

Laser-Based Characterisation of the Copper Uranyl Sulphate, Johannite

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample
2.2. Scanning Electron Microscopy with Energy Dispersive X-ray Spectroscopy (SEM-EDX)
2.3. X-ray Diffraction (XRD)
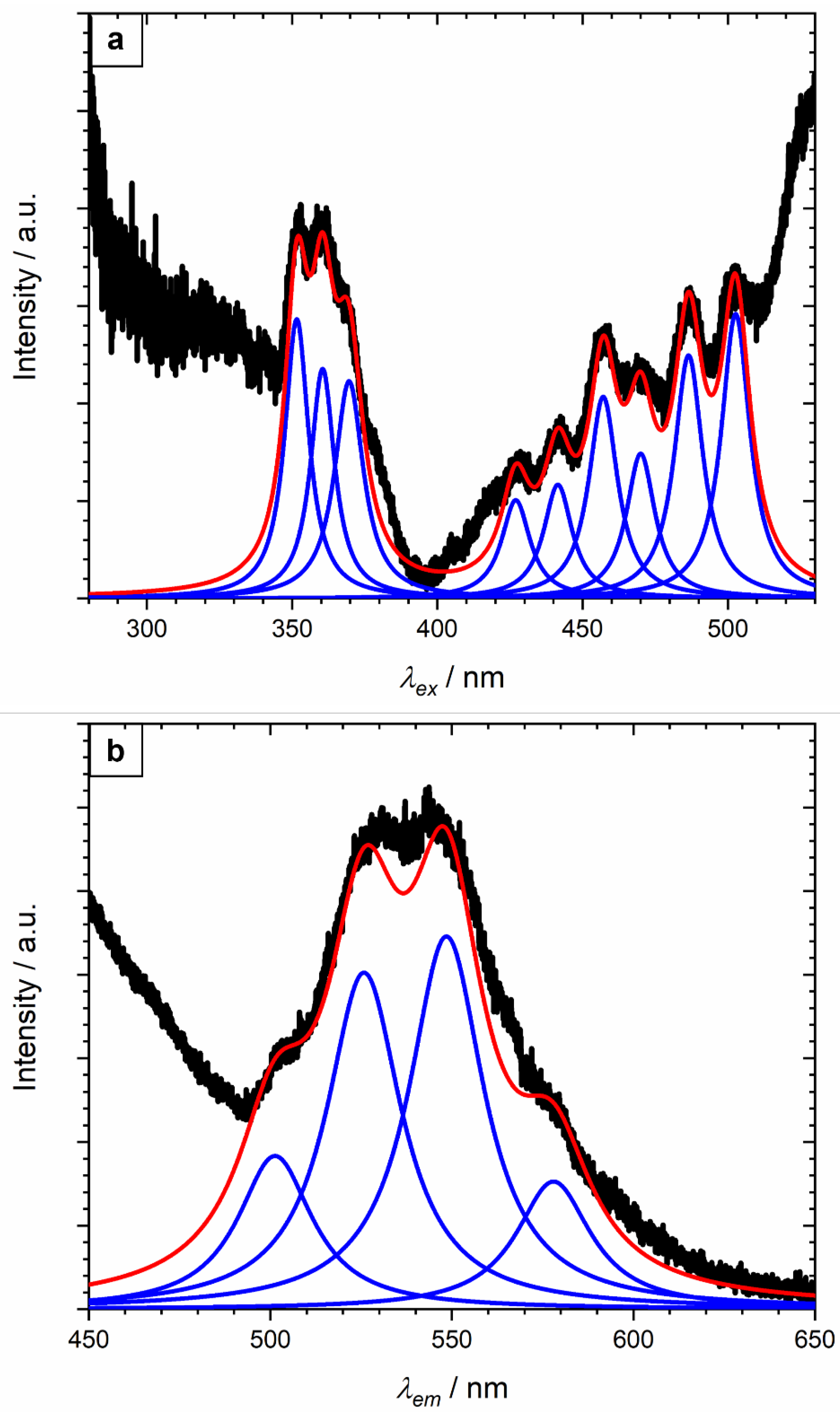
2.4. Luminescence Spectroscopy
2.5. Raman Spectroscopy
3. Results and Discussion
3.1. Composition and Phase Purity
3.2. Luminescence Spectroscopy
3.3. Raman Spectroscopy
3.3.1. Multiple-Laser Raman Spectra and Non-Raman Feature Assignment

3.3.2. Resolved 785 nm Raman Spectrum
3.3.3. Summary of Spectral Findings
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gurzhiy, V.V.; Plášil, J. Structural complexity of natural uranyl sulfates. Acta Crystallogr. B 2019, 75, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finch, R.J.; Murakami, T. Systematics and paragenesis of uranium minerals. In Uranium: Mineralogy, Geochemistry and the Environment; Reviews in Mineralogy and Geochemistry; Burns, P.C., Finch, R.J., Eds.; Mineralogical Society of America: Chantilly, VA, USA, 1999; Volume 38, pp. 91–180. [Google Scholar]
- Brugger, J.; Wallwork, K.S.; Meisser, N.; Pring, A.; Onduš, P.; Čejka, J. Pseudojohannite from Jáchymov, Musonoï and La Creusaz: A new member of the zippeite group. Am. Miner. 2006, 91, 929–936. [Google Scholar] [CrossRef]
- Frost, R.J.; Erickson, K.L.; Čejka, J.; Reddy, B.J. A Raman spectroscopic study of the uranyl sulphate mineral johannite. Spectrochim. Acta A 2005, 61, 2702–2707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugger, J.; Burns, P.C.; Meisser, N. Contribution to the mineralogy of acid drainage of uranium minerals: Marecottite and the zippeite-group. Am. Miner. 2003, 88, 676–685. [Google Scholar] [CrossRef]
- Johnson, D.B.; Hallberg, K.B. Acid mine drainage remediation options: A review. Sci. Total Environ. 2005, 338, 3–14. [Google Scholar] [CrossRef]
- Arnold, T.; Baumann, N.; Krawczyk-Bärsch, E.; Brockmann, S.; Zimmermann, U.; Jenk, U.; Weiβ, S. Identification of the uranium speciation in an underground acid mine drainage environment. Geochim. Cosmochim. Acta 2011, 75, 2200–2212. [Google Scholar] [CrossRef]
- Plášil, J.; Sejkora, J.; Škoda, R.; Škácha, P. The recent weathering of uraninite from the Červená vein, Jáchymov (Czech Republic): A fingerprint of the primary mineralization geochemistry onto the alteration association. J. Geosci. 2014, 59, 223–253. [Google Scholar] [CrossRef] [Green Version]
- Plášil, J.; Dušek, M.; Čejka, J.; Sejkora, J. The crystal stricture of rabejacite, the Ca2+-dominant member of the zippeite group. Miner. Mag. 2014, 78, 1249–1264. [Google Scholar] [CrossRef]
- Plášil, J.; Fejfarová, K.; Wallwork, K.S.; Dušek, M.; Škoda, R.Š.; Sejkora, J.; Čejka, J.Č.; Veselovský, F.; Hloušek, J.; Meisser, N.; et al. Crystal structure of pseudojohannite, with a revised formula, Cu3(OH)2[(UO2)4O4(SO4)2](H2O)12. Am. Miner. 2012, 97, 1796–1803. [Google Scholar] [CrossRef]
- Plášil, J.; Kampf, A.R.; Kasatkin, A.V.; Škoda, R.Š.; Silva, S.; Čejka, J. Meisserite, Na5(UO2)(SO4)3(SO3OH)(H2O), a new uranyl sulphate mineral from the Blue Lizard mine, San Juan County, Utah, USA. Miner. Mag. 2013, 77, 2975–2988. [Google Scholar] [CrossRef]
- Plášil, J.; Fejfarová, K.; Škoda, R.Š.; Dušek, M.; Marty, J.; Čejka, J. The crystal structure of magnesiozippeite, Mg[(UO2)2O2(SO4)](H2O)3.5, from East Saddle mine, San Juan County, Utah (U.S.A.). Miner. Petrol. 2013, 107, 211–219. [Google Scholar] [CrossRef]
- Plášil, J.; Kampf, A.R.; Kasatkin, A.V.; Marty, J. Bluelizardite, Na7(UO2)(SO4)4Cl(H2O)2, a new uranyl sulphate mineral from the Blue Lizard mine, San Juan County, Utah, USA. J. Geosci. 2014, 59, 145–158. [Google Scholar] [CrossRef]
- Kampf, A.R.; Plášil, J.; Kasatkin, A.V.; Marty, J. Belakovskiite, Na7(UO2)(SO4)4(SO3OH)(H2O)3, a new uranyl sulphate mineral from the Blue Lizard mine, San Juan County, Utah, USA. Miner. Mag. 2014, 78, 639–649. [Google Scholar] [CrossRef]
- Kampf, A.R.; Plášil, J.; Kasatkin, A.V.; Marty, J. Bobcookite, NaAl(UO2)2(SO4)4·18H2O, and wetherillite, Na2Mg(UO2)2(SO4)4·18H2O, two new uranyl sulphate minerals from the Blue Lizard mine, San Juan County, Utah, USA. Miner. Mag. 2015, 79, 695–714. [Google Scholar] [CrossRef]
- Kampf, A.R.; Kasatkin, A.V.; Čejka, J.; Marty, J. Plášilite, Na(UO2)(SO4)(OH)·2H2O, a new uranyl sulphate minerals from the Blue Lizard mine, San Juan County, Utah, USA. J. Geosci. 2015, 60, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kampf, A.R.; Plášil, J.; Olds, T.; Nash, B.; Marty, J. Ammoniozippeite, a new uranyl sulphate mineral from the Blue Lizard mine, San Juan County, and the Burro mine, San Miguel County, Colorado, USA. Can. Miner. 2018, 56, 235–245. [Google Scholar] [CrossRef]
- Rosborg, B.; Werme, L. The Swedish nuclear waste program and the long-term corrosion behaviour of copper. J. Nucl. Mater. 2008, 379, 142–153. [Google Scholar] [CrossRef]
- Olds, T.; Plášil, J.; Kampf, A.; Burns, P.; Nash, B.; Marty, J.; Carlson, S. Redcanyonite, (NH4)2Mn[(UO2)4O4(SO4)2](H2O)4, a new zippeite-group mineral from the Blue Lizard mine, San Juan County, Utah, USA. Miner. Mag. 2018, 82, 1261–1275. [Google Scholar] [CrossRef]
- King, F.; Lilja, C.; Pederson, K.; Pitkänen, P.; Vähänen, M. An update of the state-of-the-art report on the corrosion of copper under expected conditions in a deep geologic repository. In Technical Report SKB-TR-10-67; Swedish Nuclear Fuel and Waste Management Company: Stockholm, Sweden, 2010. [Google Scholar]
- Hall, D.S.; Keech, P.G. An overview of the Canadian corrosion program for the long-term management of nuclear waste. Corros. Engin. Sci. Technol. 2017, 52, 2–5. [Google Scholar] [CrossRef] [Green Version]
- Marcos, N. Native copper as a natural analogue for copper canisters. In Nuclear Waste Commission of Finnish Power Companies; Technical Report YJT-89-18; Nuclear Waste Commission of Finnish Power Companies: Helsinki, Finland, 1989. [Google Scholar]
- King, F.; Lilja, C.; Vähänen, M. Progress in the understanding of the long-term corrosion behaviour of copper canisters. J. Nucl. Mater. 2013, 438, 228–237. [Google Scholar] [CrossRef]
- Mereiter, K. The crystal structure of johannite, Cu(UO2)2(OH)2(SO4)2·8H2O [Die kristallstruktur des johannits, Cu(UO2)2(OH)2(SO4)2·8H2O]. Tschermaks Mineral. Und Petrograposche Mitt. 1982, 30, 47–57. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystal. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Harmon, R.S.; Senesi, G.S. Laser-induced breakdown spectroscopy—A geochemical tool for the 21st century. Appl. Geochem. 2021, 118, 100766. [Google Scholar] [CrossRef]
- Kato, Y.; Meinrath, G.; Kimera, T.; Yoshida, Z. A study of U(VI) hydrolysis and carbonate complexation by time-resolved laser-induced fluorescence spectroscopy (TRLFS). Radiochim. Acta 1994, 64, 107–111. [Google Scholar] [CrossRef]
- Izake, E.L. Forensic and homeland security applications of modern portable Raman spectroscopy. Forensic. Sci. Int. 2010, 202, 1–8. [Google Scholar] [CrossRef]
- Driscoll, R.J.P.; Wolverson, D.; Mitchels, J.M.; Skelton, J.M.; Parker, S.C.; Molinari, M.; Khan, I.; Geeson, D.; Allen, G.C. A Raman spectroscopic study of uranyl minerals from Cornwall, UK. RSC Adv. 2014, 4, 59137–59149. [Google Scholar] [CrossRef] [Green Version]
- Frankland, V.L.; Milodowski, A.E.; Read, D. Characterisation of meta-autunite: Towards identifying potential alteration products of spent nuclear fuel. Appl. Geochem. 2020, 123, 104792. [Google Scholar] [CrossRef]
- Lee, W.H.; Crean, C.; Varcoe, J.R.; Bance-Soualhi, R. A Raman spectromicroscopic investigation of ETFE-based radiation-grafted anion-exchanged membranes. RSC Adv. 2017, 7, 47726–47737. [Google Scholar] [CrossRef] [Green Version]
- Larsen, E.S.; Berman, H. The identity of gilpinite and johannite. Am. Miner. 1926, 11, 1–5. [Google Scholar]
- Hurlbut, C.S. Studies of uranium minerals (IV): Johannite. Am. Miner. 1950, 35, 531–535. [Google Scholar]
- Sokol, F.; Čejka, J. A thermal and mass spectroscopic study of synthetic johannite [Cu(UO2)2(SO4)2(OH)2·8H2O]. Thermochim. Acta 1992, 206, 235–242. [Google Scholar] [CrossRef]
- Ondruš, P.; Veselovský, F.; Hloušek, J.; Skála, R.; Vavřín, I.; Frýda, J.; Čejka, J.; Gabašová, A. Secondary minerals of the Jáchymov (Joachimsthal) ore district. J. Geosci. 1997, 42, 3–76. [Google Scholar]
- Anthony, J.W.; Bideaux, R.A.; Bladh, K.W.; Nichols, M.C. Handbook of Mineralogy; Mineralogical Society of America: Chantilly, VA, USA, 2001. [Google Scholar]
- Bell, J.T.; Biggers, R.E. Absorption spectrum of the uranyl ion in perchlorate media: III. Resolution of the ultraviolet band structure; some conclusions concerning the excited state of UO22+. J. Mol. Struct. 1968, 25, 312–329. [Google Scholar] [CrossRef]
- Wang, Z.; Zachara, J.M.; Yantassee, W.; Gassman, P.L.; Liu, C.; Joly, A.G. Cryogenic laser induced fluorescence characterisation of U(VI) in Hanford vadose zone pore waters. Environ. Sci. Technol. 2004, 38, 5591–5597. [Google Scholar] [CrossRef] [Green Version]
- Tits, J.; Walther, C.; Stumpf, T.; Mace, N.; Wieland, E. A luminescence line-narrowing spectroscopic study of the uranium(VI) interaction with cementitious materials and titanium dioxide. Dalton Trans. 2015, 44, 966–976. [Google Scholar] [CrossRef] [Green Version]
- Philipp, T.; Azzam, S.S.A.; Rossberg, A.; Huittinen, N.; Schmeide, K.; Stumpf, T. U(VI) sorption on Ca-bentonite at (hyper)alkaline conditions—Spectroscopic investigations of retention mechanisms. Sci. Total Environ. 2019, 676, 469–481. [Google Scholar] [CrossRef] [Green Version]
- Demnitz, M.; Hilpmann, S.; Lösch, H.; Bok, F.; Steudtner, R.; Patzschke, M.; Stumpf, T.; Huittinen, N. Temperature-dependent luminescence spectroscopic investigations of uranyl(VI) complexation with the halides F− and Cl−. Dalton Trans. 2020, 49, 7109–7122. [Google Scholar] [CrossRef]
- Frankland, V.L.; Milodowski, A.E.; Bright, J.W.G.; Read, D. The use of Raman and TRLF spectroscopy for differentiating early stage alteration products of spent nuclear fuel. Appl. Geochem. 2021, 130, 104934. [Google Scholar] [CrossRef]
- Frankland, V.L.; Rickman, S.P.; Milodowski, A.E.; Read, D. Characterisation of uranophane and boltwoodite by Raman, luminescence and laser-induced breakdown spectroscopy. Appl. Geochem. 2022, 138, 105183. [Google Scholar] [CrossRef]
- Frankland, V.L.; Milodowski, A.E.; Lawrence, R.A.; Sacchi, M.; Read, D. Characterisation of the potential spent nuclear fuel alteration product vandenbrandeite. Am. Miner. 2022, in press. [Google Scholar] [CrossRef]
- Faulques, E.; Kalashnyk, N.; Massuyeau, F.; Perry, D.L. Spectroscopic markers for uranium(VI) phosphates: A vibronic study. RSC Adv. 2015, 5, 71219–71227. [Google Scholar] [CrossRef]
- Massuyeau, F.; Perry, D.L.; Kalashynyk, N.; Faulques, E. Spectroscopic markers for uranium(VI) phosphates. Part II: The use of time-resolved photoluminescence. RSC Adv. 2017, 7, 919–926. [Google Scholar] [CrossRef] [Green Version]
- Therien, M.J. Physical chemistry: How to improve your image. Nature 2009, 458, 716–717. [Google Scholar] [CrossRef] [PubMed]
- Warrier, S.B.; Kharkar, P.S. A coumarin based chemosensor for selective determination of Cu(II) ions based on fluorescence quenching. J. Lumines 2018, 199, 407–415. [Google Scholar] [CrossRef]
- John, J.; Filipská, H.; Černichivá, K.; Beneš, P.; Semelová, M.; Bočan, J.; Geipel, G.; Kubeček, V. New TRLFS laboratory at the CTU in Prague. Czech. J. Phys. 2006, 56, D565–D568. [Google Scholar] [CrossRef]
- Vercouter, T.; Vitorge, P.; Amekraz, B.; Moulin, C. Stoichiometries and thermodynamics stabilities for aqueous sulphate complexes of U(VI). Inorg. Chem. 2008, 47, 2180–2189. [Google Scholar] [CrossRef]
- Lin, D.H.M.; Manara, D.; Lindqvist-Reis, P.; Fanghänel, T.; Mayer, K. The use of different dispersive Raman spectrometers for the analysis of uranium compounds. Vib. Spectrosc. 2014, 73, 102–110. [Google Scholar] [CrossRef]
- Ho, D.M.L.; Jones, A.E.; Goulermas, J.Y.; Tuner, P.; Varga, Z.; Fongaro, L.; Fanghänel, T.; Mayer, K. Raman spectroscopy of uranium compounds and the use of multivariate analysis for visualization and classification. Forensic. Sci. Int. 2015, 251, 61–68. [Google Scholar] [CrossRef]
- Frankland, V.L.; Bance-Soualhi, R.; Read, D. Raman analysis of meta-autunite. In Environmental Radiochemical Analysis; Evans, N., Ed.; RSC: Croydon, UK, 2019; Volume VI, pp. 79–88. [Google Scholar]
- Taga, M.; Kono, T.; Yamashita, N. Photoluminescence of the properties of gypsum. J. Miner. Petrol. Sci. 2011, 106, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Frost, R.L.; Plášil, J.; Čejka, J.; Sejkora, J.; Keefe, E.C.; Bahfenne, S. Raman spectroscopic study of the uranyl mineral pseudojohannite Cu6.5[(UO2)4O4(SO4)2]2(OH)5·2.5H2O. J. Raman Spectrosc. 2009, 40, 1816–1821. [Google Scholar] [CrossRef] [Green Version]
- Frost, R.L.; Čejka, J.; Ayoko, G.A.; Weier, M.L. Raman spectroscopic and SEM analysis of sodium-zippeite. J. Raman Spectrosc. 2007, 38, 1311–1319. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, J.R.; Cooney, R.P. On the determination of uranium oxygen bond lengths in dioxouranium(VI) compounds by Raman spectroscopy. J. Mol. Struct. 1989, 193, 295–300. [Google Scholar] [CrossRef]
- Badger, R.M. Between the internuclear distances and force constants of molecules and its application to polyatomic molecules. J. Chem. Phys. 1935, 3, 710–714. [Google Scholar] [CrossRef]
- Jones, L.H. Determination of U-O bond distance in uranyl complexes from their infrared spectra. Spectrochim. Acta 1959, 15, 409–411. [Google Scholar] [CrossRef]
- Frankland, V.L.; Milodowski, A.E.; Read, D. Characterisation of andersonite by Raman, luminescence and laser-induced breakdown spectroscopy. Appl. Geochem. 2022, 142, 105353. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λex/nm | λem/nm | Non-Raman Features from the 457 nm Spectrum/nm |
|---|---|---|
| 351.6 (vw) | - | 495.9 (m) |
| 360.5 (vw) | 501.3 (vw) | 502.1 (m) |
| 369.6 (vw) | - | 517.0 (s) |
| 427.0 (vw) | 525.8 (vw) | 523.9 (m) |
| 441.5 (vw) | - | 540.0 (m) |
| 457.1 (vw) | 548.5 (vw) | 547.8 (w) |
| 470.0 (vw) | 578.0 (vw) | - |
| 486.5 (vw) | ||
| 502.7 (vw) |
| Assignment | ν (This Study, NMW 67.154GR.69)/cm−1 | ν (Johannite [4])/cm−1 | ν (Johannite [29])/cm−1 |
|---|---|---|---|
| ν3(SO4)2− | 1224 (m,b) | - | - |
| 1184 (m,b,sh) | - | - | |
| 1147 (w,b,sh) | 1147 | - | |
| 1100 (w,sh) | 1100 | - | |
| 1090 (w) | 1090 | 1095 | |
| ν1(SO4)2− | 1043 (w) | 1042 | 1045 |
| ν3(UO2)2+ | - | 948 | - |
| ν1(UO2)2+ | 834 (s) | - | 836 |
| - | 812 | - | |
| - | 788 | - | |
| - | 756 | - | |
| ν4(SO4)2− | 625 (vw) | - | - |
| - | 539 | - | |
| - | - | 488 | |
| ν2(SO4)2− | - | 481 | - |
| 447 (w,b) | - | - | |
| - | 384 | - | |
| 351 (w,b) | - | 352 | |
| ν2(UO2)2+ | - | 277 | - |
| 244 (vw) | - | 244 | |
| 202 (vw) | 205 | 203 | |
| Lattice vibrations | 128 (vw) | - | - |
| 84 (vw) | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frankland, V.L.; Milodowski, A.E.; Read, D. Laser-Based Characterisation of the Copper Uranyl Sulphate, Johannite. Minerals 2022, 12, 1419. https://doi.org/10.3390/min12111419
Frankland VL, Milodowski AE, Read D. Laser-Based Characterisation of the Copper Uranyl Sulphate, Johannite. Minerals. 2022; 12(11):1419. https://doi.org/10.3390/min12111419
Chicago/Turabian StyleFrankland, Victoria L., Antoni E. Milodowski, and David Read. 2022. "Laser-Based Characterisation of the Copper Uranyl Sulphate, Johannite" Minerals 12, no. 11: 1419. https://doi.org/10.3390/min12111419
APA StyleFrankland, V. L., Milodowski, A. E., & Read, D. (2022). Laser-Based Characterisation of the Copper Uranyl Sulphate, Johannite. Minerals, 12(11), 1419. https://doi.org/10.3390/min12111419