3.1. ICP-MS Results
The interpretation of the chemical results obtained by ICP-MS followed a statistical procedure following observations on compositional data by Aitchison (1982), Aitchison (2008), Buxeda i Garrigós (1999), and Buxeda i Garrigós and Kilikoglou (2003) [
19,
38,
39,
40]. R (Core Team 2019) [
41] was the software employed for all the transformations, statistical analyses and data visualization and the routines used are published in a reproducible manner elsewhere. The statistical procedure in this case consisted of using ratios of logarithms obtained by dividing all the components of the dataset by the component that introduced the lowest chemical variability to the entire set of ceramics (see Martín-Fernández et al., (2015) [
42] for a thorough discussion on the use of log-ratio principles). In the present study, chemical changes, enrichment and leaching were tested using principal component analysis. Moreover, the compositional heterogeneity was evaluated calculating the compositional variation matrix (MCV), which provides information about the variability introduced by each component into the dataset. It is noted that the analytical variance should originate from natural sources and not from experimental or analytical errors. Since the tungsten carbide cell used to mill the ceramics was a potential contaminator, Co and Ta were removed from the statistical procedure. The reason is that Co is a known binder of tungsten alloys and usually occurs along with Ta traces [
43]. Furthermore, Sn, Ni, Cu and Zn were also removed, the first because it showed values under the quantification limit and the rest because they showed low repeatability, which could not have been related to any specific alteration, suggesting that an analytical error could have occurred.
In
Figure 2 the compositional variation matrix of the 69 ceramics is shown. The y-axis (τ.
i) represents the calculated value for the log-ratio variation for each element in the dataset. Therefore, it provides an estimation of each element contribution to the total variation [
40].
Figure 2a shows that CaO was the component that introduced the highest variability to the 69 ceramics dataset, something that was expected because the clays were selected in the function of their CaO amount. Moreover,
Figure 2b,c present the MCV of 23 PT and PF samples (twenty test-pieces and three control-pieces), showing that the components that introduced the highest variability were Pb and Na
2O. Additionally,
Figure 2d presents the MCV of 23 AP samples, and in this case, Na
2O was the component that introduced the highest variability in addition to CaO.
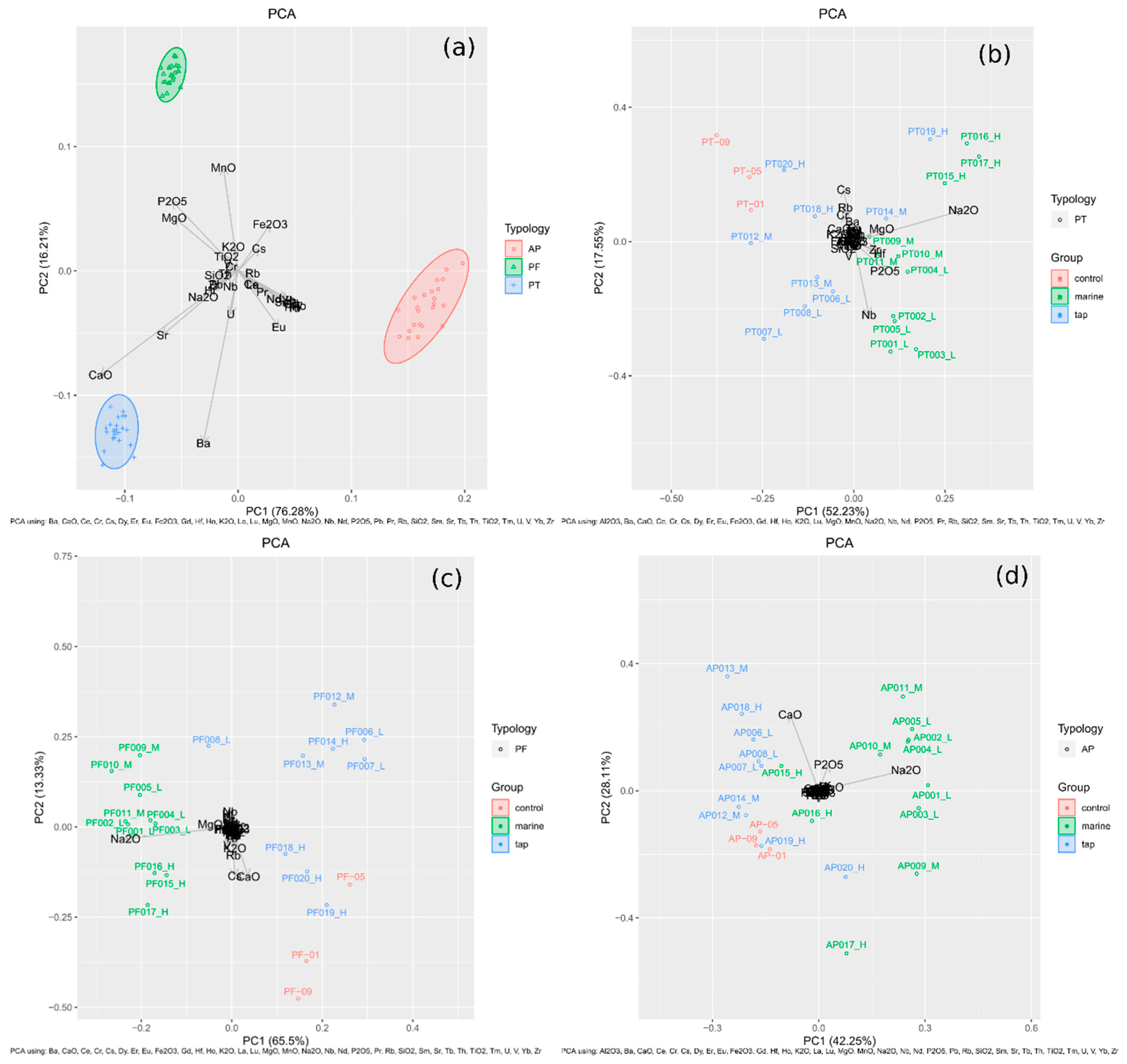
In addition, in
Figure 3, four PCAs (Principal Component Analysis) are shown, with samples labelled as L, M and H; L means that those samples were fired at low temperatures (850 °C), M at medium firing temperatures (950 °C) and H at high firing temperatures (1100 °C).
Figure 3a presents the PCA of 69 samples, in which a clear differentiation of the three type of pastes can be seen. On the one hand, the raw material of PT samples contained more CaO than the other samples (around 14%, as is presented in
Supplementary Materials (Table S1), so it was expected that PT samples would orient themselves that way. In addition, PT samples seemed to contain more Sr, Ba and Na
2O too. As can be seen in
Figure 2, Na
2O was the component adding more variation in the three pastes, but
Figure 3a shows that PT pastes were those that contained more Na
2O. Ceramics can be enriched in sodium because this element is present in both terrestrial and marine environments [
6], or due to the formation of analcime or precipitation of halite in the paste [
4,
10]. Barium and strontium can also be absorbed in the surfaces or barium can be precipitated as barite (BaSO
4) and strontium as carbonate, sulphate, borate, phosphate or halide [
6]. Moreover, PF samples contained more MgO, MnO and P
2O
5 than the other samples, while AP samples contained more Cs, Rb, Fe
2O
3 and trace elements, related to their micaceous paste. It is common to detect an enrichment of magnesium in ceramics buried in marine environments [
3,
6]. Manganese also may accumulate on ceramics during burial, and they have the tendency to precipitate as oxides [
6]. In addition, phosphorous is commonly identified, adsorbed in ceramics buried in both terrestrial and marine environments [
11,
12,
13,
35].
Figure 3b shows the PCA of PT samples, for which Pb has been omitted because there was no specific reason for its high variability. In this PCA, the three marine samples and one tap sample fired at high temperatures (1100 °C) were the samples that contained the most Na
2O. Additionally, samples fired at the lowest temperature were those that contained more Nb, suggesting that this type of less vitrified samples absorb more Nb. Moreover, control-pieces are nearer to tap water samples than to marine samples, suggesting that more changes occurred in the marine samples. Regarding the
Figure 3c,d, they show the PCA of PF and AP samples (Pb has been omitted in PF PCA). In both PCAs, the remarkable fact was that tap samples seemed to contain more CaO, and, in the case of PF, the samples were those fired at the highest temperature. A deeper discussion will be given when the XRD analyses and SEM–EDS analyses are explained.
3.3. XRD Results
First, raw clays were analyzed by XRD for comparison purposes (
Table 4).
Regarding PT samples, there were some mineralogical differences between the PT control-pieces, which were due to the firing alteration processes, as they were fired at different temperatures (
Table 5).
The first clear difference was the disappearance of illite (clay mineral) with the increase in firing temperature. This phenomenon occurs because the clay minerals lose the water they contain, so their lattice structure collapses and the clay minerals start to break down, forming new silicates [
9]. Although kaolins were not present in the ceramic, they probably were present in the raw material. The dihydroxylation of kaolinite occurs at 400–500 °C and metakaolinite is formed. Then, at around 900 °C, alumina-rich spinel (MgAl
2O
4) starts to form from this metakaolinite and at 1000–1100 °C it crystallizes into mullite (3Al
2O
3·2SiO
2). Mullite exists in needles or rod-like crystals, reinforcing and strengthening the fired piece [
9,
46,
47]. The presence of mullite is interesting in this context because, in general, when small amounts of carbonates are present (<10%) in high-calcareous clays, the mullite formation is inhibited and Ca-silicates are formed. However, depending on the concentration of Al
2O
3 in the samples, and if the amount of carbonates is higher than 10%, mullite can be formed. El Ouahabi and collaborators (2015) [
48] identified mullite in a high-calcareous ceramic, whose Al
2O
3 concentration was 14.6%. The PT ceramics had 14.2–15.8% Al
2O
3 content, which justifies the presence of mullite in these ceramics. The diffractogram of PT-09 control-piece, in which mullite was present, is shown in
Supplementary Materials (Figure S1).
Additionally, calcite was present in the sample fired at lower temperature, and it disappeared in the other samples, while gehlenite (Ca
2Al
2SiO
7) and diopside (Ca(Mg,Fe)Si
2O
6) appeared. The decrease in calcite is due to its decomposition at approximately 600–900 °C, forming lime (CaO) and carbon dioxide gas (CO
2), according to the following reaction [
9,
20,
23,
49].
Then, the CaO reacts, depending on the firing temperature, the duration of the firing, the granulometry of the carbonate fragments or their crystalline structure, with the clay minerals present in the body to form Ca-silicates and aluminosilicates such as gehlenite, anorthite (CaAl
2Si
2O
8) (which increased with the firing temperature in the control-pieces) and diopside [
23]. This is why diopside, plagioclase (e.g., anorthite, albite) and gehlenite appeared in the control-pieces fired at higher temperatures.
In addition, Heimann and Maggetti (1981) [
2] described the reactions for the alterations of gehlenite to form anorthite at temperatures close to and above 1000 °C.
These reaction paths can explain the increment of anorthite and the decrease in the amount of gehlenite in PT-09 (fired at 1100 °C). Additionally, the amount of hematite also increased with temperature. The reason for this could be that hematite may crystallize due to the decomposition of calcite in the presence of quartz and Fe-bearing clay minerals [
50]. The carbonates decompose to produce calcium silicates reacting with the amorphous phase. Thus, hematite can nucleate and grow.
When it comes to the XRD results of marine PT test-pieces (
Table 5), samples fired at 850 °C (PT001, PT002, PT003), had a greater amount of calcite and gehlenite in PT001 (3 months of burial). The decomposition reaction paths of calcite and gehlenite due to the post-depositional period could support the findings of their lower amounts in the pieces with longer deposition periods [
2,
20,
49]. The amounts of Qz, Pl, Kfs and Ilt were similar in all the samples, but the presence of gypsum (CaSO
4·2H
2O) in PT002 and PT003 marked the most important difference between these samples. The presence of gypsum is associated with the pyrite formation in reductive marine environments or to the dissolution of calcite and reaction with sulphate, in an acid environment [
4]; however, these were not the case because the marine environment of the experiment was oxidizing and both environments were basic. Therefore, the presence of gypsum might be associated with the raw material, although in very small amounts, given the lack of the successful identification of such a compound in their diffractograms, thus, the most probable source was the precipitation of sulfate from the seawater. Additionally, for samples fired at 950 °C (PT009, PT010, PT011), calcite appeared in PT009, and the amount of Qz, Pl, Gh, Hem, Kfs and Ilt were similar in all the samples. Calcite should not appear in PT009 because it was not present in the control-piece PT-05. However, the CaO that did not react could have recarbonated to CaCO
3 [
8,
20,
49,
51] and at a basic pH (higher than 7–8) it precipitated [
6]. First, it reacts with the water to give calcium hydroxide (Ca(OH)
2, portlandite) and, then, portlandite reacts with CO
2 leading to the formation of calcite, according to the next reaction path [
49].
Gehlenite appeared in a smaller amount than in the corresponding control-piece (PT-09), probably due to the alteration of gehlenite to form anorthite. In addition, mullite was not present in PT017, possibly making visible the fact that factors such as the firing of ceramics in the kiln were subjected to variations. It could be that the amount of carbonates in this particular sample was lower than in the rest of the samples due to the mentioned variations, so mullite was not formed [
48].
Finally, in samples fired at 1100 °C (PT015–017), analcime (NaAlSi
2O
6·H
2O) appeared in all of them. Buxeda and collaborators (2002b) recorded in calcareous ceramics fired at temperatures above 1000 °C, an increase in the concentration of Na and sometimes of Mg and Sr, and a decrease in the concentration of K and Rb, under certain burial conditions [
10,
16]. Several studies [
10,
19,
21] have documented the formation of analcime mineral, also known as analcite, as the reason for these facts. Buxeda and colleagues (2002b) explain from the observations of Picon (1976) that, depending on the firing temperature, a glassy phase is formed in the ceramics [
10,
24]. This glassy phase contains a large part of the overall K of the shard, which is leached with Rb from the ware during the burial because of the instability of the glassy phase under certain weathering conditions. Then, analcime crystallizes from the remaining silicate material by the fixation of Na from the burial environment [
2,
10,
15,
19,
22]. This fact can be seen in the ceramics immersed in marine water, with high concentration of Na, which explains why analcime is found in those ceramics but not in the similar ceramics immersed in tap water. Moreover, this fact is in accordance with the PCA shown in
Figure 3b, in which those marine samples fired at 1100 °C were the samples that contained the most Na
2O.
Regarding the XRD results of tap water PT test-pieces (
Table 5), no change was identified in the phases of the samples of different burial periods, probably because tap water is not as aggressive as marine water. Wollastonite was the new phase that appeared in PT pieces, specifically in PT019. This mineral is a high-temperature secondary phase, which crystallizes from Ca-rich clays [
52].
Regarding the PF test- and control-pieces,
Table 6 shows the mineralogical differences that occurred in the control-pieces due to the firing alteration processes.
These mineralogical changes were very similar to the changes that occurred in PT control-pieces. The differences were the appearance of mullite in PT samples but not in PF samples as well as the presence of cristobalite, spinel and larnite in PF samples (unlike in PT samples). The alteration paths for the calcite, gehlenite, hematite, plagioclase and diopside mineral phases are the same as those explained in the PT control-pieces section. Moreover, cristobalite, a polymorph of quartz, appears in PF-09 (1100 °C), due to the inversions that occur in quartz at high temperatures and, in addition, larnite, a calcium silicate (Ca
2SiO
4), and alumina-rich spinel (MgAl
2O
4), appeared due to high-temperature alterations of clay minerals [
9]. The diffractogram of PF-09 control-piece, in which larnite was present, is published in
Supplementary Materials (Figure S1).
When it comes to the XRD results of the marine PF test-pieces (
Table 6) fired at 850 °C (PF001–003), the amount of calcite and gehlenite was greater in PF001 (3 months of immersion) than it occurred in PT samples fired at 850 °C for the reasons explained above. Additionally, for samples fired at 950 °C (PF009–011), calcite appeared in PF009 due to the recarbonation of CaO, as in the PT009 sample, and the amounts of Qz, Pl, Gh, Hem, Di, Kfs and Ilt were similar in all the samples. Finally, for samples fired at 1100 °C, plagioclase and hematite were higher in PF017 (18 months of burial). The reason for the greater amount of plagioclase could be, as has been aforementioned, the alteration of gehlenite to form anorthite. Qz, Spl and Di were in similar amount to the rest of the samples and Ilt disappeared. Crs was not detected in these samples as it may have not developed during firing, and larnite appeared in PF015 and PF017 but not in PF016.
Results of the PF test-pieces immersed in tap water were almost the same as those of the PT test-pieces, thus, very few changes were identified in the phases of the samples of different burial periods (
Table 6).
Regarding the micaceous (AP) test- and control-pieces,
Table 7 shows that there were some mineralogical differences between the AP control-pieces due to the firing alteration processes. The first clear difference was the presence of mullite in high-fired ceramics, as in the PT pieces. In addition, the amount of hematite increased with temperature. This phenomenon also occurred with PT and PF samples. Similarly, corundum (Al
2O
3) appeared in AP-09, due to high firing temperatures.
When it comes to the XRD results of marine and tap water AP test-pieces, it is noted that there were no significant differences either in the phases (that is, all the samples had the same phases as their corresponding control-pieces) or between samples with different immersion periods. The existing differences between the samples were those that corresponded to the firing temperatures, shown in
Table 7.
3.4. SEM–EDS Results
Regarding the SEM–EDS analyses for the control-pieces of the three type of samples, first, the control-pieces fired at 850 °C (PT-01, PF-01 and AP-01) showed an early initial vitrification in accordance with their firing temperature, due to the appearance of isolated smooth-surfaced areas (
Figure 5a–c). Additionally, PT-05, PF-05 and AP-05 (fired at 950 °C) showed a medium vitrification (
Figure 5d–f) and those fired at 1100 °C (PT-09, PF-09 and AP-09) showed a continuous vitrification (
Figure 5g–i) due to the formation of a continuous smooth vitrified layer over the whole fractured surface [
44]. In PF, mineral phases of different sizes were identified. By EDS analyses it is known that these mineral phases were mainly composed of Si, O, Ca, Fe, Mg, Al, K and, sometimes, Ti, suggesting, according to XRD results, the presence of quartz, potassium feldspar, illite, calcite, hematite, gehlenite, plagioclase, cristobalite and diopside, among others. Furthermore, the EDS analyses of the PT control-pieces revealed the high presence of calcium and magnesium in the three control samples. These elements can appear in the form of carbonates, oxides and silicates, among others. Additionally, S and Cl were present in PT-05, demonstrating that those elements had their origin in the raw material. In this case, S may be associated with gypsum, supporting the presence of gypsum in the XRD analyses of PT002 and PT003 test-pieces. Finally, it was observed that AP pieces contained long and plane mineral phases, which formed a laminar structure within the ceramic paste, likely from the mica group. The pieces fired at 850 °C kept this laminar structure, while the samples fired at 1100 °C did not so much because they showed a continuous, smooth vitrified layer. EDS analyses showed that these mineral phases were mainly composed of Si, O, Fe, K, Al, Mg, Ca, and, sometimes, Ti, suggesting, in agreement with XRD results, the presence of quartz, potassium feldspar, illite-muscovite, and hematite, among others. It was also noted that Ca and Mg seemed to be related. Additionally, the EDS analyses of AP-05 showed, as in AP-01, mineral phases composed of Fe, Ca, K, Mg, C, Al, Ca, O, Si, Ti and, in some cases, S and Mn, in agreement with the presence of quartz, potassium feldspar, illite and mullite, according to XRD analyses. Although calcium carbonates and silicates were not identified by XRD, probably due to their lower amount, some aggregates were recognized by SEM–EDS, but in lower amounts than in PT and PF samples.
Additionally, the SEM and EDS analyses of test-pieces were very diverse and mineral forms such as needles, flakes, sponges and long and short prisms, were found, mostly in PT and PF samples. Regarding the SEM–EDS analyses of AP marine and tap water test-pieces, it was noted that, in comparison to PT and PF pieces, AP pieces showed much fewer aggregates inside their pastes. Contrary to what would be expected, in AP, more secondary phases were identified in samples fired at high temperatures than in those at low temperatures, even though the pastes fired at low temperatures were less porous. Perhaps the reason for this is that the laminar structure that the pastes contain makes it difficult for solutions from the environment to access, and therefore, may block the formation of the secondary phases. On the contrary, it is likely that the vitrification of AP samples helps to form canals through which solutions from the environment can access.
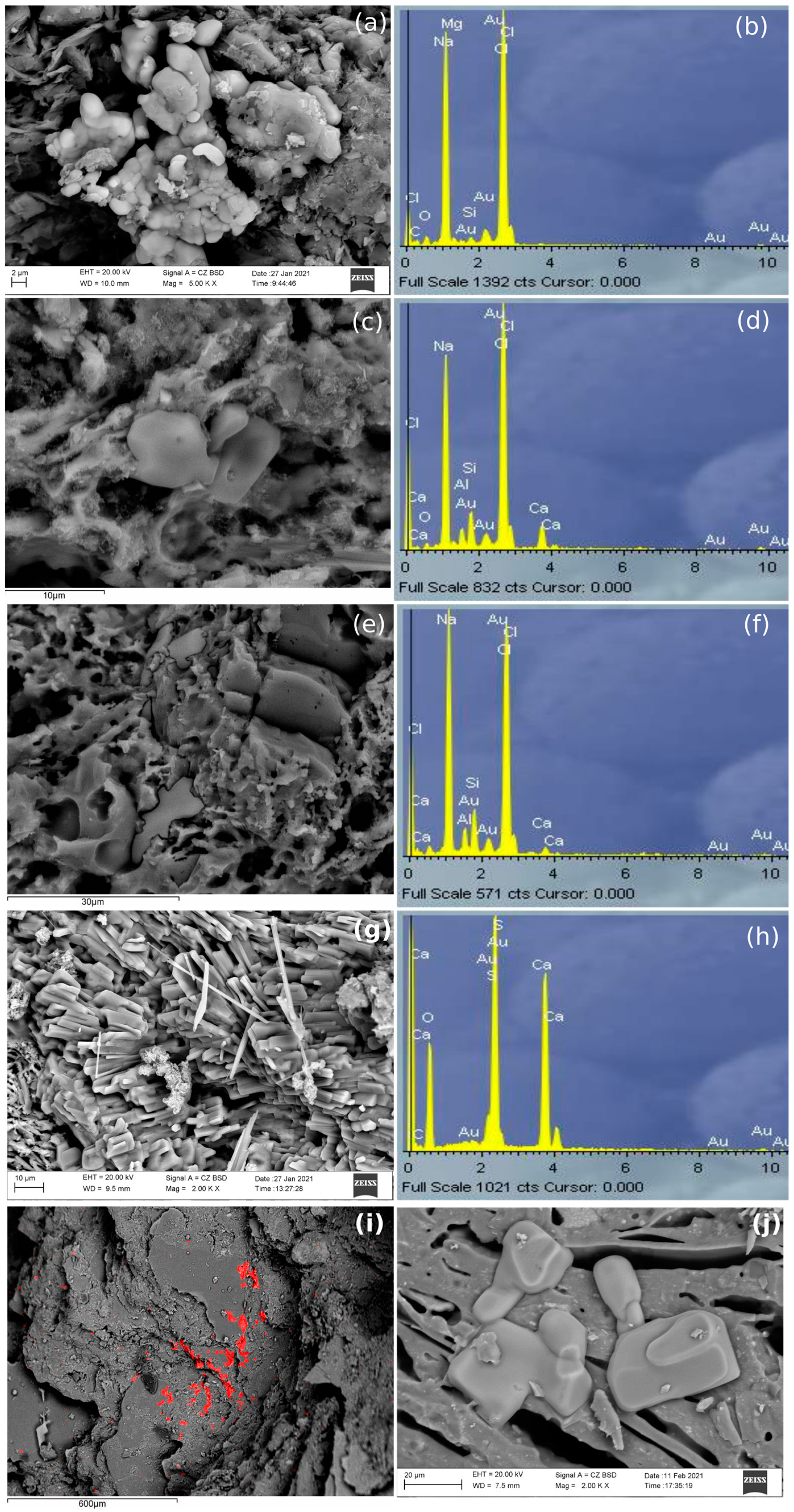
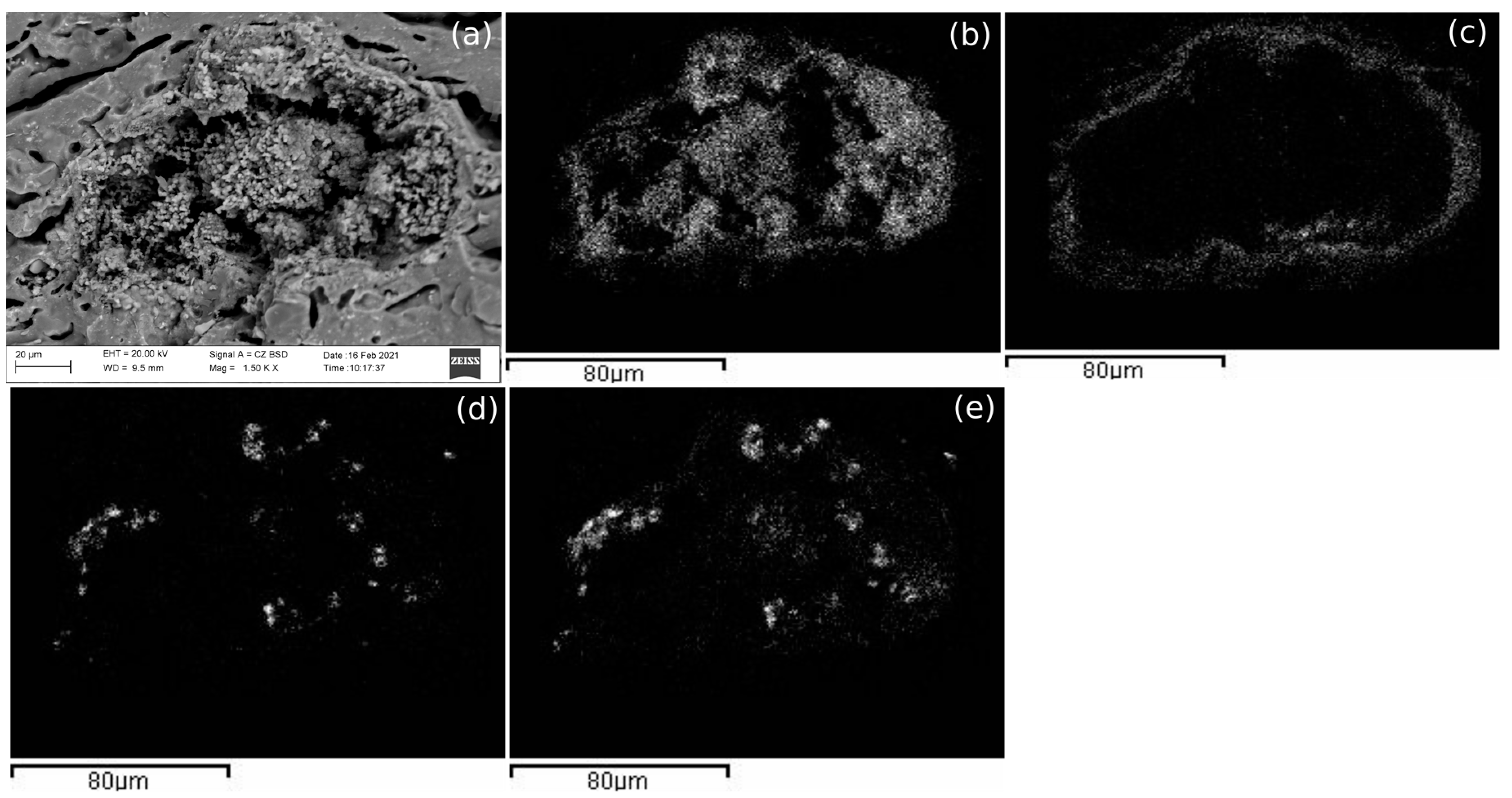
Marine samples of the three pastes showed some crystallizations containing Na, Cl and S (
Figure 6). These crystallizations could correspond to NaCl (halite) and calcium sulphate, both in most of the cases precipitated from seawater. However, there is a possibility that gypsum in some PT and AP samples was coming from the raw material because S was identified by EDS analyses in control-pieces. While in PT and PF samples, it seems that these aggregates were formed in cavities, in AP they seemed to rest on top of large mineral phases, suggesting that the movement of the water could have been of laminar flux instead of percolation. However, as an exception, marine AP samples fired at 1100 °C showed NaCl aggregates in the cavities formed in the vitrified pastes. However, AP samples submerged in tap water and fired at 850 °C and 950 °C did not show any newly formed aggregate, being similar to the AP control-pieces, since tap water does not contain enough salts to penetrate in the pastes.
Additionally,
Figure 7 shows one of the crystallizations identified in both PF and PT samples submerged in marine and tap water environments. These crystallizations mostly appeared in PF tap water samples, but were also present in marine and tap PT samples. They were similar to sponges and were composed mainly of Ca and F, appearing principally in cavities. In addition, Mn was identified in the extremities of this aggregate, as well as elements such as Ca, O, C, Al and Si (probably related to calcium silicates) around these cavities.
According to Thomas and collaborators (1977) [
53], fluorine is an element that can be present in raw clays. García-Ten and collaborators (2006) [
54] explain that, during the ceramics firing, the fluorine ion replaces OH
− groups present in the crystalline structure of mica and other clay minerals (such as illite or montmorillonite). This reaction leads to the formation of hydrofluoric acid (HF) and silicon tetrafluoride (SiF
4), among others. These compounds are released during firing between 500–700 °C. However, in high-calcareous ceramics, HF can react with calcite to form fluorite (CaF
2). Additionally, De Bonis and collaborators (2014) [
55] reported newly formed cuspidine [Ca
4Si
2O
7(F,OH)
2] at the edges of silicates (such as feldspars and quartz) in contact with fluorite-based carbonates, in ceramics fired from 850 °C to 1100 °C. The authors proposed the next reaction path for cuspidine formation.
Therefore, the sponge-like structures of these samples may be cuspidine. The presence of Mn in the extremities of the Ca-F aggregate could suggest that, in these cases, the newly formed mineral might be a mineral from the cuspidine group containing Mn, such as normandite [NaCa(Mn,Fe)(Ti,Nb,Zr)(Si2O7)OF]. These newly formed structures may have their origin in calcite. Calcite decomposed to CaO leaving cavities, so cuspidine or normandite may have formed in these cavities. Moreover, it has been noted that it seems that there were more cavities in the tap water samples than in seawater samples. The reason could be that there is more fluorine in tap water environment, so more calcite reacted with fluorine to form CaF2. Additionally, in some cases, these aggregates did not show the same structure; the reason could be their different grade of crystallization. Finally, although AP test-pieces contained F in their EDS analyses, the sponge-like structures were not present. In comparison with what we saw using qualitative SEM–EDS analyses for AP, PT and PF samples, PF and PT samples had much more calcium-containing minerals, so the changes in the pastes were notable. In the case of AP pieces, as they did not contain much calcium, most probably the fluorine of the raw material or of the water solution was unable to react with the calcite.
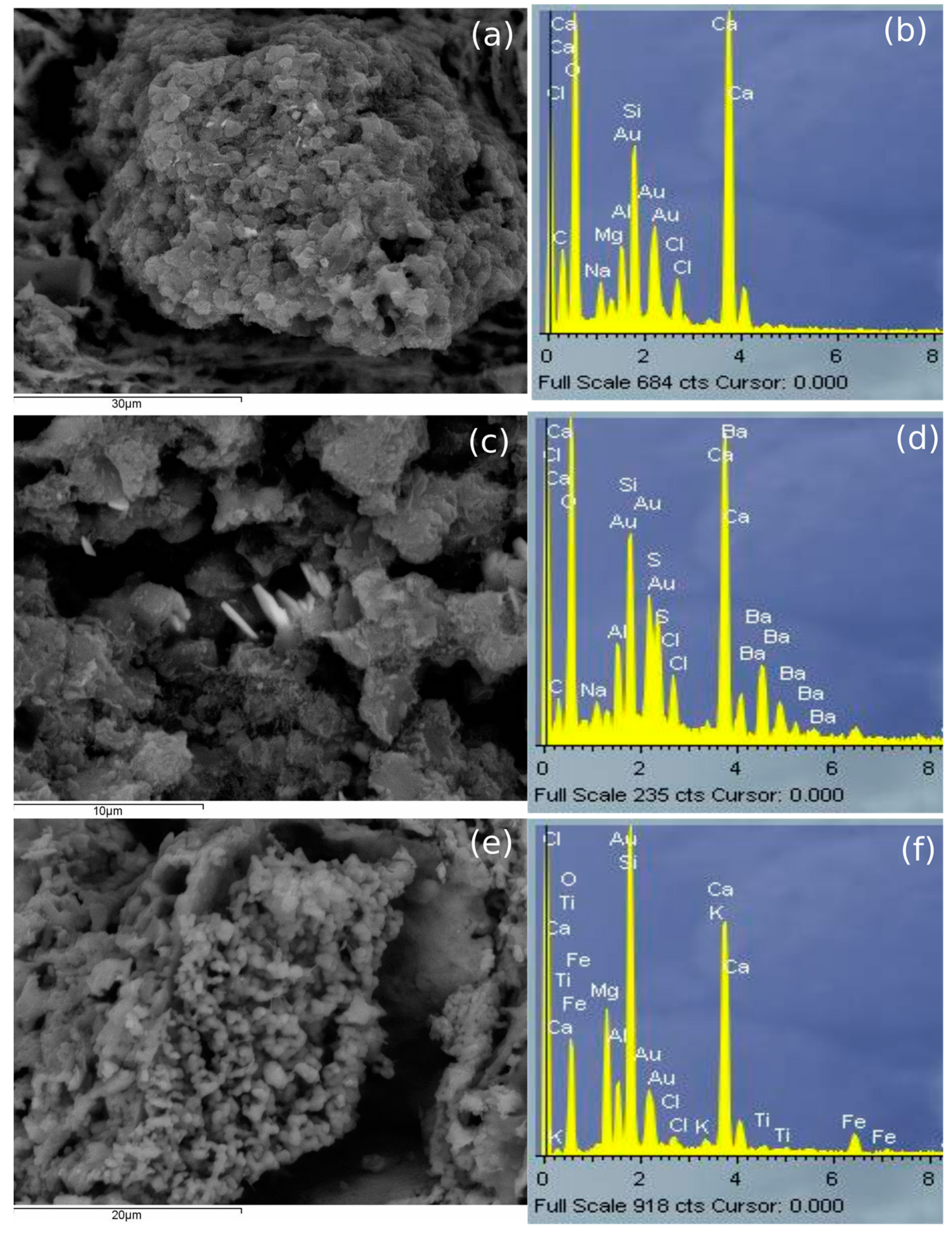
Moreover, aggregates of calcium carbonate with salts such as NaCl and MgCl
2 (present in the environment) and barium sulphate (probably barite, as commented in
Figure 3a), were also identified in PT010 sample (
Figure 8) as well as a structure that seemed to be formed by Mg and calcium hydroxide or carbonate, in PT016. In addition, calcium containing phases may have existed in AP samples but in such little amounts that was difficult to see them. The exceptions appeared in AP011 and AP014, in which calcium silicate and calcium related to Mg were identified, probably with their origin in calcite and phyllosilicates. Finally, AP samples submerged in the marine environment and fired at 1100 °C showed calcium carbonate or silicate aggregates.
Figure 9 shows an alteration in the paste of one of the AP samples fired at 1100 °C; Mg and O were homogeneously distributed and in the same place there were also some parts composed of NaCl (coming from seawater). This aggregate was surrounded by Ca (which suggests it had its origin in dolomite or in calcite). AP samples submerged in tap water and fired at 1100 °C also showed the same type of aggregates of calcium carbonate or silicates.
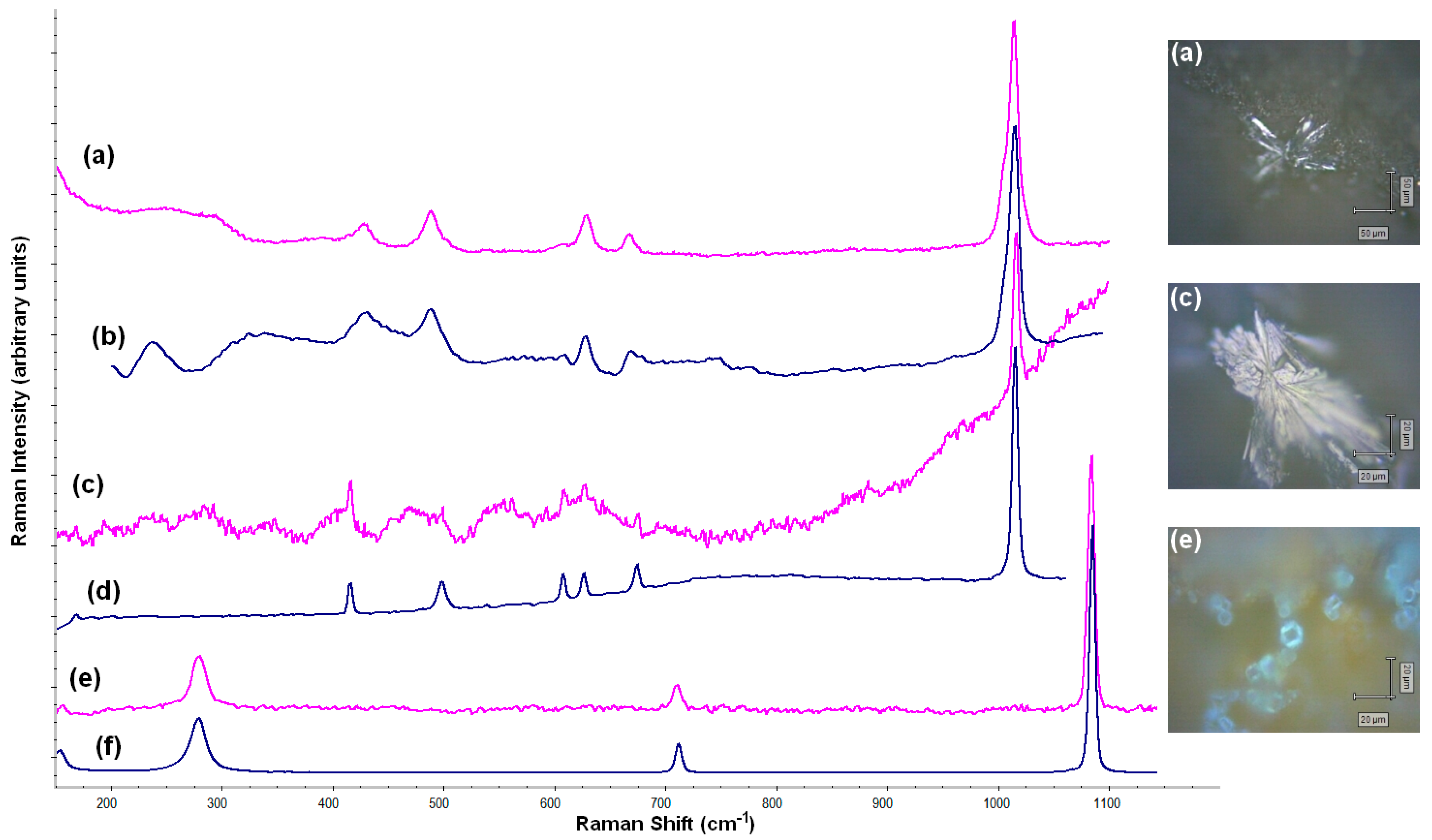
Apart from the paste analyses, the surfaces of PF010 and PF014 samples were analyzed by Raman spectroscopy (
Figure 10). The results demonstrated that NaCl and different forms of calcium sulphate were salts that precipitated from the seawater and they rested on the surface of the ceramic. It has been concluded that the aciculate crystals of different forms were calcium sulphate; the former (a) corresponded to anhydrite (CaSO
4), while (c) corresponded to calcium sulphate hemihydrate (bassanite) [CaSO
4·
(H
2O)]. Probably, there was a mixture of different phases of CaSO
4-H
2O system, as Prieto-Taboada and collaborators (2014) explained [
56]. Additionally, calcite was identified in the paste of PF014 by Raman (e), supporting the hypothesis of the recarbonation of calcite samples fired at 950 °C and 1100 °C.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}