designated by assigning an R or S label to a dummy atom placed at the centre of the tetrahedron, and then assigning priorities in the usual Cahn-Ingold-Prelog manner [32]. Thus, in 15 the (Ph2As)Co(CO)2 vertex has priority 1, the (Ph2P)Co(CO)2 vertex has priority 2, and the capping carbon atom has the lowest priority.) The rearrangement necessitates concomitant scrambling of the carbonyl ligands, but the barrier to this secondary process was independently measured to be only 10.5 kcal·mol−1, and so poses no problem.
2.3.4. Tris-Chelate Complexes M(L-L)3
Complexes such as tri(acetylacetato)chromium(III) or the tris(ethylenediamine)cobalt(III) cation possess D3 symmetry and are chiral. When the latter compound is partnered by a chiral anion, e.g., tartrate, the diastereomers can be separated by fractional crystallization, and the enantiomeric cations can each be obtained subsequently in optically pure form. In some cases, systems of this general type racemize more rapidly than can be conveniently monitored by polarimetry or other related techniques, Nevertheless, such processes can be readily followed by NMR by incorporating a potentially diastereotopic substituent to probe the chirality. As noted above, it is not necessary to work with optically pure compounds since each isomer exhibits its own individual NMR spectrum.
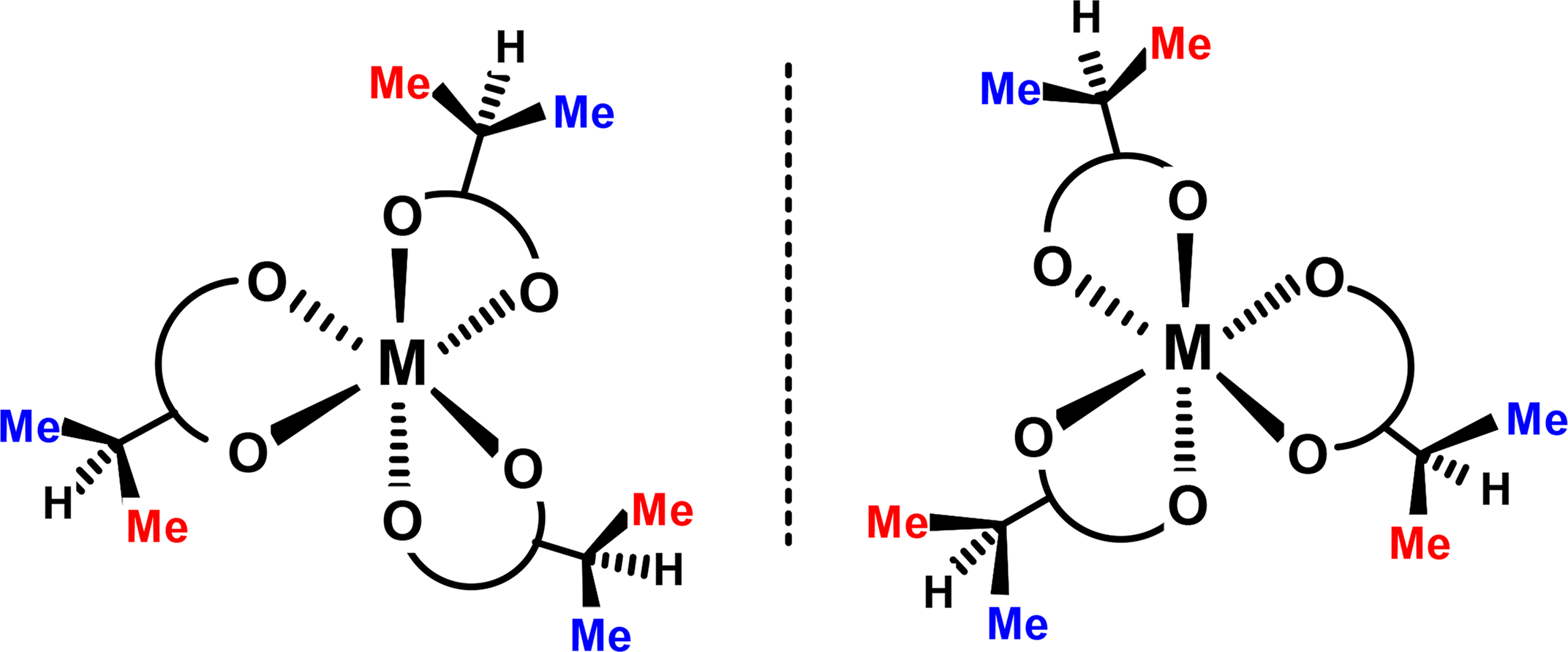
An important early contribution by Jurado and Springer [33] involved the dynamic behaviour of tri(acetylacetato)aluminium, which adopts D3 symmetry. Incorporation of isopropyl groups, shown schematically in 16, lowers the molecular symmetry to C3 and provides diastereotopic methyls to monitor the rate of racemization (Figure 5). Typically, at ambient temperature, the 60 MHz 1H NMR spectrum of 16 in the methyl region appears as two sets of doublets indicating that exchange between the enantiomers is still slow on the NMR time-scale. However, above 120 °C, these peaks coalesce and sharpen, eventually yielding a single well-defined doublet, and the barrier was determined by line-shape analysis to be 22 kcal·mol−1.
Of course, in this pioneering study carried out more than 40 years ago, the available spectrometer operated at 60 MHz for protons [33]. Nevertheless, this was appropriate since on a modern system (operating at 500 MHz or greater) peak coalescence would only be observable at a much higher temperature, probably beyond the boiling point of any normal NMR solvent, and possibly above the decomposition point of the complex. This approach has since been widely used to probe the stereodynamics of other tris-chelate complexes, as well as many related systems of the type (L-L)2MX2 and (L-L)2MXY. The various possible mechanisms of loss of stereochemical integrity, either dissociative, or via an internal rearrangements such as the Ray-Dutt or Bailar twist, have been comprehensively discussed in a now classic review and a long series of publications by Serpone and Bickley [34,35].
2.3.5. Inversion of Corannulene
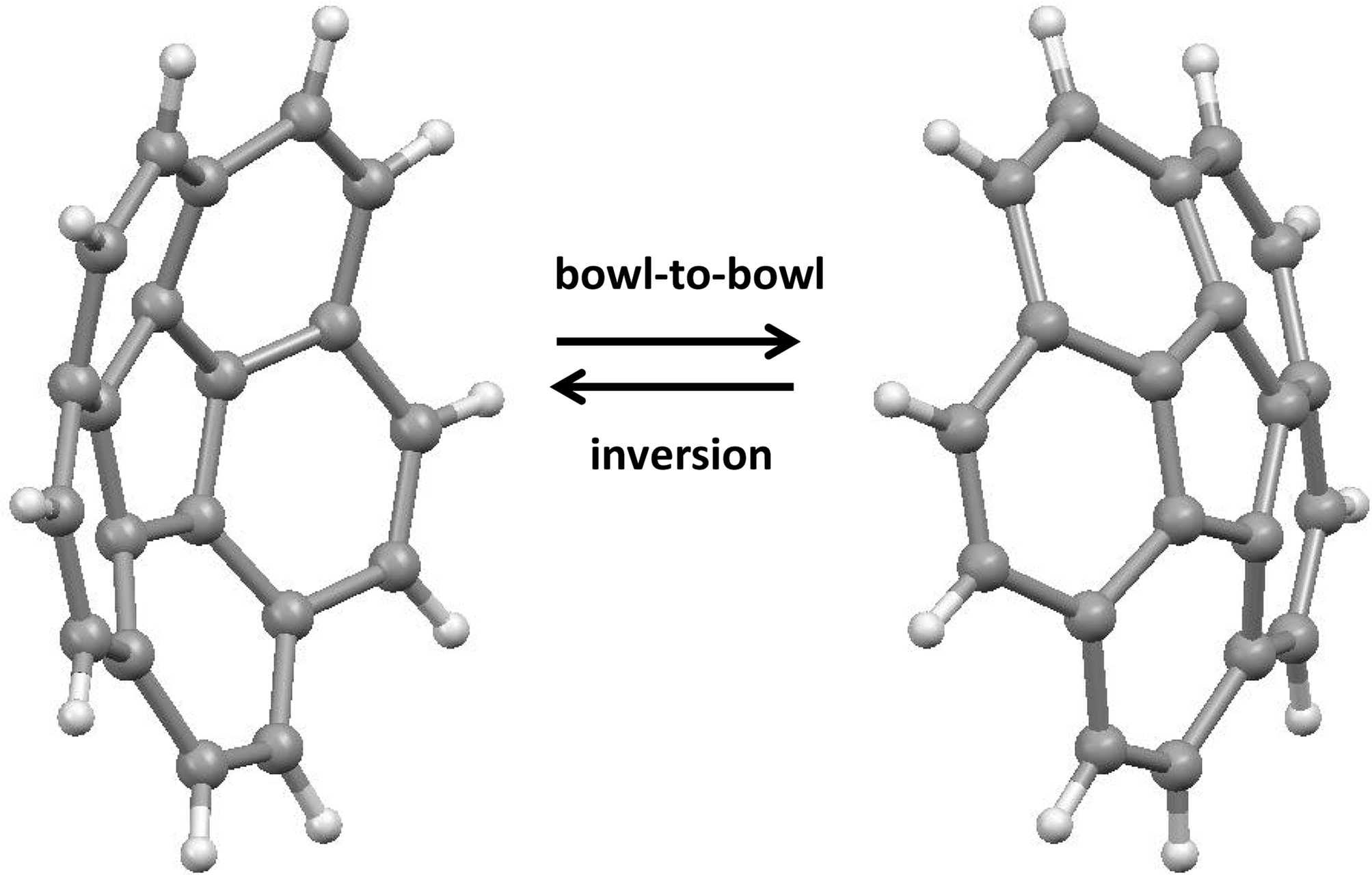
Corannulene, C20H10, was first prepared in minuscule yields by Barth and Lawton in a herculean 17-step process [36]; its non-planar twenty-carbon framework is a substantial substructure of the fullerene C60. Subsequently, in a synthetic tour de force, Scott devised a much shorter and more efficient route [37], and Siegel has since developed the synthesis to the point where corannulene is now available in kilogram quantities [38], thus allowing the study of its own dynamic behavior as well as that of numerous derivatives. The barrier to umbrella-like bowl-to-bowl inversion, illustrated in Figure 6, involves the interconversion of C5v symmetric entities via a planar D5h transition state.
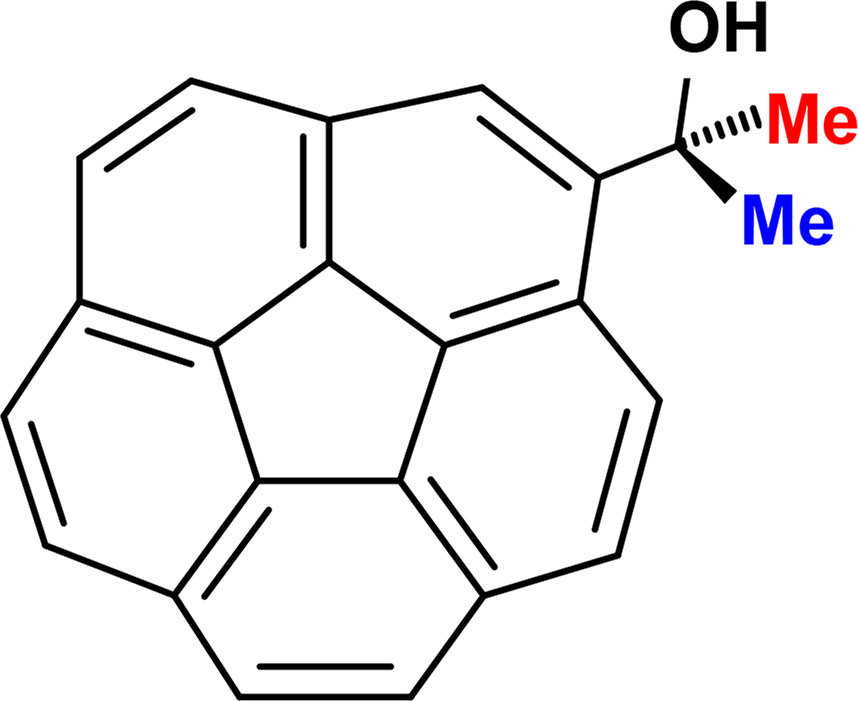
The activation energy for this process was first experimentally determined from variable-temperature NMR spectroscopic measurements on racemic corannulenyl-dimethylcarbinol (Figure 7). In the slow exchange regime, the diastereotopic methyl groups of the Me2COH substituent exhibit separate resonances, but they are equilibrated when the C20 skeleton adopts a planar geometry; the measured value of 10.2 kcal·mol−1 [39] may be compared to the DFT-calculated value for corannulene itself of 10.5 kcal·mol−1 [40].
2.4. Hindered Rotation of Aryl Groups in Organic Systems
2.4.1. Pentafluorophenyl Rings
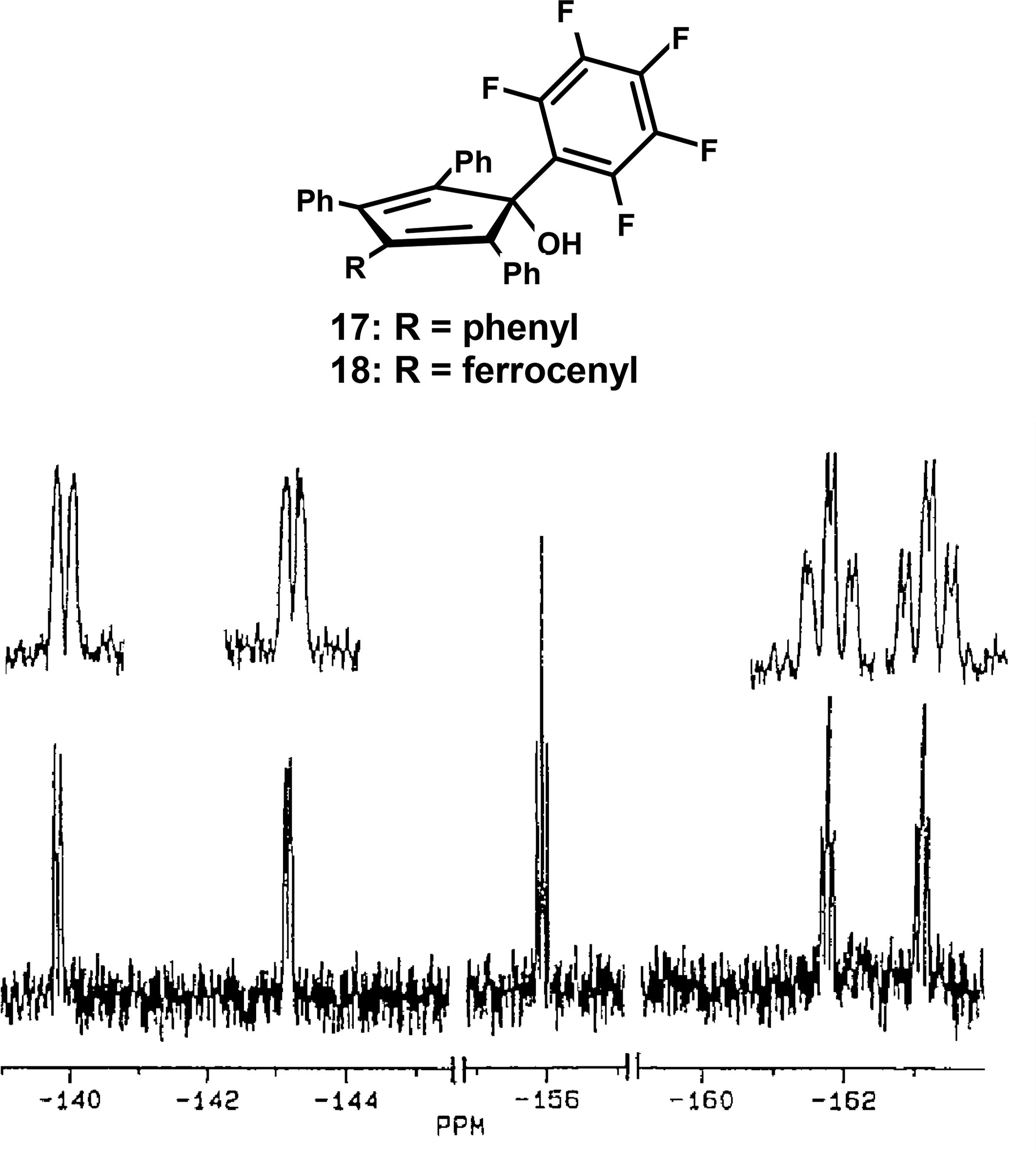
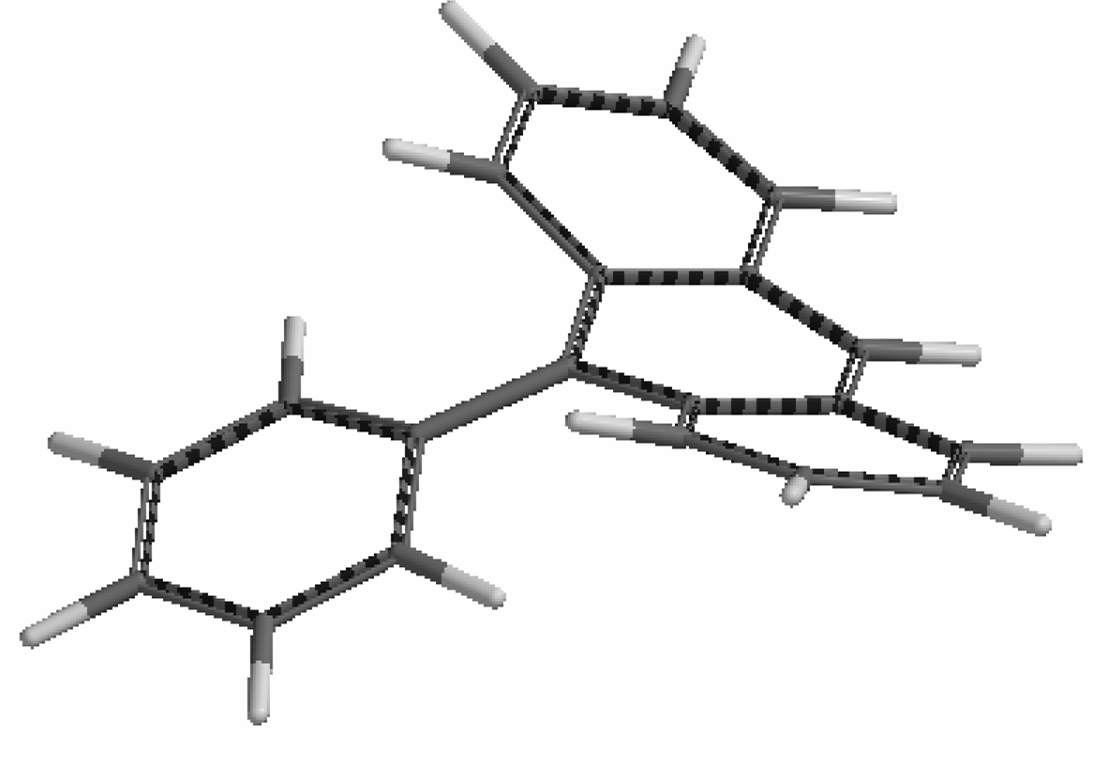
We have already encountered barriers to phenyl rotations in systems of the type (CnPhn)MLx, where the π-bonded organometallic substituent breaks the Dn symmetry of the free ligand by differentiating between the faces of the central ring. However, there other cases where the aryl group is sufficiently sterically crowded to exhibit hindered rotation without the need to incorporate an additional bulky group. For example, the reaction of pentafluorophenyl-lithium with tetracyclone yields (among other isomers) 5-pentafluorophenyl-1,2,3,4-tetraphenylcyclopentadien-5-ol, 17. The X-ray crystal structure of 17 reveals the crowded environment of the pentafluorophenyl ring such that it is oriented orthogonal to the plane of the central ring, thus splitting the degeneracy of the ortho, and also the meta fluorines. In this system, slowed rotation of the pentafluorophenyl ring is evident even at room temperature, as indicated by the appearance of five 19F NMR resonances (Figure 8)—a phenomenon greatly enhanced by the much larger chemical shift dispersion exhibited by fluorine nuclei in aromatic rings [41] compared to that shown by protons in the analogous situation. Slowed rotation of the fluorinated ring in the ferrocenyl-substituted system 18 is also readily apparent, with a barrier of 20 kcal·mol−1 in each case [42].
2.4.2. Simple Phenyl Rings
A more challenging problem arose when it was predicted from DFT calculations [43,44] that the barrier to phenyl rotation in 9-phenylanthracene, 19, was also 20 kcal·mol−1. The steric hindrance arises because the two ring systems cannot become coplanar without bringing the phenyl ortho hydrogens too close to H(1) and H(8) of the anthracene skeleton. The DFT-calculated transition state revealed a non-planar, stepped, ladder-like arrangement, as shown in Figure 9.
The intrinsic C2v, or D2h symmetry of 9-phenyl anthracene, 19, or 9,10-diphenylanthracene, 20, respectively, renders the pairs of ortho and meta CH positions equivalent and so does not allow the rotational process to be monitored by variable-temperature NMR spectroscopy. Hence, it was necessary to break the symmetry, but to do so in such a fashion so as not to perturb the molecular geometry significantly. This was accomplished in two ways. Initially, 9,10-bis(3-chlorophenyl)anthracene, 21, and 9,10-bis(3-fluorophenyl)anthracene, 22, were synthesized and their variable-temperature NMR spectra were acquired. In the chlorophenyl case, 21, the phenyl ring carbons exhibited multiple 13C resonances indicating clearly the presence of syn (C2v) and anti (C2h) rotamers; the barrier to their interconversion was found to be 21 kcal·mol−1 [45]. Moreover, in 22, not only can one see two CF environments in the fluorine NMR spectrum in solution, but also the X-ray crystal structure revealed the presence of equal quantities of syn and anti isomers; the structure of anti-22 appears as Figure 10, and reveals that the fluorophenyls adopt an angle of 85° with the plane of the anthracene thus deviating only slightly from the perpendicular orientation.
The next step was to investigate the rotational barrier of an unsubstituted phenyl. This requires that the symmetry of the environment of the phenyl has to be broken rather than the symmetry of the phenyl itself. To this end, 9-(1-naphthyl)-10-phenylanthracene, 23, was prepared and characterized by X-ray crystallography; the molecule adopted a structure such that the phenyl and naphthyl rings made dihedral angles of 80° and 88°, respectively, with the plane of the anthracene. Gratifyingly, the 600 MHz 1H NMR spectrum of 23 exhibited five resonances for the phenyl ring (Figure 11), even at room temperature, and peak coalescence behaviour yielded a phenyl rotational barrier of 21 kcal·mol−1 [40]. Once again, judicious symmetry breaking allowed the detection and measurement of the barrier to a hidden molecular rearrangement process.
Analogously, the barrier to rotation of the substituent in 9-(3-indenyl)anthracene was evaluated via a variable-temperature NMR study of 9,10-di-(3-indenyl)anthracene, 24, that also exhibits hindered rotation (barrier of 23 kcal·mol−1) and exists as syn and anti rotamers both in solution and in the solid state [46].

3. Rotations and Migrations in Indenyl Systems
3.1. Indenyl and Ethylene Rotations in (Indenyl)bis(ethylene)rhodium(I)
The use of NMR spectroscopy to measure rotational barriers in metal–alkene complexes dates back 50 years to the classic report by Cramer [47] in which he noted that the alkene protons in (η5-C5H5)Rh(C2H4)2, 25, which has effective C2v symmetry, could be distinguished by their relative orientations with respect to the cyclopentadienyl ring. Thus the “outside” (δ 2.86) and “inside” (δ 1.03) protons could, in principle, be interconverted by a formal rotation about an axis joining the rhodium to the centre of the carbon-carbon double bond. Variable-temperature NMR studies elucidated this barrier to alkene rotation as 15 kcal·mol−1 [48].

In contrast, in the analogous indenyl complex (η5-C9H7)Rh(C2H4)2, 26, the corresponding barrier was reported to be considerably lower, 10.5 kcal·mol−1. It was suspected that the ability of the indenyl ligand to slide from an η5- to an η3-bonding mode, thus generating aromatic character in the six-membered ring, could be relevant [49]. The 60 MHz 1H NMR spectrum of 26 shows, in the ethylene region, a single resonance (doublet split by 103Rh) at +50 °C, which splits into two multiplets at −90 °C. This was rationalized in terms of slowed ethylene rotation on the NMR time-scale with a barrier of 10.3 kcal·mol−1 [50].
However, one must exercise some caution since in 26 the symmetry is lowered to CS, and there are four, not two, proton environments in each C2H4 ligand. Complete equilibration of these protons requires not only rotation about the alkene-rhodium axis, but also rotation of the Rh(C2H4)2 fragment about the metal-indenyl axis. As shown in Scheme 12, the two rotation mechanisms interconnect different proton environments. Specifically, rotation about the metal-indenyl vector interconverts A1 and B2, B1 and A2, C1 and D2, and D1 and C2. In contrast, alkene rotation interconverts A1 and C1, B1 and D1, A2 and C2, and B2 and D2. Thus, the A1 proton can gain access only to the B2, C1 and D2 positions, and it is only the presence of a molecular mirror plane that renders A1 equivalent to A2 (and thence to B1, C2 and D1). In the absence of a molecular mirror plane the eight protons will fall into two non-interconvertible sets of four. In this case, each of these two sets of four is made up of a trans-related pair of protons from different ethylenes; we do not simply have diastereotopic ethylenes each providing a set of four interconvertible proton environments.
To monitor these two rotational processes independently, it is necessary to break the molecular mirror plane, as in (1-methylindenyl)bis(ethylene)rhodium(I), 27, which, at 165 K on a 500 MHz spectrometer, indeed exhibits four 1H NMR resonances in the ethylene “outside” region (3.2–2.2 ppm) and, likewise, four more in the “inside” region (1.5–0.8 ppm); each of these absorptions is, of course, doublet split by 103Rh with JRh-H ∼ 2.5 Hz. Experimentally, the initial coalescence from 165 K to 200 K interchanges proton environments only within the “outside” and within the “inside” regions. This is ascribed to rotation of the Rh(C2H4)2 fragment about the metal-indenyl axis, and yields a barrier of 8.5 ± 0.4 kcal·mol−1. Subsequently, as one approaches room temperature, coalescence between the “inner” and “outer” sets leads ultimately to two resonances, and the barrier was found to be 10.5 ± 0.5 kcal·mol−1. This latter process must be assigned to ethylene rotation which clearly has a higher barrier than indenyl rotation, showing that these two rearrangements are not correlated [51].
Overall, one can conclude that the incorporation of a methyl group into the five-membered ring in 27 not only breaks the mirror symmetry, thus allowing independent monitoring of indenyl and alkene rotations, but also maintains the separation of the eight ethylene protons into two sets of four. This has an interesting consequence for the 13C spectra of 27 which exhibit four peaks at 165 K, two peaks at 220 K and a single resonance at 280 K when the combination of the two rotation processes allows each carbon access to all three other sites. In particular, we note that the two peaks observed at 200 K do not arise from individual ethylenes, but rather each peak comprises a pair of carbons from different ethylenes but related by the two-fold rotation about the indenyl-rhodium axis, as illustrated in Scheme 13 [51].
An analogous study on bis-(trimethylphosphine)(1-methylindenyl)rhodium(I), 28, in which the 31P nuclei are diastereotopic, allowed direct evaluation of the barrier to rotation of the Rh(PMe3)2 fragment about the indenyl-rhodium axis as 11.2 kcal·mol−1 [52].

Parenthetically, one should note an elegant extension of these concepts by the Siegel/Baldridge group whereby alkene rotation and migration of a metal-diene across the corannulene surface has been investigated [53]. As noted in Section 2.3.5, corannulene undergoes rapid bowl-to-bowl inversion; however, when substituted at the 1,3,5,7,9 positions, the two forms are enantiomeric. Coordination of a cationic (η4-norbornadiene)rhodium moiety to one of the arene rings breaks the C5 symmetry of the corannulene and also renders inequivalent the four vinylic hydrogens of the norbornadiene.
The onset of rotation about the metal-arene axis raises the local symmetry of the norbornadiene to C2, but the overall molecular symmetry remains as C1. Migration of the diene-rhodium fragment around the peripheral benzo rings creates a dynamic C5 symmetry for the corannulene unit but the norbornadiene retains its two-fold character (Scheme 14). When the norbornadiene is itself homochiral, as in (R,R)-2,5-dimethyl-bicyclo[2.2.1]hepta-2,5-diene, the result is a dynamic resolution of the equilibrating enantiomers of the sym-pentasubstituted corannulenes [53].
3.2. Silatropic Shifts over Indenyl Surfaces
Molecules of the type (indenyl)2SiX2, where X = Me, Cl or MeO, are important because of their relevance to stereospecific polymerizations of alkenes [54]. In particular, their dynamic behaviour and the chiral character of their zirconium complexes play a role in determining the tacticity of the polymers [55]. Molecules such as (indenyl)2SiMe2 can exist not only as a d,l mixture, 29, but also as the meso isomer 30. These isomers are readily distinguished since in the chiral C2-symmetric isomers the methyl substituents are equivalent, whereas in the meso isomer these substituents lie in the molecular mirror plane and are non-equivalent. At elevated temperatures these diastereomers undergo interconversion and a [1,3] silicon migration process was proposed [54], despite the fact that 1,5-silatropic shifts are Woodward-Hoffmann allowed [4].

This rearrangement was reinvestigated in a single selective inversion NMR experiment which revealed cross-peaks between the proton attached to the sp2 carbon at C(3) and the proton attached to C(1), the sp3-carbon adjacent to silicon; it also shows cross-peaks between the d,l-methyl resonance of 29 and both methyls of meso isomer 30 [56]. Thus the mechanism clearly involves overall migration of silicon between the C(1) and C(3) sites.
However, orbital symmetry considerations favour a [1,5] suprafacial sigmatropic shift which would require the intermediacy of an iso-indene (Scheme 15) with the attendant loss of aromatic character, an issue that has been extensively addressed computationally [57,58]. In this case, one is not trying to break symmetry, but instead attempting to prove the existence of a mirror-symmetric intermediate.
This was subsequently verified by chemically trapping the iso-indene intermediate in a Diels-Alder reaction with tetracyanoethylene (TCNE) as in Scheme 16; in this case, successive [1,5]-suprafacial silatropic shifts and [4+2] cycloadditions led to the double Diels-Alder adduct 31 [56].
In a subsequent study, this rearrangement was extended to tri(indenyl)methylsilane, 32, that exists as RRR, RRS, RSS and SSS isomers in the ratio 1:3:3:1, respectively, giving rise to a total of eight H(2) environments, and a total of sixteen H(1) and H(3) environments. The 3:1 ratio of silicon environments in 32 is very clearly shown in Figure 12 which depicts its two-dimensional 1H-29Si shift-correlated NMR spectrum. The unit intensity 29Si resonance connects only to the protons in the RRR and SSS isomers, whereas the triple intensity peak correlates only with the other combinations.
Considering the consequences of successive [1,5] silatropic shifts across the surface of a single indenyl moiety, such a process would invert the chirality of one indenyl ring and take a molecule of configuration RRR into its RRS stereoisomer. This rearrangement process takes the three equivalent H(2) protons of the RRR isomer (in which all three ring environments can be designated R) and moves them into three new indenyl ring environments, labelled R(1), R(2) and S(3). A second migration interconverts the enantiomorphic RRS and RSS molecules, thus taking the indenyl rings labelled R(1), R(2) and S(3) into R(3), S(2) and S(1) environments, respectively. Finally, the third step regenerates a homotopic molecule, SSS, in which all three indenyl ring environments are identically S (Scheme 17).
Figure 13 depicts a cube onto which can be mapped all the permitted exchange pathways for the H(2) environments. Each exchange along a cube edge requires two successive [1,5]-shifts of a Me(indenyl)2Si moiety across the surface of the remaining indenyl ring; the net result is equivalent to a [1,3]-shift. Note that: (i) the transformations R(1) → S(1) and R(2) → S(2) are invisible since they involve isochronous sites; and (ii) the exchanges R(1) → R(2) and S(1) → S(2) require the traversal of two edges. Evidently, the S → R interchange requires silatropic migrations across all three rings (or three edges of the cube).
These predictions were verified in a 2D-EXSY experiment that revealed strong cross-peaks between directly connected H(2) environments, but much weaker cross-peaks for two-step processes, as exemplified in Figure 14. Interestingly, each migration also interconverts an H(1) and H(3) site, and these sixteen environments can be mapped onto a tesseract (a 4-dimensional hypercube) [59]. Once again, the subtle changes in symmetry during the multi-step rearrangement process are readily monitored by modern NMR techniques. The syntheses, reactivity and dynamics of η1-indenyl derivatives of main group and transition elements has been comprehensively reviewed [60].
3.3. Cyclopentadienyl(dicarbonyl)iron Migration across an Indenyl Surface
While silatropic shifts across the indenyl skeleton can be monitored either directly by peak coalescence at elevated temperatures (110 °C), or indirectly by selective inversion techniques, the corresponding migration of the cyclopentadienyl(dicarbonyl)iron fragment in (η5-C5H5)Fe(CO)2(η5-C9H7), 33, is a more challenging task. The molecule is temperature-sensitive and, when heated, readily loses the carbon monoxide ligands to form benzoferrocene, 34 (Scheme 18). However, a 2D-EXSY experiment at 45 °C (Figure 15) clearly proves that overall [1.3]-migration occurs. Moreover, the intermediacy of the iso-indene, 35, was once again verified by Diels-Alder trapping with tetracyanoethylene [61].
4. The Dynamic Behaviour of Triptycenes
4.1. Triptycenes as Molecular Bevel Gears
The triptycene unit, with its triple-bladed paddlewheel architecture, has long attracted the attention of researchers seeking to synthesize nano-scale analogues of gears, brakes and other types of machinery. These studies focussed not merely on the iconic aspects whereby the molecule resembled its macro-scale counterpart in appearance, but rather as analogic models that exhibited the same dynamic behaviour as the larger scale object [14,15]. Typically, ditriptycyl ether, 36, the di-indenyl system, 37, and the bibenzimidazole, 38, which each bear adjacent triptycyl units, all exhibit rapid cogwheeling which requires the interaction of tightly meshed gears [62,63].

4.2. Triptycenes as Components of Molecular Brakes
A particularly ingenious experiment involving the incorporation of a triptycyl unit in a molecular brake has been reported by Kelly; in this case, a bipyridyl substituent was attached at C(9) and, initially, the triptycyl fragment in 39 was free to rotate. However, complexation of both bipyridyl nitrogens to a metal (normally Hg2+) forces a pyridine ring hydrogen in between two of the blades of the paddlewheel and breaks its three-fold symmetry, as shown in Scheme 19 [64].
An attempt to develop an organometallic molecular brake was described by Stephens and Richards who synthesized [(9-triptycylethynyl)cyclopentadienyl](tetraphenylcyclobutadiene)cobalt, 40. It was hoped that rotation of the paddlewheel would be gated by the bulky C4Ph4 ligand; unfortunately, however, there was no spectroscopic evidence to support such a scenario [65].

A different approach to breaking the three-fold symmetry of the triptycene as a component of a molecular brake was based upon the migration of an organometallic moiety [66]. The haptotropic shift of a metal carbonyl or other organometallic fragment over the surface of an indene is a well-established occurrence [67]. Typically, deprotonation of the [(η6-indene)Mn(CO)3]+ cation, 41, induces migration of the tricarbonylmanganese moiety from the six-membered to the five-membered ring, as in 42 (Scheme 20).
This phenomenon was exploited to bring about the reversible η6- to η5-haptotropic shift of a tricarbonylchromium moiety across an indenyl framework, from 43 to 44 (Scheme 21), so as to block the rotation of the triptycyl paddlewheel and thus activate the first organometallic molecular brake. The migration of the bulky organometallic group over a distance of approximately two Å breaks the three-fold symmetry of the triptycyl unit and renders two of the blades no longer equivalent to the third, as clearly indicated in the 1H and 13C NMR spectra of 44. Analogous compounds of Mn(CO)3 and Re(CO)3 were also prepared; as for the chromium system, the NMR data (Figure 16) again exhibited 2:1 splitting of the paddlewheel blades in the manganese and rhenium η5-complexes [68].
The structure of the molecular brake was independently established via X-ray crystallography that unequivocally revealed the positioning of the metal tripod such that a carbonyl ligand is tightly embedded within a valley between two of the triptycene blades. This is illustrated in Figure 17 which presents space-fill views both from the side and from below the paddlewheel [68].
4.3. Hindered Rotations in Ferrocenyl-Triptycenes
The ability of an (η5-cyclopentadienyl)M(CO)3 unit to hinder rotation of triptycene when directly attached to C(9) prompted an investigation of the closely related ferrocenyl analogues. In 9-ferrocenyltriptycene, the rotational barrier was found to be 17 kcal·mol−1, but 9,10-diferrocenyltriptycene, 45, was found to exist as co-existing, slowly interconverting meso and d,l rotamers in which the ferrocenyl substituents were eclipsed and staggered, respectively (Scheme 22). These isomers are readily differentiated since in the meso (C2v) case the mirror-related benzo rings give rise to two sets of CH environments, whereas in the C2-symmetric rac isomer one sees four such resonances in both the 1H and 13C NMR regimes. The structure of one of the racemic rotamers, whereby the two ferrocenyls are aligned with different valleys, appears as Figure 18. A 2D-EXSY study revealed that the exchange process occurs by successive rotations of a single ferrocenyl substituent, akin to a set of molecular dials. Thus, interconversion of the d and l rotamers requires two rotations, passing via the meso intermediate [69].
5. Concluding Remarks
Broken symmetry is at the core of many physical and chemical phenomena, ranging from the generation of mass by the Higgs boson, to the predominance of matter over anti-matter, to the chiral nature of so many natural products, and the right-handed character of the DNA double helix. Applying these concepts to molecular stereochemistry and dynamics, we have seen how judiciously broken symmetry can provide information about the mechanisms and energetics of otherwise undetectable rearrangement processes.
Evidently, in a short review one can only provide representative examples of some of the many ways ingenious chemists have tackled this problem. These include: (a) replacement of one molecular fragment by another, e.g., CO by CS or NO, diphos by arphos, phenyl by naphthyl, (C5H5)Ni by Co(CO)3, etc.; (b) incorporation of a diastereotopic unit, such as isopropyl, to probe the chirality of the system; (c) addition of a substituent to break mirror symmetry, e.g., a 1-methyl group in the indenyl framework, or a π-bonded organometallic fragment to discriminate between faces of a planar cyclic system; or (d) monitoring the behaviour of a system containing numerous stereocentres, as in the molecular dynamics of tri(indenyl)methylsilane.
In recent decades, advances in NMR technology have given us immensely higher field strengths, a plethora of new pulse sequences, multi-dimensional spectra, etc., but classical variable-temperature measurements are still a valuable component of our arsenal of investigative techniques. One should, however, not undervalue the contributions of the synthetic chemist in this regard; it is relatively easy to draw a hypothetical molecule with substituents appropriate for an NMR study, but more of a challenge to devise a synthetic route and to prepare an analytically pure sample. Nevertheless, we can be confident that symmetry breaking will endure as an important technique for our understanding of complex molecular dynamics, and we look forward with anticipation to even more elegant revelations in the coming years.
Acknowledgments
Many years of generous financial support from the Natural Sciences and Engineering Research Council of Canada (NSERC), from the Petroleum Research Fund, administered by the American Chemical Society, and from Science Foundation Ireland (SFI) are gratefully acknowledged.
Conflicts of Interest
The author declares no conflict of interest.
References and Notes
- Helgaker, T.; Jasuński, M.; Garbacz, P.; Jackowski, K. The NMR indirect nuclear spin-spin coupling constant of the HD molecule. Mol. Phys 2012, 110, 2611–2617. [Google Scholar]
- Oth, J.F.M.; Müllen, K.; Gilles, J.-M.; Schröder, G. Comparison of 13C and 1H magnetic resonance resonance spectroscopy as techniques for the quantitative investigation of dynamic processes. The Cope rearrangement in bullvalene. Helv. Chim. Acta 1974, 57, 1415–1433. [Google Scholar]
- Koritsanszky, T.; Buschmann, J.; Luger, P. Topological analysis of experimental electron densities. 1. The different C–C bonds in bullvalene. J. Phys. Chem 1996, 100, 10547–10553. [Google Scholar]
- Woodward, R.B.; Hoffmann, R. The Conservation of Orbital Symmetry; Verlag Chemie, GmbH: Weinheim, Germany, 1970; p. 126. [Google Scholar]
- Maricq, M.M.; Waugh, J.S.; Fletcher, J.L.; McGlinchey, M.J. Anisotropic ring-carbon shifts in arene chromium tricarbonyl complexes. J. Am. Chem. Soc 1978, 100, 6902–6904. [Google Scholar]
- Hunter, G.; Iverson, D.J.; Mislow, K.; Blount, J.F. Conformational variability in hexaethylbenzene π-complexes. Crystal and molecular structure of tricarbonyl(hexaethylbenzene)chromium(0) and dicarbonyl(hexaethylbenzene)(triphenylphosphine)chromium(0). J. Am. Chem. Soc 1978, 100, 6902–6904. [Google Scholar]
- McGlinchey, M.J.; Bougeard, P.; Sayer, B.G.; Hofer, R.; Lock, C.J.L. Solid state 13C and high resolution 1H and 13C NMR spectra of dicarbonyl(hexaethylbenzene)thiocarbonylchromium(0): A reaffirmation of slowed tripodal rotation. Chem. Commun 1984, 789–790. [Google Scholar]
- Mailvaganam, B.; Frampton, C.S.; Sayer, B.G.; Top, S.; McGlinchey, M.J. An X-ray crystallographic and high field NMR study of [(C6Et6)Cr(CO)2NO]+ BF4− and of [(C6Et6)Cr(CO)(CS)NO]+ BF4−: Steric inhibition of tripodal rotation. J. Am. Chem. Soc 1991, 113, 1177–1185. [Google Scholar]
- McGlinchey, M.J. Slowed tripodal rotation in arene-chromium complexes: Steric and electronic barriers. Adv. Organomet. Chem 1992, 34, 285–325. [Google Scholar]
- Downton, P.A.; Mailvaganam, B.; Frampton, C.S.; Sayer, B.G.; McGlinchey, M.J. Unequivocal proof of slowed chromium tricarbonyl rotation in a sterically crowded arene complex: An X-ray crystallographic and variable-temperature high field NMR study of (C6Et5COCH3)Cr(CO)3. J. Am. Chem. Soc 1990, 112, 27–32. [Google Scholar]
- Kilway, K.V.; Siegel, J.S. Effects of transition-metal complexation on the stereodynamics of persubstituted arenes. Evidence for steric complementarity between arene and metal tripod. J. Am. Chem. Soc 1992, 114, 255–261. [Google Scholar]
- Brydges, S.; McGlinchey, M.J. Conformations and threshold rotational mechanisms of C5Ar5 and C5Ar4X propellers: A structure correlation and computational study. J. Org. Chem 2002, 67, 7688–7698. [Google Scholar]
- Brydges, S.; Britten, J.F.; Chao, L.C.F.; Gupta, H.K.; McGlinchey, M.J.; Pole, D.L. The structure of a seven-bladed propeller: C7Ph7+ is not planar. Chem. Eur. J 1998, 4, 1199–1203. [Google Scholar]
- Iwamura, H.; Mislow, K. Stereochemical consequences of dynamic gearing. Acc. Chem. Res 1988, 21, 175–182. [Google Scholar]
- Brydges, S.; Harrington, L.E.; McGlinchey, M.J. Sterically hindered organometallics: Multi-n-rotor (n = 5, 6 and 7) and the search for correlated rotations. Coord. Chem. Rev 2002, 233–234, 75–105. [Google Scholar]
- Field, L.D.; He, T.; Humphrey, P.; Masters, A.F.; Turner, P. Manganese complexes of the pentaphenylcyclopentadienyl ligand. Polyhedron 2006, 25, 1498–1506. [Google Scholar]
- Field, L.D.; Lindall, C.M.; Masters, A.F.; Clentsmith, G.K.B. Pentaarylcyclopentadienyl complexes. Coord. Chem. Rev 2011, 255, 1733–1790. [Google Scholar]
- Li, L.; Decken, A.; Sayer, B.G.; McGlinchey, M.J.; Brégaint, P.; Thépot, J.-Y.; Hamon, J.-R.; Lapinte, C. Multiple fluxional processes in chiral molecules: The barriers to aryl and tripodal rotation in (C5Ph5)Fe(CO)(PhMe2P)CHO and (C5Ph5)Fe(CO)2Br. Organometallics 1994, 13, 682–689. [Google Scholar]
- Note, however, the following interesting report: Labuta, J.; Ishihara, S.; Ariga, K.; Hill, J.P. Dynamic processes in prochiral solvating agents (pro-CSAs) studied by NMR spectroscopy. Symmetry 2014, 6, 345–367.
- Gloaguen, B.; Astruc, D. Chiral pentaisopropylcyclopentadienyl and pentakis(1-ethylpropyl)cyclopentadienyl complexes: One-pot synthesis by formation of 10 carbon-carbon bonds from pentamethylcobalticinium. J. Am. Chem. Soc 1990, 112, 4607–4609. [Google Scholar]
- McGlinchey, M.J.; Girard, L.; Ruffolo, R. Cluster-stabilized cations: Syntheses, structures, molecular dynamics and reactivity. Coord. Chem. Rev 1995, 143, 331–381. [Google Scholar]
- Schilling, B.E.R.; Hoffmann, R. M3L9(ligand) complexes. J. Am. Chem. Soc 1979, 101, 3456–3467. [Google Scholar]
- Edidin, R.T.; Norton, J.R.; Mislow, K. Evidence for tilted ground-state structures and fluxionality in Co3(CO)9CCHR+. Organometallics 1982, 1, 561–562. [Google Scholar]
- Hallgren, J.E.; Eschbach, C.S.; Seyferth, D. The carbatricobalt decacarbonyl cation. A novel acylating agent. J. Am. Chem. Soc 1972, 94, 2547–2549. [Google Scholar]
- D’Agostino, M.F.; Mlekuz, M.; Kolis, J.W.; Sayer, B.G.; Rodger, C.A.; Halet, J.-F.; Saillard, J.-Y.; McGlinchey, M.J. 13C NMR and EHMO studies on Seyferth’s [Co3(CO)9CCO]+ cluster: To bend or not to bend. Organometallics 1986, 5, 2345–2350. [Google Scholar]
- Shriver, D.; Kaesz, H.D.; Adams, R.D. The Chemistry of Metal Cluster Complexes; VCH: New York, NY, USA, 1990. [Google Scholar]
- Mlekuz, M.; Bougeard, P.; Sayer, B.G.; McGlinchey, M.J.; Peng, S.; Marinetti, A.; Saillard, J.-Y.; Ben Naceur, J.; Mentzen, B.; Jaouen, G. Syntheses, crystal structures and DNMR studies on the mixed clusters CpNiFe(CO)3(RC≡CR')M, (M = CpNi, Co(CO)3, Mo(CO)2Cp): Some comments on the acetylene rotation process. Organometallics 1985, 4, 1123–1130. [Google Scholar]
- Schilling, B.E.R.; Hoffmann, R. Dependence of equilibrium geometry and rearrangement modes on electron count in one class of trinuclear complexes of acetylene. Acta Chem. Scand. Ser. B 1979, 33, 231–232. [Google Scholar]
- Hoffmann, R. Building bridges between inorganic and organic chemistry. Angew. Chem. Int. Ed. Engl 1982, 21, 711–724. [Google Scholar]
- Stohrer, W.-D.; Hoffmann, R. Bond stretch isomerism and polytopal rearrangements in (CH)5+, (CH)5− and (CH)4CO. J. Am. Chem. Soc 1972, 94, 1661–1668. [Google Scholar]
- Sutin, K.A.; Kolis, J.W.; Mlekuz, M.; Bougeard, P.; Sayer, B.G.; Quilliam, M.A.; Faggiani, R.; Lock, C.J.L.; McGlinchey, M.J.; Jaouen, G. Arphos and diphos complexes of Co2(CO)6MC-CO2-i-Pr [M = Co(CO)3, (C5Me5)Mo(CO)2]: X-ray crystal structure and NMR fluxionality. Organometallics 1987, 6, 439–447. [Google Scholar]
- El Hafa, H.; Cordier, C.; Gruselle, M.; Besace, Y.; Jaouen, G.; McGlinchey, M.J. An NMR study of the dynamic behavior of [(2-propynylbornyl)Mo2(CO)4Cp2]+ BF4−: Non-fluxional Mo-Co clusters as the key to understanding the mechanism of the formation of metal-stabilized cations. Organometallics 1994, 13, 5149–5156. [Google Scholar]
- Jurado, B.; Springer, C.S., Jr. Direct measurement of enantiomerization of labile aluminium(III) β-diketonates. Chem. Commun 1971, 85–87. [Google Scholar]
- Serpone, N.; Bickley, D.G. Kinetics and mechanisms of isomerization and racemization processes of six-coordinate chelate complexes. Prog. Inorg. Chem 1972, 17, 391–566. [Google Scholar]
- Serpone, N.; Bickley, D.G. Configurational rearrangements in cis-M(AA)2X2, cis-M(AA)2XY, and cis-M(AB)2X2 complexes 10. The cis-M(AB)2X2 system—Diastereotopic probe on the X-ligands. Inorg. Chim. Acta 1982, 57, 211–216. [Google Scholar]
- Barth, W.E.; Lawton, R.G. The synthesis of corannulene. J. Am. Chem. Soc 1971, 93, 1730–1745. [Google Scholar]
- Scott, L.T.; Cheng, P.-C.; Hashemi, M.M.; Bratcher, M.S.; Meyer, D.T.; Warren, H.B. Corannulene. A three-step synthesis. J. Am. Chem. Soc 1997, 119, 10963–10968. [Google Scholar]
- Borchardt, A.; Fichicello, A.; Kilway, K.V.; Baldridge, K.K.; Siegel, J.S. Synthesis and dynamics of the corannulene nucleus. J. Am. Chem. Soc 1992, 114, 1921–1923. [Google Scholar]
- Scott, L.T.; Hashemi, M.M.; Bratcher, M.S. Corannulene bowl-to-bowl inversion is rapid at room temperature. J. Am. Chem. Soc 1992, 114, 1920–1921. [Google Scholar]
- Biedermann, P.U.; Pogodin, S.; Agranat, I. Inversion barrier for corannulene. A benchmark for bowl-to-bowl inversions in fullerene fragments. J. Org. Chem 1999, 64, 3655–3662. [Google Scholar]
- On a 500 MHz NMR spectrometer, fluorine spectra are run at 471 MHz; since 19F nuclei in aromatic rings span a chemical shift range greater than 20 ppm, peak separation can exceed 10,000 Hz.
- Gupta, H.K.; Stradiotto, M.; Hughes, D.W.; McGlinchey, M.J. Reactions of C6F5Li with tetracyclone and 3-ferrocenyl-2,4,5-triphenylcyclopentadienone: A 19F and X-ray crystallographic study of hindered pentafluorophenyl rotations. J. Org. Chem 2000, 65, 3652–3658. [Google Scholar]
- Nori-shargha, D.; Asadzadeha, S.; Ghanizadehb, F.-R.; Deyhimic, F.; Aminic, M.M.; Jameh-Bozorghi, S. Ab initio study of the structures and dynamic stereochemistry of biaryls. J. Mol. Struct. THEOCHEM 2005, 717, 41–51. [Google Scholar]
- Nowak, W.; Wierzbowska, M. A theoretical study of geometry and transition moment directions of flexible fluorescent probes—acetoxy derivatives of phenylanthracene. J. Mol. Struct. THEOCHEM 1996, 368, 223–234. [Google Scholar]
- Nikitin, K.; Ortin, Y.; Müller-Bunz, H.; McGlinchey, M.J. Restricted rotation in phenyl-anthracenes: A prediction fulfilled. Org. Lett 2011, 13, 256–259. [Google Scholar]
- Nikitin, K.; Fleming, C.; Müller-Bunz, H.; Ortin, Y.; McGlinchey, M.J. Severe Energy Costs of Double Steric Interactions: Towards a Molecular Clamp. Eur. J. Org. Chem 2010, 5203–5216. [Google Scholar]
- Cramer, R. Olefin coordination compounds of rhodium. The barrier to rotation of coordinated ethylene and the mechanism of olefin exchange. J. Am. Chem. Soc 1964, 86, 217–222. [Google Scholar]
- Cramer, R.; Kline, J.B.; Roberts, J.D. Bond character and conformation equilibration of ethylene- and tetrafluoroethylene-rhodium complexes from nuclear magnetic resonance spectra. J. Am. Chem. Soc 1969, 91, 2519–2524. [Google Scholar]
- Caddy, P.; Green, M.; O’Brien, E.; Smart, L.E.; Woodward, P. Reactions of coordinated ligands. Part 22. The reactivity of bis-(ethylene)(indenyl)rhodium in displacement reactions with olefins, dienes and acetylenes. J. Chem. Soc. Dalton Trans 1980, 962–972. [Google Scholar]
- Eshtiagh-Hosseini, H.; Nixon, J.F. Ethylene displacement reactions of indenylbis(ethylene)rhodium. J. Less-Common Met 1978, 61, 107–121. [Google Scholar]
- Mlekuz, M.; Bougeard, P.; Sayer, B.G.; McGlinchey, M.J.; Rodger, C.A.; Churchill, M.R.; Ziller, J.W.; Kang, S.-K.; Albright, T.A. X-ray crystal structure and molecular dynamics of (indenyl)bis(ethylene)rhodium(I): 500 MHz NMR spectra and EHMO calculations. Organometallics 1986, 5, 1656–1663. [Google Scholar]
- Marder, T.B.; Calabrese, J.C.; Roe, D.C.; Tulip, T.H. The slip-fold distortion of π-bound indenyl ligands—Dynamic NMR and X-ray crystallographic studies of (η-indenyl)RhL2 complexes. Organometallics 1987, 6, 2012–2014. [Google Scholar]
- Bandera, D.; Baldridge, K.K.; Linden, A.; Dorta, R.; Siegel, J.S. Stereoselective coordination of C5-symmetric corannulene derivatives with an enantiomerically pure [Rh(nbd*)] metal complex. Angew. Chem. Int. Ed 2011, 50, 865–867. [Google Scholar]
- Chen, Y.-X.; Rausch, M.D.; Chien, J.C.W. Indenyl- and fluorenylsilanes: Synthesis and thermal diastereomerization. Organometallics 1993, 12, 4607–4612. [Google Scholar]
- Ellis, W.W.; Hollis, T.K.; Odenkirk, W.; Whelan, J.; Ostrander, R.; Rheingold, A.L.; Bosnich, B. Synthesis, structure, and properties of chiral titanium and zirconium complexes bearing biaryl strapped substituted cyclopentadienyl ligands. Organometallics 1993, 12, 4391–4401. [Google Scholar]
- Rigby, S.S.; Girard, L.; Bain, A.D.; McGlinchey, M.J. The molecular dynamics of d,l- and meso-bis(indenyl)dimethylsilane: A re-examination of the mechanism of interconversion by using single selective inversion NMR. Organometallics 1995, 14, 3798–3801. [Google Scholar]
- Rigby, S.S.; Gupta, H.K.; Werstiuk, N.H.; Bain, A.D.; McGlinchey, M.J. The barriers to trimethylsilyl migrations in indenes and benzindenes: Silatropic shifts via aromatic transition states. Polyhedron 1995, 14, 2787–2796. [Google Scholar]
- Rigby, S.S.; Gupta, H.K.; Werstiuk, N.H.; Bain, A.D.; McGlinchey, M.J. Do aromatic transition states lower barriers to silatropic shifts? A synthetic, NMR spectroscopic, and computational study. Inorg. Chim. Acta 1996, 251, 355–364. [Google Scholar]
- Stradiotto, M.; Rigby, S.S.; Hughes, D.W.; Brook, M.A.; Bain, A.D.; McGlinchey, M.J. A multidimensional NMR study of tris(indenyl)methylsilane: Molecular dynamics mapped onto a hypercube. Organometallics 1996, 15, 5645–5652. [Google Scholar]
- Stradiotto, M.; McGlinchey, M.J. η1-Indenyl complexes of main group and transition elements: Syntheses, structures and molecular dynamics. Coord. Chem. Rev 2001, 219–221, 311–378. [Google Scholar]
- Stradiotto, M.; Hughes, D.W.; Bain, A.D.; Brook, M.A.; McGlinchey, M.J. The fluxional character of (η5-C5H5)Fe(CO)2(η1-C9H7): Evidence for the [4+2] cycloaddition of a metal-substituted isoindene with tetracyanoethylene. Organometallics 1997, 16, 5563–5568. [Google Scholar]
- Nikitin, K.; Müller-Bunz, H.; Ortin, Y.; Risse, W.; McGlinchey, M.J. Twin triptycyl spinning tops: A simple case of molecular gearing with dynamic C2 symmetry. Eur. J. Org. Chem 2008, 3079–3084. [Google Scholar]
- Franz, D.K.; Linden, A.; Baldridge, K.K.; Siegel, J.S. Molecular spur gears comprising triptycene rotators and bibenzimidazole stators. J. Am. Chem. Soc 2012, 134, 1528–1535. [Google Scholar]
- Kelly, T.R. Progress toward a rationally designed molecular motor. Acc. Chem. Res 2001, 34, 514–522. [Google Scholar]
- Stephens, A.M.; Richards, C.J. A metallocene molecular gear. Tetrahedron Lett 1997, 38, 7805–7808. [Google Scholar]
- Harrington, L.E.; Cahill, L.; McGlinchey, M.J. Towards an organometallic molecular brake with a metal foot-pedal: The synthesis, dynamic behavior and X-ray crystal structure of (9-indenyl)triptycene-Cr(CO)3. Organometallics 2004, 23, 2884–2891. [Google Scholar]
- Oprunenko, Y.F. Inter-ring haptotropic rearrangements in pi complexes of transition metals with polycyclic aromatic ligands. Russ. Chem. Rev 2000, 69, 683–704. [Google Scholar]
- Nikitin, K.; Müller-Bunz, H.; Ortin, Y.; McGlinchey, M.J. A molecular paddle-wheel with a sliding organometallic latch: Syntheses, X-Ray crystal structures and dynamic behaviour of [η6–2-(9-triptycyl)-indene]Cr(CO)3, and of [η5–2-(9-triptycyl)-indenyl]M(CO)3, M = Mn, Re. Chem. Eur. J 2009, 15, 1836–1843. [Google Scholar]
- Nikitin, K.; Müller-Bunz, H.; Ortin, Y.; Muldoon, J.; McGlinchey, M.J. Molecular dials: Hindered rotations in mono- and di-ferrocenyl anthracenes and triptycenes. J. Am. Chem. Soc 2010, 132, 17617–17622. [Google Scholar]









































© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
McGlinchey, M.J. Symmetry Breaking in NMR Spectroscopy: The Elucidation of Hidden Molecular Rearrangement Processes. Symmetry 2014, 6, 622-654. https://doi.org/10.3390/sym6030622
McGlinchey MJ. Symmetry Breaking in NMR Spectroscopy: The Elucidation of Hidden Molecular Rearrangement Processes. Symmetry. 2014; 6(3):622-654. https://doi.org/10.3390/sym6030622
Chicago/Turabian StyleMcGlinchey, Michael J. 2014. "Symmetry Breaking in NMR Spectroscopy: The Elucidation of Hidden Molecular Rearrangement Processes" Symmetry 6, no. 3: 622-654. https://doi.org/10.3390/sym6030622
APA StyleMcGlinchey, M. J. (2014). Symmetry Breaking in NMR Spectroscopy: The Elucidation of Hidden Molecular Rearrangement Processes. Symmetry, 6(3), 622-654. https://doi.org/10.3390/sym6030622