4.1. Water-Rock Interactions in the Komatiite–H2O–NO3− System
Fluid composition and secondary mineral assemblages provide information on the water-rock interactions that include hydrogen generation and nitrate reduction. To aid the interpretation of reactions occurring during the experiments, saturation indexes of solid materials were calculated based on the measured fluid composition (
Table 6). A list of potential water-rock reactions is also shown in
Table 7.
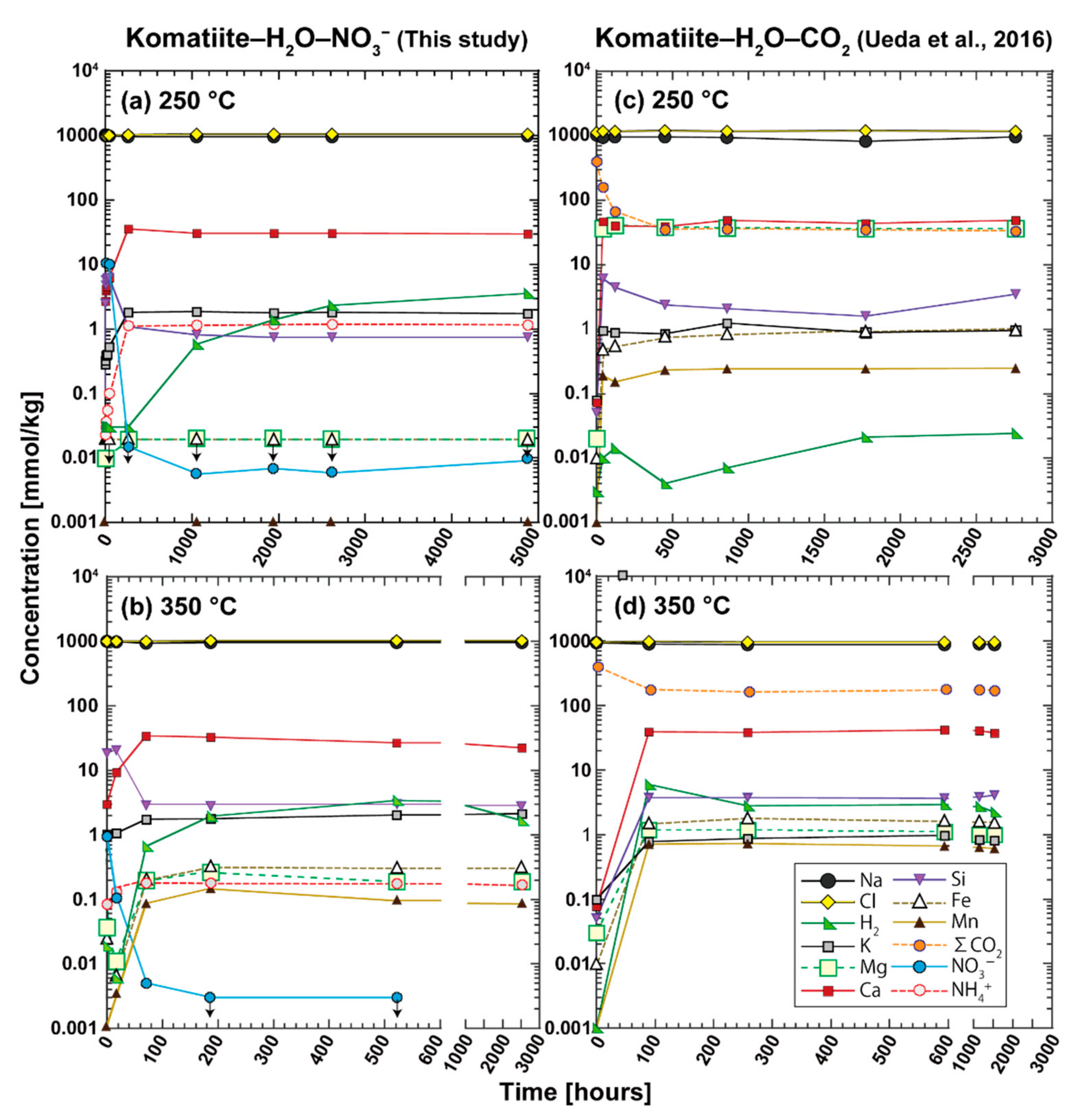
In experiment N250, the concentration of dissolved Si (SiO
2(aq)) increased to the maximum value (6 mmol/kg) during the first 8 h at 250 °C and then, remained at the maximum value from 8 h to 44 h (
Figure A1). The maximum concentration of SiO
2(aq) was 15% of the SiO
2(aq) concentration predicted from the solubility of amorphous silica (
Table 6), but it was ~100 times higher than the SiO
2(aq) concentration under olivine dissolution at temperatures from 230 °C to 320 °C and apH
in-situ of 8 [
66]. Moreover, the positive SI values of forsterite and fayalite after the first 44 h (i.e., supersaturation with respect to forsterite) indicates that olivine dissolution had been inhibited in the first 44 h (
Table 6). Therefore, the water-rock interactions that mainly occurred from 8 h to 44 h are predicted to be the dissolution of komatiite glass and the precipitation of secondary silicates. Given the low solubility of Al
2O
3 [
67] and the fact that the sampled fluids had low concentrations of Mg and Fe, the precipitation of secondary silicates may have largely occurred through reactions involving solid phases of komatiite glass (rxns. 1–3 in
Table 7). This inference is supported by the stable pH
in-situ value in the first 44 h because reactions 1 to 3 did not affect pH
in-situ much as compared with silicate formation from dissolved species (rxns. 6–8).
The time interval between 44 h to 264 h was characterized by the decrease in SiO
2(aq) concentrations from the maximum value to the steady state value and the decrease in pH
in-situ from 7.7 to 6.2. The decrease in the SiO
2(aq) concentration suggests the completion of the dissolution of the SiO
2 component in komatiite glass and the precipitation of secondary silicates. In particular, the decrease in pH
in-situ requires the precipitation of dissolved Ca as Ca-bearing silicates (saponite, andradite, Ca-pyroxene) (rxns. 1, 4, 5) because the dissolved Ca concentration was high at the milli-molal level, while the dissolved concentrations of Mg and Fe were low over the same time interval. The precipitation of Ca-bearing silicates was consistent with the positive SI values of andradite and diopside (CaMgSi
2O
6) over the same time interval (
Table 6). On the other hand, this was apparently inconsistent with the increase in the dissolved Ca concentration from 6 to 36 mmol/kg. This discrepancy can be resolved if the decrease in pH
in-situ promoted the dissolution of the CaO component in glass (rxn. 15). Because andradite precipitation involves iron oxidation and H
2 generation (rxn. 4), this reaction probably induced nitrate reduction between 44 h to 264 h (
Section 4.2). The oxidation of the FeO component in komatiite glass also possibly contributed to H
2 generation and nitrate reduction between 44 h to 264 h (rxns. 13–14).
The last interval between 264 h to 4871 h was characterized by the decrease in the Ca concentration to the steady state value (30 mol/kg; 266 h to 1054 h) and the increase in the H
2 concentration to 2 mmol/g. The decrease in the Ca concentration was attributed to the formation of andradite because the SI value of andradite was positive until 1054 h, whereas the SI value of diopside became negative after 264 h (
Table 6). This suggests that andradite formation had produced H
2 until 1054 h. On the other hand, dissolution of fayalite, as inferred from the saturation of fluid with respect to fayalite since 1054 h (SI = 0), may have produced H
2 since 1054 h by the oxidation of the FeO component in olivine (FeO
ol) with water to form magnetite and/or Fe
III-bearing serpentine (rxns. 13–14) [
68].
In experiment N350, the SiO
2(aq) concentration in the first 17 h at 350 °C had the maximum value (20 mmol/kg), that is 63% of the SiO
2(aq) concentration predicted from the solubility of amorphous silica (
Figure A1). Over the same time interval, pH
in-situ decreased from 7.1 to 5.8, whereas the Ca concentration increased to 10 mmol/kg. As discussed above, this suggests the dissolution of komatiite glass coupled with the formation of secondary silicates, which includes the precipitation of dissolved Ca as Ca-bearing silicates (saponite, Ca-pyroxene) (rxns. 1–5).
Subsequently, the SiO2(aq) concentration decreased from the maximum value to the steady state value (1 mmol/kg) from 17 h to 71 h. The decrease in the SiO2(aq) concentration was accompanied by a decrease in pHin-situ. Thus, this time interval was probably characterized by the complete consumption of the SiO2 component in komatiite glass and the precipitation of secondary silicates, including Ca-bearing silicates, which is consistent with the positive SI value of diopside at 17 h.
A main feature of the time interval between 71 h to 2515 h was the increase in the H2 concentration to 3 mmol/kg. The SI values of secondary minerals predicted that H2 generation during the entire interval of N350 can be driven by the oxidation of dissolved and solid phases of ferrous iron (Fe2+(aq), FeOrock = FeOglass, FeOol) to form magnetite and/or FeIII-bearing serpentine (rxns. 13–14). Because nitrate reduction to ammonia was completed by 71 h, H2 produced from iron oxidation may have been completely and/or partially consumed to reduce nitrate before 71 h.
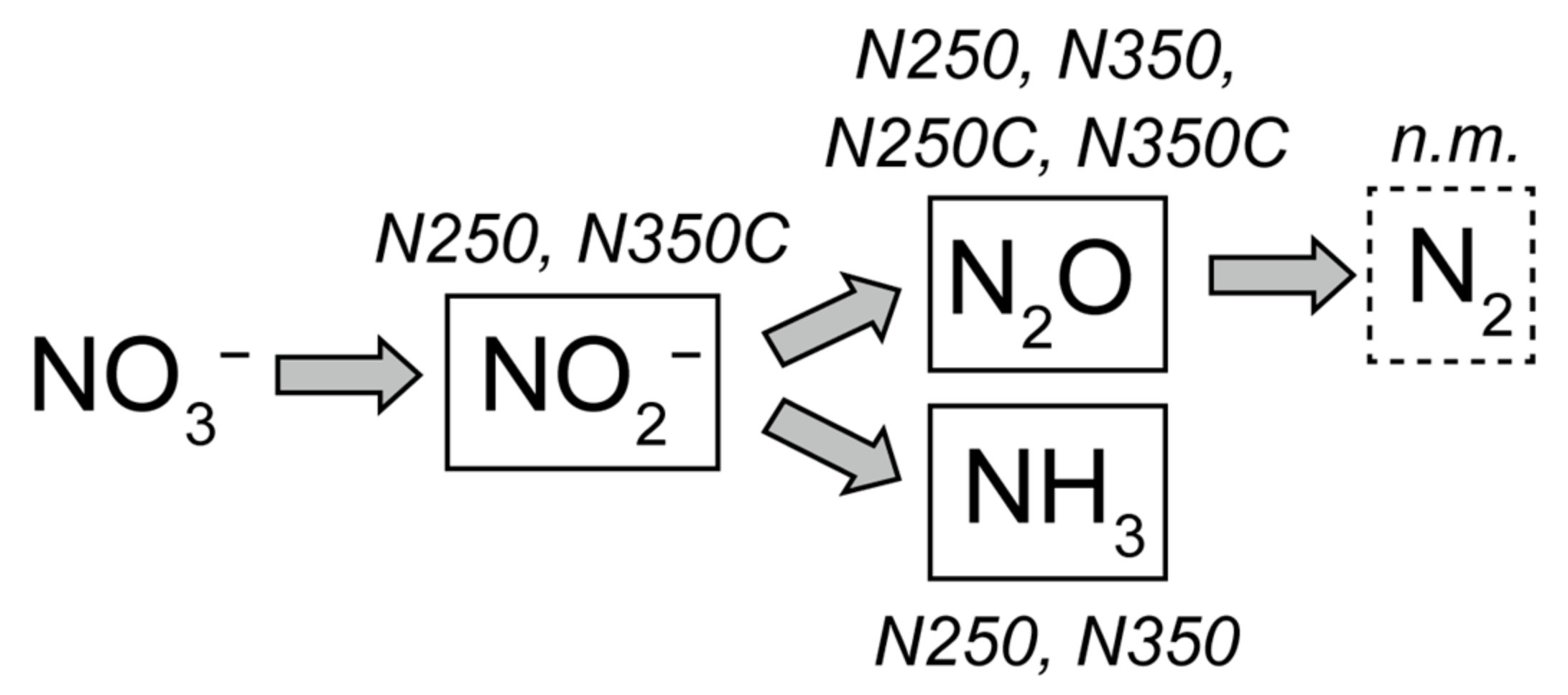
4.2. Mechanisms of Nitrate Reduction to Ammonia and Denitrification in the Komatiite–H2O–NO3− System
This study strongly suggests that reactions in komatiite cause the complete reduction of nitrate to N
2 and NH
3 with a ratio of N
2(aq)/NH
3(aq) of 4.5 at both 250 °C and 350 °C, under pressures of 500 bars (
Figure 8). The thermodynamic equilibrium of dissolved nitrogen species in the N–H–O system was evaluated first as a possible explanation for the N
2(aq)/NH
3(aq) ratio to characterize the mechanism of nitrate reduction in hydrothermal environments.
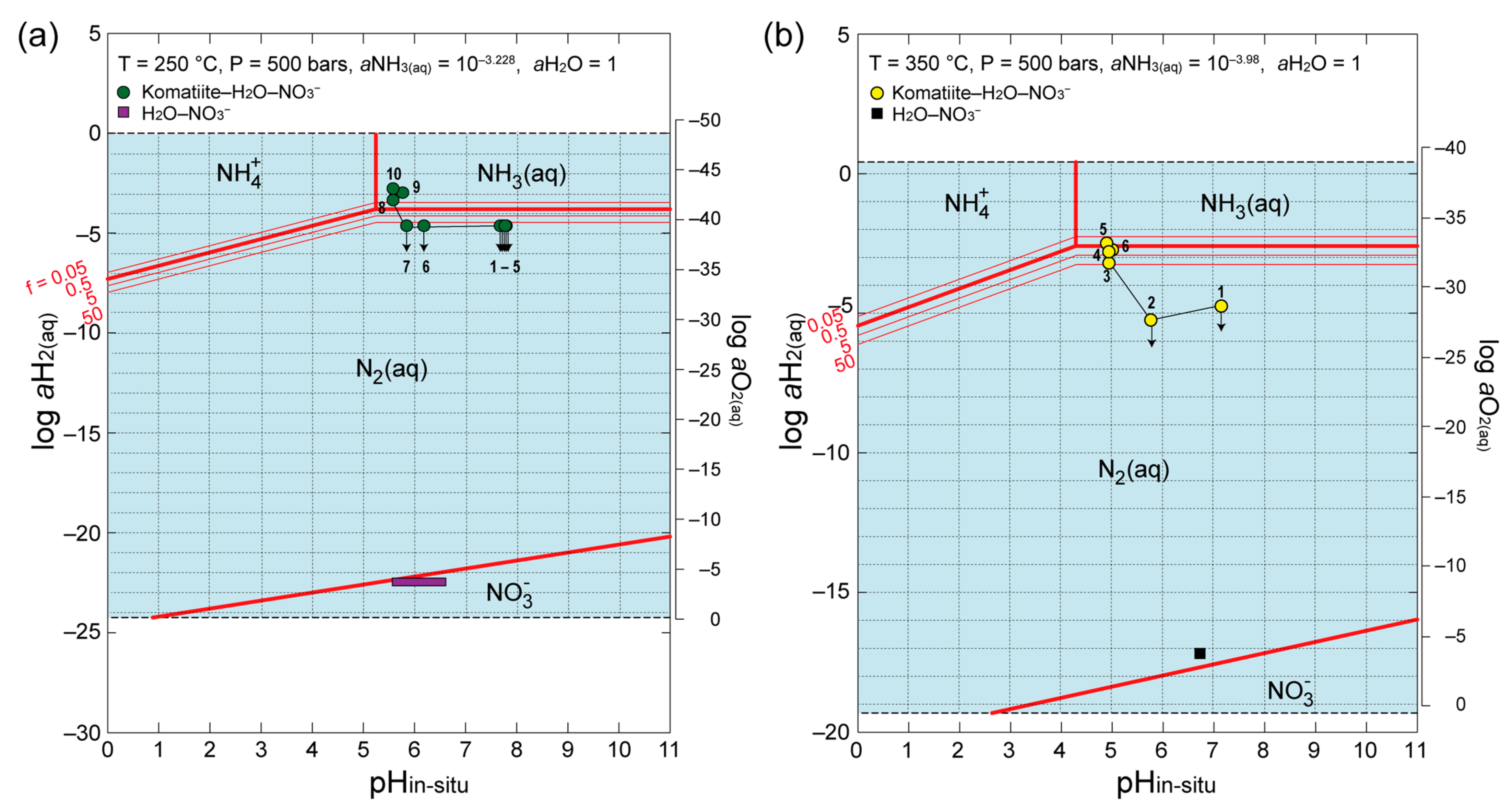
Figure 9a,b show the pH
in-situ and
aH
2(aq) (or
aO
2(aq)) states of water with theoretically predicted stability fields of dissolved nitrogen species in thermodynamic equilibrium at 250 °C and 350 °C, 500 bars, respectively. The stability fields are drawn based on the activity of NH
4+ in the final sampled fluid (
aNH
3(aq) = 10
−3.228 and 10
−3.98 at 250 °C and 350 °C, respectively). Bold red lines in the diagrams indicate the pH
in-situ and
aH
2(aq) values at which the activity ratios of two nitrogen species (NH
3(aq)–NH
4+, NH
3(aq)–N
2(aq), NH
4+–N
2(aq), and N
2(aq)–NO
3−) equal one in thermodynamic equilibrium. The pH
in-situ and
aH
2(aq) values were plotted for all sampled fluids in the komatiite–H
2O–NO
3− and H
2O–NO
3− systems in these diagrams to compare the dominant nitrogen species in the experimental products with those from the thermodynamic prediction. Given that the very low H
2 concentrations of sampled fluids in the H
2O–NO
3− systems were difficult to measure, the presumed
aH
2(aq) values were plotted in the diagrams assuming that sub-milli molal O
2 in the initial solutions was not consumed during the experiments at 250 °C and 350 °C (e.g., dissolved O
2 concentration is 0.2 mmol/kg in an air saturated seawater).
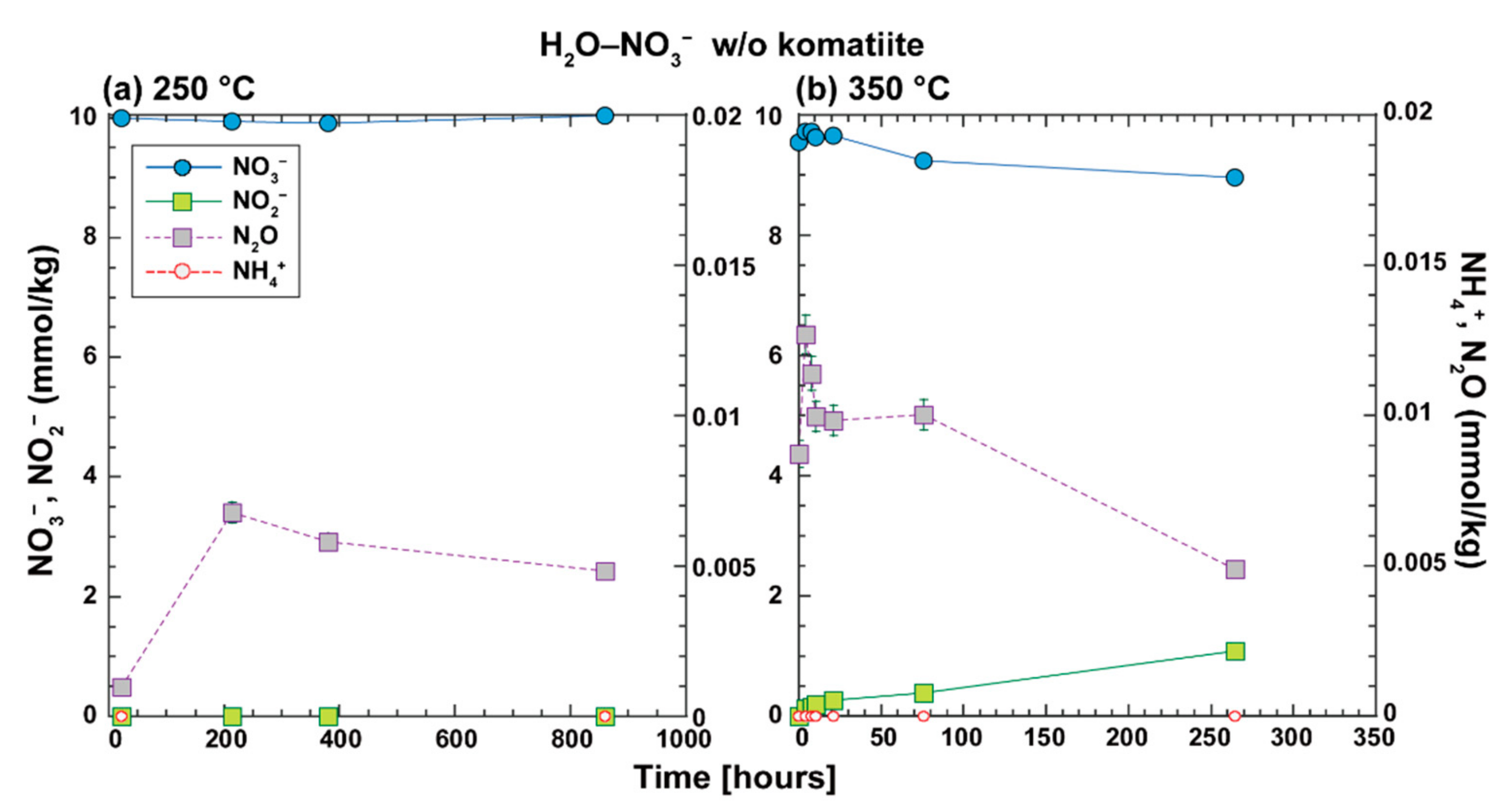
In the H
2O–NO
3− system, sampled fluids at 250 °C and 350 °C were predicted to locate the stability fields of nitrate and N
2, respectively (purple and black boxes in
Figure 9). In these experiments, nitrate at 250 °C was unreacted for 862 h, whereas nitrate at 350 °C was partially reduced for 264 h to nitrite and N
2O, which were expected to be reduced to N
2 via chemo-denitrification. The nitrate chemistry observed in the experiments is consistent with that predicted from the thermodynamic equilibrium of dissolved nitrogen species in the N–H–O system.
In the komatiite–H
2O–NO
3− systems, sampled fluids at 250 °C and 350 °C evolved to have higher
aH
2(aq) values than that in the H
2O–NO
3− systems, which are more favorable for ammonium production (green and yellow circles in
Figure 9). The N
2(aq)/NH
3(aq) ratios in thermodynamic equilibrium at the experimental
aH
2(aq) and pH
in-situ conditions are estimated to be less than 50 for fluid samples taken during the interval of nitrate reduction at 250 °C (the first 264 h; N250-1–N250-6), whereas they increase from less than 5,000,000 to 50 for fluid samples taken during the first 264 h at 350 °C (N350-1–N350-3). Therefore, theoretical prediction assuming thermodynamic equilibrium of nitrogen species in N–H–O system underestimates the amount of NH
3(aq) produced from nitrate in the komatiite–H
2O–NO
3− systems by a factor of 10 and 10–1,000,000.
This suggests that nitrate reduction proceeds at the site of H2 generation where local aH2(aq) is higher than aH2(aq) of bulk fluids (e.g., heterogeneous reaction at the solid surface). It is also possible that nitrate reduction is mainly driven by a reductant other than H2.
Ferrous iron components in komatiite (FeO
rock) are potential candidates for reaction sites to reduce nitrate. A general H
2 generation reaction during serpentinization is written as iron oxidation reactions 14 and 15 in
Table 7 [
68]. In the komatiite–H
2O–NO
3− systems at 250 °C, the H
2 generation reaction also occurs during andradite formation as discussed in
Section 4.1 (rxn. 4). As discussed in
Section 4.1, nitrate reduction in experiments N250 and N350 occurred during the time interval that was thermodynamically favorable to andradite formation (at 250 °C) and iron oxidation to form magnetite and/or Fe
III-bearing serpentine (at both 250 °C and 350 °C). Thus, we suggest the following reactions as nitrate reduction in the komatiite–H
2O–NO
3− system (see
Appendix A for details).
The reactions 19 to 24 represent nitrate reduction by both H2 and FeII at the site of serpentinization and andradite formation. They probably contribute to a lower N2(aq)/NH3(aq) ratio in the komatiite–H2O–NO3– system than expected due to the thermodynamic equilibrium of dissolved nitrogen species in N–H–O system at 250 °C and 350 °C, 500 bars.
4.3. H2 Generation Ability of Komatiite-Hosted Hydrothermal System in Early Earth
Previous simulations of komatiite hydrothermal alteration in the early ocean [
49,
50] typically considered two types of experiments, i.e., those under CO
2-bearing and CO
2-free conditions. These contrasting conditions not only reflect geological records (such as strongly carbonated komatiites and carbonate-free, serpentinized komatiites) [
69], but also the two types of fluid circulation paths. The first type addresses the direct reactions with Hadean–Archean seawater containing high concentration of ΣCO
2 [
50]; this is similar to sulfate in modern seawater introduced directly through faults into reaction zones at the base of seafloor hydrothermal systems, forming sulfate minerals that strongly affect the redox state of hydrothermal fluid, e.g., [
70,
71].
The second type assumes reactions between komatiite and CO
2-free fluid in a reaction zone where CO
2 in downwelling seawater has been mostly precipitated as carbonate minerals in the surrounding rocks (e.g., recharge zone) before the fluid reaches reaction zones [
49]. Previous studies revealed that the formation of carbonate minerals in the reaction zone suppressed H
2 generation given that most of iron originally contained in igneous phases was incorporated directly into carbonate minerals as Fe(II) without going through oxidation and reduction processes in water that generate H
2 [
50]. The experiments in this study were a simulation of the CO
2-free hydrothermal alteration of komatiite in a reaction zone, which has higher reduction abilities than those in CO
2-bearing systems.
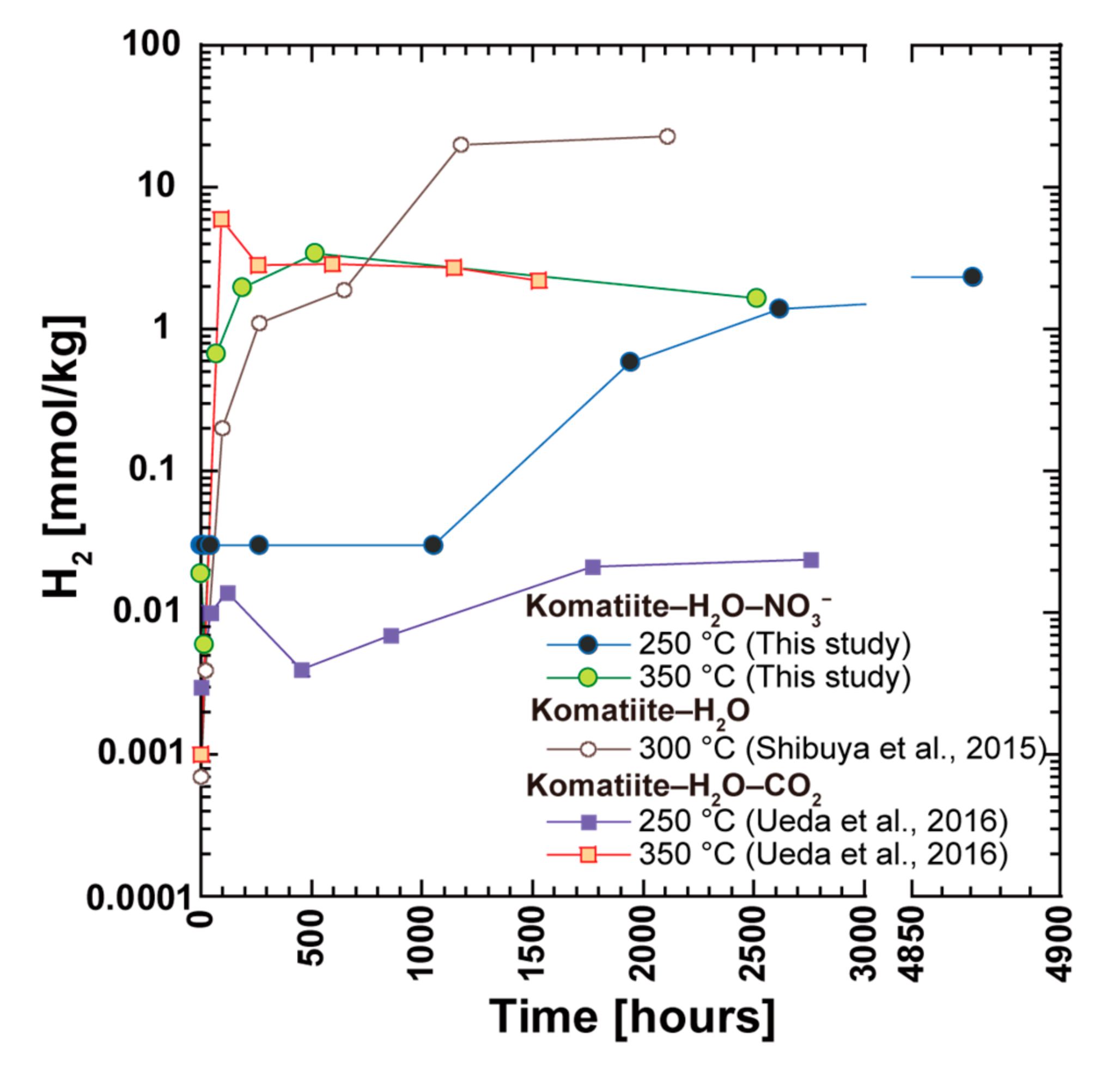
In this case, a certain amount of H
2 generation in the fluids was expected in the CO
2-free experiments. However, the H
2 concentrations in fluid in N350 is 1.7 mmol/kg, which is almost identical to or lower than that in CO
2-bearing experiments at 350 °C. On the contrary, the H
2 concentration in N250 is 100 times higher than that of the CO
2-bearing experiments at 250 °C (
Table 3). This indicates that the reduction ability of komatiite was partially used for the reduction of nitrate during the experiments, as discussed in
Section 4.2. Accordingly, the potential H
2 concentrations were calculated for the case where nitrate was not included in the initial solutions to assess the maximum H
2 generation ability of komatiite during its alteration at 250 °C and 350 °C. Using the N
2(aq)/NH
3(aq) ratio of 4.5 in experiments N250 and N350, the presumed mass balance reaction of nitrate reduction by H
2(aq) that is generated from the reaction of ferrous iron with water can be expressed as
This reaction implies that the reduction of nitrate in the initial fluid requires 2.6 times H
2(aq), suggesting that an additional 26 mmol/kg and 2.6 mmol/kg of H
2(aq) were potentially generated in N250 and N350, respectively. Certainly, these values should be upper limits for H
2 concentrations given than nitrate can be reduced by ferrous iron (
Section 4.2.). However, the potential H
2 concentrations in N250 and N350 and the results of the previous CO
2-free experiment at 300 °C (H
2 = 23 mmol/kg) [
49] suggests that the optimal temperature range for H
2 generation is 250–300 °C during the serpentinization of Al-depleted komatiite in CO
2-free conditions. This temperature range is comparable to the optimal temperature range for H
2 generation during the serpentinization of peridotites inferred from thermodynamic constraints [
68,
72].
4.4. Roles of Komatiite-Hosted Hydrothermal System on Pre-Biotic Chemistry
Komatiite-hosted hydrothermal systems have influenced pre-biotic chemistry and affected the sustainability of primitive microbial communities through the production of H2, alkalinization of fluids, and ammonia production in the komatiite–H2O–NO3− system.
H
2 is a key molecule for pre-biotic chemistry and energy metabolism. Theoretical predictions for the thermodynamic state of pre-biotic chemical evolution have highlighted that an H
2-rich hydrothermal environment was energetically advantageous for the synthesis and preservation of amino and fatty acids [
73]. Further, the most plausible energy metabolisms to support the emergence and early evolution of life are considered to be H
2-driven hydrogenotrophic methanogenesis/acetogenesis and/or methanotrophic acetogenesis, e.g., [
43,
45,
46]. On the basis of fluid chemistry and mineralogy in the komatiite–H
2O–CO
2 system, Ueda et al. (2016) have suggested that H
2-rich fluid might not have been produced by the hydrothermal alteration of komatiite under CO
2-bearing conditions at temperatures ≤ 250 °C [
50]. This implies that hydrothermal systems on the deep seafloor (more than 1000 m below the sea surface) were favorable for the sustainability of primitive microbial communities as high hydrostatic pressure was necessary to create high-temperature fluids. In contrast, the steady-state H
2 concentrations in the komatiite–H
2O–NO
3– system at 250 °C were 2.3 mmol/kg for experiment N250 and could be as high as 26 mmol/kg if the initial nitrate concentration is lower than 10 mmol/kg (
Section 4.3). Such hydrogen concentrations are comparable to those in high-temperature hydrothermal fluids venting from a modern, peridotite-hosted system that harbors H
2-based chemosynthetic microbial communities, including methanogens (H
2 = 3–16 mmol/kg) (e.g., Kairei and Rainbow Fields) [
74,
75]. Thus, H
2-based, primitive microbial communities in the Hadean ocean may have developed in komatiite-hosted hydrothermal systems of lower fluid temperature (down to 250 °C) than previously thought.
Alkaline fluids were necessary for primordial metabolisms driven by chemiosmosis and electrochemistry at the vent–ocean interface (i.e., hydrothermal mounds), where hydrothermal fluids mixed with acidic and/or neutral, CO
2-rich Hadean seawater [
76,
77,
78]. For example, the selective synthesis of alanine from pyruvate and ammonia (>30% in yield) occurs in water with pH of 9–11 at 70 °C in the presence of iron oxyhydroxides [
76]. Certain steps in the reductive tricarboxylic acid cycle are promoted by FeS-Fe
0 assemblages that can be produced electrochemically in alkaline water with pH of 10 at 100 °C in the presence of 1 mmol/kg H
2 [
78]. This study shows that the pH of fluid in the komatiite–H
2O–NO
3− system becomes more alkaline than that in the komatiite–H
2O–CO
2 system at 250 °C (pH
25°C = 8.7 and 6.0, respectively) (
Table 3). Thus, hydrothermal fluids generated in the komatiite–H
2O–NO
3– system may have been more favorable to certain types of primordial metabolisms at the vent–ocean interface where the reaction zone temperature was around 250 °C. In contrast, when the temperature of the reaction zone rises over 350 °C, the hydrothermal fluids in the komatiite–H
2O–CO
2 system become more favorable for primordial metabolisms as the destabilization of carbonate minerals above 350 °C stimulates the alkalinization effect by ΣCO
2 in the aqueous fluid, producing an alkaline pH in the CO
2-bearing fluid [
48,
50,
79].
Ammonia is produced from nitrate at a high yield (10%) in the komatiite–H
2O–NO
3– system both at 250 °C and 350 °C (
Table 1). Ammonia is a key reactant for synthesizing amino and nucleic acids, and previous research successfully produced organic nitrogen compounds (amides, amino acids, and nitriles) by reacting ammonia with a variety of organic compounds (e.g., methane, formic acid, oxalic acid, and pyruvate) under limited hydrothermal conditions with and without minerals [
76,
80,
81,
82,
83]. As the fluid residence time from the onset of the high-temperature (>200 °C) reaction to venting at the seafloor (several years for present-day hydrothermal systems) [
84,
85] is much longer than the time required for complete reduction of nitrate at ≥250 °C (<300 h) (
Table 1), a stable supply of ammonia to hydrothermal environments could be expected for the komatiite–H
2O–NO
3− system in the Hadean ocean. Although the actual concentration of nitrate in the early oceans (thus, ammonia concentration in hydrothermal fluids) may have been lower than that used in our experiments, the stable supply of ammonia may have allowed abiotic synthesis of organic nitrogen compounds at least in localized environments that can concentrate ammonia such as pore spaces of clay mineral clusters in altered komatiite crusts, and interiors of iron oxyhydroxides in hydrothermal mounds (e.g.,
Figure 6g). However, further studies are necessary to explore pathways for the synthesis of organic nitrogen compounds that are resistant to the fluctuation of environmental conditions (pH, temperature and coexisting mineral type) that the hydrothermal vent environments probably encountered.
4.5. Implications for Biological Nitrogen Metabolism, Phytosynthetic Activity, and the Global Nitrogen Cycle in the Early Earth
The stable and high-yield production of ammonia in the komatiite–H
2O–NO
3− system is an important process not only for pre-biotic chemistry in the hydrothermal environments but also for controlling ammonia concentrations in the early global ocean that probably influenced biological nitrogen metabolism, the development of photosynthetic ecosystems, and global nitrogen cycle in the early Earth. Approximations of the ammonia production rate in the komatiite-hosted hydrothermal systems in the early ocean, F
NH3,ko (mol/y), suggest that biological activity had started in hydrothermally influenced local environments, but was not likely widespread throughout the global surface ocean. The F
NH3,ko (mol/y) value can be estimated from the following relation:
where Y
NH3,ko is the yield of ammonia by thermochemical nitrate reduction in the komatiite-hosted hydrothermal systems, F
H2O is the high-temperature (≥250 °C) fluid flux from the komatiite-hosted hydrothermal systems (kg/y), and C
NO3 is the nitrate concentration in seawater (mol/kg). Our experiments suggest that Y
NH3,ko value is 0.1, whereas numerical modeling by Laneuville et al. (2018) indicated that C
NO3 in early seawater was dependent upon atmospheric nitrate production rate and had a range from 10
−6 to 10
−2 mol/kg [
86], cf. [
87,
88]. The F
H2O value is initially approximated below, followed by the F
NH3 value.
In the modern ocean, hydrothermal fluid flux is considered proportional to the annual production rate of oceanic crust at fast-spreading ridges [
89]. Numerical modeling also suggests that plate velocity has not changed much throughout Earth’s history [
90,
91]. Thus, the high-temperature fluid flux from the komatiite-hosted hydrothermal systems can be approximated as follows:
where P
ko is the annual komatiite production rate (km
3/y) and k is a coefficient that relates high-temperature fluid flux to P
ko (kg/km
3). Field observations suggest that the thickness of the Archean oceanic crust was approximately three times greater than modern equivalents owing to the higher degree of partial melting [
41,
42]. Further, numerical modeling suggests that the global spreading rate of Archean oceanic crust (the product of plate velocity and plate boundary length: km
2/year) was approximately three times higher than that at present [
90]. Thus, the annual production rate of Archean oceanic crust was estimated to be 180 km
3/y, that is, nine times greater than at present [
92]. If the production ratio of oceanic plateau to oceanic crust at mid-oceanic ridges in the Archean was identical to that at present (0.05), the P
ko value was estimated to be 10 km
3/y in the Archean, which is nine times greater than that of the present oceanic plateau (1.1 km
3/year) [
93]. Given that high-temperature (>250 °C) fluid flux in the modern ocean is estimated to be 0.2–3 × 10
13 kg/y, e.g., [
94,
95,
96], k is calculated to be 10
11–10
12 kg/km
3. Collectively, the F
H2O,ko and F
NH3,ko values of the Archean ocean are estimated to be 10
12–10
13 kg/y and 10
5–10
10 mol/y, respectively (
Table 8). Given higher heat flux from Earth’s interior in the Hadean than Archean, the F
H2O,ko and F
NH3,ko values of the Hadean ocean are considered to be greater than the Archean ocean.
The F
NH3,ko value provide insights into the evolution of biological nitrogen metabolism. It has to be noted that our estimate of the F
NH3,ko value is based on simple equations and several assumptions, thus allowing the F
NH3,ko value to have a wide range from 10
5 to 10
10 mol/y. Nonetheless, our estimate indicates that the ammonia concentration in the early oceans had increased to 0.01–1000 μmol/kg from 4.4 Ga to 3.4 Ga if ammonia consumption rate in the ocean (e.g., biological assimilation) was much lower than the ammonia production rate. Microorganism can assimilate ammonia at the micro-to-milli molal levels for growth. For example, a half saturation constant for ammonia uptake has been reported to be 4–74 μmol/kg for bacteria isolated from different habitats [
97]. Thus, if nitrate concentration in the early oceans was greater than 10 μmol/kg, the long-term production of ammonia through thermochemical nitrate reduction for the first billion years might have allowed the subsequent development of an early biosphere in the global surface ocean. In contrast, if nitrate concentration in the early oceans was less than 1 μmol/kg, komatiite-hosted hydrothermal activity would not have contributed to the accumulation of ammonia in the global ocean. In this case, the onset of biological nitrogen fixation (N
2 → NH
3) was required for the development of early biosphere, consistent with genomic, physiologic, and geological records that are compatible with nitrogen fixation by the last universal common ancestor or methanogen in hydrothermal environments at 3.5 Ga [
4,
5,
98,
99], cf. [
100]. To determine which scenario is more appropriate for the evolutional history of nitrogen metabolism, future work is required to estimate nitrate concentration in early seawater from geological records. Nitrogen isotopic characterization of abiotically produced ammonia is also important to determine the source of nitrogen isotopic compositions in Archean sedimentary rocks of marine origin, which have been interpreted as evidence for biological nitrogen fixation in surface oceans at 3.2 Ga [
101] on the basis of nitrogen isotopic similarity to N
2 in the Archean atmosphere (ca. 0‰) [
10,
102,
103,
104].
Results of our experiments also suggest that 90% of nitrate nitrogen introduced to the komatiite-hosted hydrothermal system had probably been converted to N
2, resulting in a larger input of N
2 to the early ocean than that of ammonia. The higher input of N
2 may have stabilized the partial pressure of N
2 (pN
2) in the atmosphere and climate in an era of low solar luminosity as the increase in pN
2 would have increased the warming effect of existing greenhouse gases in an anoxic atmosphere (CO
2, CH
4) [
105]. The approximate global N
2 flux from thermochemical nitrate reduction in early Archean seafloor hydrothermal systems, including both komatiite- and basalt-hosted systems, is F
N2 (mol/y), which can be compared to the N
2 fluxes from other processes to develop a quantitative assessment of the contribution of thermochemical nitrate reduction to pN
2 in the early Archean.
The estimated F
N2 value in the early Archean includes the estimate of the N
2 production rate in the komatiite-hosted hydrothermal system in the early Archean, F
N2,ko (mol/y) using the following equation:
where Y
N2,ko is the yield of N
2 by thermochemical nitrate reduction in the komatiite-hosted hydrothermal systems. By substituting 0.45, 10
12–10
13 kg/y, and 10
−6–10
–2 mol/kg for Y
N2,ko, F
H2O,ko, and C
NO3 in Equation (28), respectively, F
N2,ko is calculated to be 10
6–10
11 mol/y. If the production ratio of oceanic plateau to oceanic crust at mid-oceanic ridges in the Archean was identical to that at present (0.05), high-temperature fluid flux from the basalt-hosted hydrothermal system at Archean mid-oceanic ridges is one order magnitude larger than F
H2O,ko. Accordingly, F
N2 is estimated to be 10
7–10
12 mol/y (i.e., 10
8–10
13 gN/y) in the early Archean, if the yield of N
2 by thermochemical nitrate reduction in the basalt-hosted system Y
N2,mo had been equal or greater than Y
N2,ko, which is an appropriate assumption due to lower potentials of H
2 and NH
3 generation in basalt-hosted system than komatiite-hosted system.
When comparing N
2 fluxes from major processes, the F
N2 value in the early Archean is quantitatively important. Major processes that supply N
2 to the present atmosphere are biological denitrification in the global ocean (NO
3− → N
2) (1–3 × 10
14 gN/y) [
106], degassing from the MORB source mantle (3 × 10
10 gN/y) [
107] and degassing by arc volcanism (3 × 10
11 gN/y) [
108]. In the early Archean, geological records suggest that biological denitrification was not yet underway, whereas degassing fluxes of N
2 from the MORB source mantle and via arc volcanism could have been one order of magnitude greater than today as they change proportionally with the annual production rate of oceanic crust. Therefore, N
2 flux by thermochemical nitrate reduction in the global hydrothermal systems would have been greater than that from the Earth’s interior if nitrate concentration in Archean seawater had been greater than 1000 μmol/kg.
In summary, the approximate N
2 fluxes to the atmosphere in the early Archean from hydrothermal systems by thermochemical nitrate reduction and from the Earth’s interior were 10
8–10
13 gN/y and 10
12 gN/y, respectively. Meanwhile, fluid inclusions in the Archean rock samples indicate that the abundance of N
2 in the Archean atmosphere was 2–12 × 10
21 gN, which is 0.5–3 times the present atmospheric N
2 [
10,
103,
104,
109]. Therefore, the residence time of atmospheric N
2 in the early Archean is calculated to be 10
8 y or greater. For comparison, the residence time of atmospheric N
2 at present is 10
7 years due to the high rates of biological nitrogen fixation and biological denitrification in the global ocean (1–3 × 10
14 gN/y) [
106]. Although pN
2 and its secular variation during Earth history have not yet been estimated precisely, our simple estimate implies that pN
2 is stable for ≥10
8 years in the early Archean if the global nitrogen cycle was driven primarily by abiotic nitrogen processes in the atmosphere and seafloor hydrothermal systems. Together with the emission of hydrogen and methane gases during serpentinization, this point should be considered when evaluating the role of hydrothermal activity in the long-term climate of the early Earth.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









