1. Introduction
5-aminosalicylic acid (5-ASA) is an anti-inflammatory drug widely used in the treatment of different diseases, such as Crohn’s diseases, chronic bowel ulcerative colitis, and proctitis [
1,
2,
3,
4]. The consumption of 5-ASA is a growing market, worth an estimated US
$1.5 billion in the USA only [
5]. For the treatment of the Crohn’s disease and chronic bowel ulcerative colitis disease, 5-ASA is administered orally [
6], while for the treatment of proctitis it is administered rectally [
7,
8]. When the drug is administered orally it is rapidly absorbed in the stomach and in the small intestine. However, for the treatment of these diseases, the drug adsorption at the level of the large intestine and the colon is very important [
9].
The consumption of high doses of pharmaceutical drugs can produce side effects and resistance problems. The design and development of new modified drug delivery systems is attracting more research attention, with the aim of finding improved therapeutic strategies to reduce the frequency of drug administration and increasing the efficiency of the bioactive drugs [
10]. Different nanoparticulated materials have been proposed for modifying drug delivery. The use of clay minerals as carriers for these systems appears as a low-cost and biocompatible alternative [
11,
12,
13,
14]. Several studies on the interaction between drugs and different types of clays used as nanocarriers have been performed [
15,
16]. Halloysite nanotubes have been proposed as a natural vehicle for the dosage and modified release of several drugs. Halloysite nanotubes have been recently studied as nanocarriers for the controlled release of drugs [
17,
18]. Specifically, several experimental studies have been carried out to study the interaction between 5-ASA and halloysite nanotubes [
19,
20,
21,
22].
Halloysite, Al
2Si
2O
5(OH)
4·nH
2O, is a multilayer nanotubular clay mineral resulting from the wrapping of 1:1 layers of kaolinite with dimensions of 500–1000 nm in length and 15–100 nm in inner diameter [
23,
24,
25]. Halloysite nanotubes are common excipients in pharmaceutical products and can modify drug bioavailability. They can retain organic molecules and, after administration, release the retained bioactive compounds under controlled conditions [
10,
26,
27,
28]. Therefore, halloysite nanotubes can be used as excipients to achieve colon-targeted drug delivery systems of the 5-ASA drug.
After the experimental studies, theoretical studies need to be carried out in order to explain the chemical interactions that occur in the adsorption processes of 5-ASA inside the halloysite nanotube, and in the slow release of the drug [
9]. Hence, a theoretical study of the interactions between clay minerals, in particular halloysite, and 5-ASA drug is proposed, as halloysite nanotubes represent a good candidate for the modified release of 5-ASA.
Therefore, the aim of this work is to perform molecular modeling studies by methods based on atomistic force fields for molecular modeling and quantum mechanics calculations to predict the interactions between the drug 5-ASA and the halloysite nanotube excipient, in order to explain the use of the excipient for colon-targeted drug delivery systems.
2. Methodology and Models
The halloysite structure was generated from the atomic coordinates of a slice of a halloysite from previous work [
29], with the stoichiometry Al
2Si
2O
5(OH)
4 and chirality (19,0). Periodic boundary conditions were applied to create a periodical crystal structure. For the transformation of the initial slice to a nanotube, the
c axis cell parameter was optimized, obtaining
d(O–Si) = 1.67 Å and
c = 9.05 Å. To avoid interactions between vicinal nanotubes, periodical cell parameters
a = 50 Å,
b = 50 Å were applied (
Figure 1). Then, a cylinder halloysite with an internal diameter of 27 Å was obtained with an external sheet of the tetrahedral siloxane and one internal sheet of Al oxide-hydroxide [
18]. Although the internal diameter of a natural halloysite nanotube is around 15–50 nm, our model can be a good scenario to reproduce the interactions of the adsorption process at the molecular level.
Hence, our halloysite nanotube unit cell has the formula Al
76Si
76O
190(OH)
152 with 646 atoms. Its optimization was performed with quantum mechanical calculations by using Density Functional Theory (DFT) within the Castep code [
30], with the generalized gradient approximation (GGA), the Perdew–Burke–Ernzerhof (PBE) correlation exchange functional, and a cut off energy of 300 eV [
30]. The optimization was performed with energy and force convergence tolerances of 5.0 × 10
−6 eV/atom and 0.01 eV/Å, respectively.
Starting from this optimized structure, a 1 × 1 × 2 supercell was generated of halloysite, Al
152Si
152O
380(OH)
304, with 1292 atoms to avoid intermolecular interactions between adsorbates of vicinal cells. The atomic charges were optimized with the Charge Equilibration (QEq) method, maintaining the structure electrically neutral [
30] (
Table 1).
Moreover, two conformers of 5-ASA (F1 and F2) were considered, taking into account the relative orientation of the carboxylic group with respect to the hydroxyl substituent. The hydroxyl O atom of the carboxylic substituent can be oriented to the H atom of the hydroxyl substituent (conformer F1), or the carbonyl group can be oriented towards the H atom of the hydroxyl substituent (conformer F2) (
Figure 2). Both conformers were optimized in a periodic box by using Castep and empirical interatomic potentials with the Compass force field (FF), which provided good results in previous studies [
31,
32]. For non-bonding interactions in the FF calculations, the coulomb and van der Waals interactions were calculated by the Ewald method with a cut-off of 12 Å. The optimization was performed with energy and force convergence tolerances of 2.0 × 10
−5 kcal/mol and 0.001 kcal/mol/Å, respectively. The atomic charges provided by the forcefield were used (
Figure 2).
Different solid models were generated with the 1 × 1 × 2 halloysite nanotube supercell and the 5-ASA conformers applying periodic boundary conditions. Preliminary Monte Carlo simulated annealing calculations based on Compass FF of the adsorption of 5-ASA on halloysite were performed, exploring different orientations of adsorbate with respect to the mineral surface. In the adsorption, both conformers of 5-ASA (F1 and F2) were placed in the most stable positions of the internal surface of halloysite according to the previous Monte Carlo simulations. The adsorbed complexes were optimized, maintaining the fixed mineral structure apart from the H atoms with the Compass force field. Dry conditions without the presence of water molecules were used to avoid additional interactions that can hide the actual adsorbate–surface interactions.
In addition, a model of halloysite was prepared with the presence of 1126 water molecules placed inside the inner zone of nanotube, filling the whole internal space and surrounding the external surface of the nanotube. The amount of water molecules was considered for an average density close to 1 g/cm3 for the water zones for avoiding overpressured situations. The atomic charges of the Simple Point Charge (SPC) water model was used for the water molecules included in the model, with charges of −0.82 for the oxygen atoms and +0.41 for the hydrogen atoms.
The adsorption energy (
Eadsorption) was calculated according to the equation:
where
E(halloysite + 5-ASA) is the energy of the adsorption complex,
Ehalloysite is the energy of the halloysite model, and
E5-ASA is the energy of the drug molecule.
3. Results and Discussion
Firstly, both conformers of 5-ASA (F1 and F2) were optimized, isolated in a periodical box of 30 Å × 30 Å × 30 Å to avoid intermolecular interactions. In both conformers all substituents maintained coplanarity with the aromatic ring. The intramolecular hydrogen bond is 2.75 Å in F1 and 2.81 Å in F2. The F1 conformer (
Figure 2a) is 2.4 kcal/mol more stable than the F2 one (
Figure 2b). However, CASTEP calculations of both conformers showed that F2 is 4.4 kcal/mol more stable than F1. This indicates that our FF does not properly estimate the intramolecular hydrogen bond of F2. Nevertheless, this energy difference is very small in comparison to the adsorption energies, described below.
In the halloysite structure, several OH groups can be distinguished: those oriented perpendicular to the surface; those oriented parallel to the surface; and those of the internal part oriented to the siloxane surface. The H and O atoms of these groups have different atomic charges due to their different electrostatic interactions.
Then, the adsorption of the 5-ASA conformers in the space of halloysite (
Figure 1) was studied to determine the most important interaction sites with the mineral. In the Al
152Si
152O
380(OH)
304 model, each 5-ASA conformer was placed in three different orientations in the nanotube.
3.1. Adsorption of the Conformer F1 of 5-ASA on Halloysite
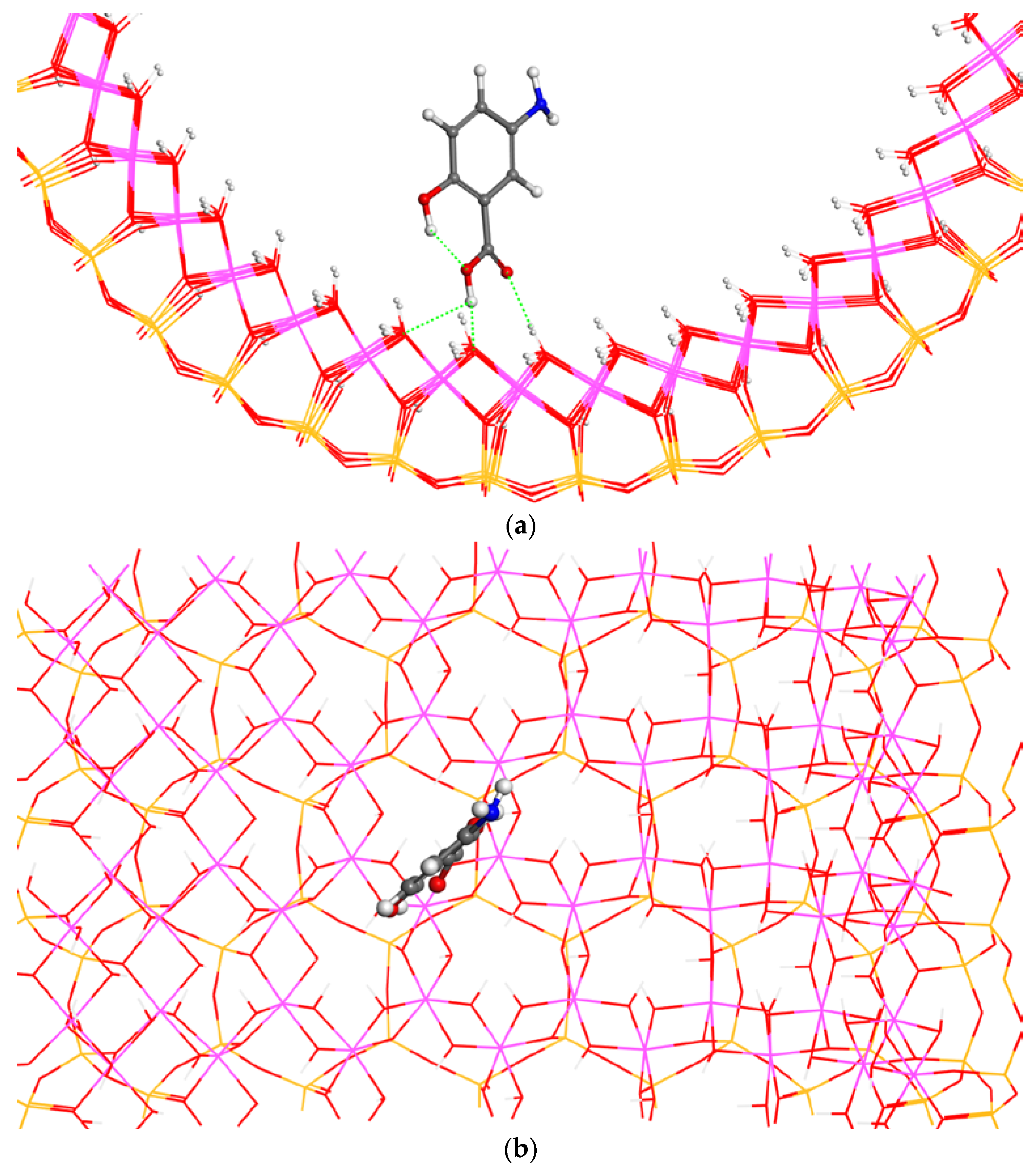
The adsorption of the F1 conformer placed parallel to the surface with a crossing orientation in the 1 × 1 × 2 halloysite supercell was studied, where the axis formed by the amino and hydroxyl substituents in the 5-ASA molecule is perpendicular with respect to the cylinder axis,
c axis, of halloysite. After the optimization (
Figure 3), the adsorbate maintained the parallel disposition with respect to the mineral surface and the crossing orientation. The adsorbate-surface interactions were mainly electrostatic ones between the negatively charged O atoms of the carbonyl and hydroxyl groups of 5-ASA and the positively charged H atoms of the surface aluminol groups with
d(C=O…HOAl) = 1.86 Å and
d(CC(H)O…HOAl) = 1.87 Å distances; between the H atoms of the amino group and the O atoms of surface aluminol groups with the
d(CNH…O(H)Al) = 2.27, 2.34, 2.45 Å distances; and between the amino N atoms and the surface O atoms with
d(CN…HOAl) = 2.49 Å. The O atoms of the 5-ASA substituents do not break the coplanarity with the aromatic ring except the carboxylic group, probably due to the repulsion with the surface O atoms. The carboxylic group is slightly tilted, maintaining the planarity of the moiety with the carbonyl O atom oriented towards one surface H atom and the hydroxyl group oriented against the surface due to repulsions with surface O atoms. The non-bonding
d(O…H) distances are short, but they cannot be considered strong H bonds because the (C)O…HO(Al) angles are smaller than 120°. Nevertheless, these small distances indicate strong electrostatic interactions. The amino H atoms approaching the surface break the coplanarity with the aromatic ring, and the NH…OAl interactions can be considered as weak hydrogen bonds with NH…O(Al) angles close to 180°. Moreover, the intramolecular hydrogen bond of 5-ASA molecule is maintained strong with
d(CCOH…O(H)CO) = 1.74 Å. The adsorption energy of the complex was −26.23 kcal/mol (
Table 2), indicating that this adsorption is energetically favorable.
On the other hand, when the F1 conformer of 5-ASA is placed in a parallel disposition with respect to the mineral surface with a different orientation, the axis formed by the hydroxyl and amino substituents in the 5-ASA molecule has a parallel orientation with respect to the
c axis of the halloysite nanotube. The optimization of this adsorption complex in the 1 × 1 × 2 halloysite supercell showed that the adsorbate maintained the parallel disposition with respect to the mineral surface, but with a twisted orientation of the molecule with respect to the surface where the axis formed by the carboxylic substituents and the aromatic ring was perpendicular to the
c axis of halloysite (
Figure 4). The adsorption energy was −26.49 kcal/mol (
Table 2). This energy is similar to the former one, indicating that the adsorbate-surface interactions are similar when the adsorbate is in a parallel disposition with respect to the surface and the relative orientation of the adsorbate does not significantly change the energetically favorable adsorption. The main non-bonding interatomic distances between adsorbate and surface are similar to those of the former adsorption complex:
d(C=O…HOAl) = 1.80 Å,
d(CC(H)O…HOAl) = 2.23 Å,
d(O=COH…O(H)Al) = 2.17 Å,
d(CNH…O(H)Al) = 2.38 Å, and
d(CN…HOHAl) = 2.36 Å. On the other hand, the intramolecular hydrogen bond in the 5-ASA molecule with
d(CCOH…O(H)CO) = 1.72 Å is stronger than that in the isolated molecule.
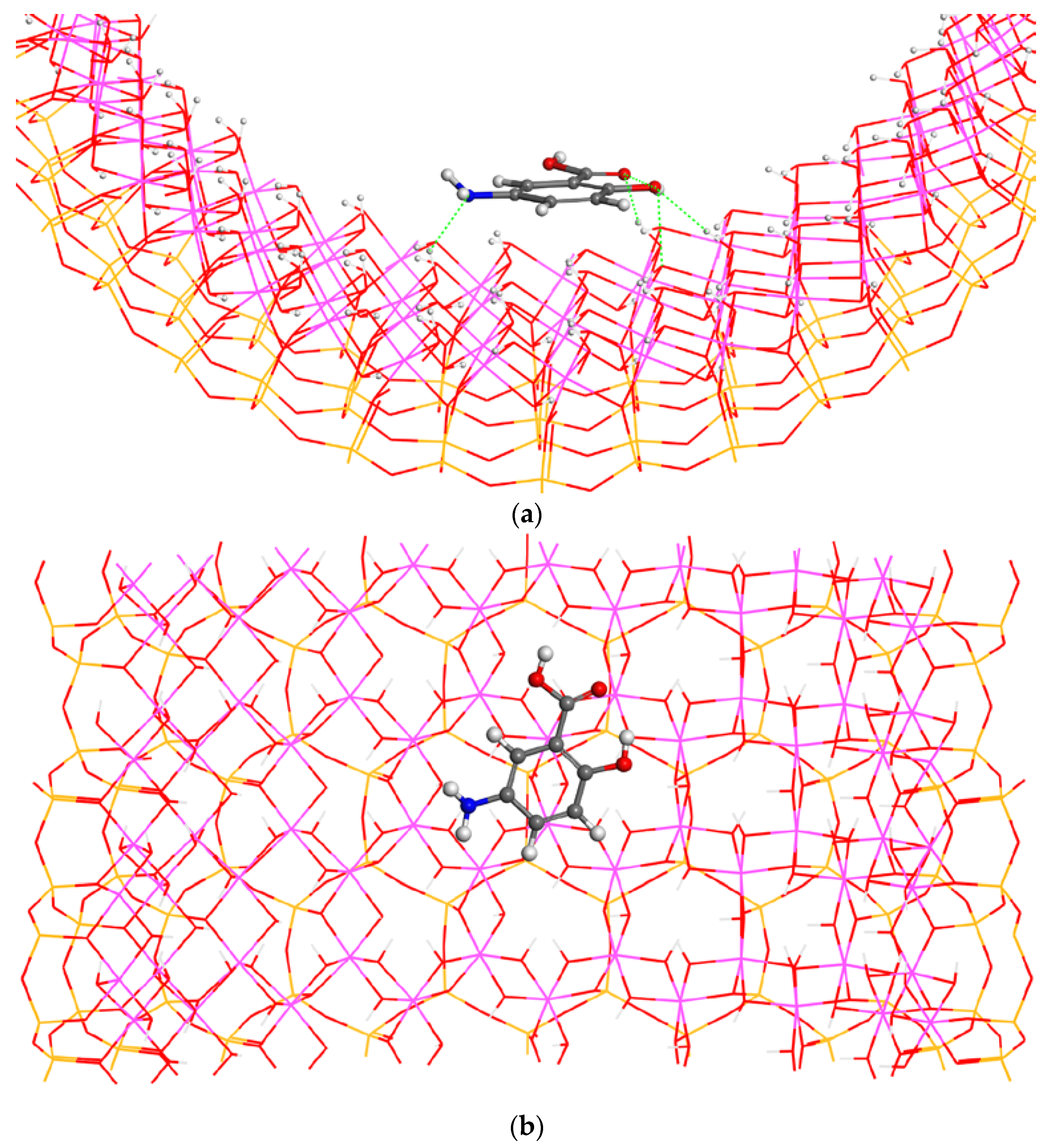
Another possible orientation of the adsorbate with respect to the mineral surface is that with the axis formed by the carboxylic group and aromatic ring of the 5-ASA molecule in a perpendicular plane with respect to the surface and the carboxylic group oriented towards the surface. The optimization of this adsorption complex maintained the perpendicular orientation of 5-ASA (
Figure 5). The intramolecular hydrogen bond is maintained with the same strength with
d(CCOH…O(H)CO) = 1.73 Å. The main interatomic adsorbate-surface distances were
d(C=O…HOAl) = 1.80 Å and
d(O=COH…O(H)Al) = 1.51 Å. These two distances show both strong hydrogen bonds with OHO angles close to 180°. The adsorption energy of this complex is −16.48 kcal/mol. This is significantly lower than the above adsorption models. This means that, in spite of both strong hydrogen bonds, this adsorption complex does not have the same electrostatic interactions of the hydroxyl and amino groups with the mineral surface as observed in above models. Besides, this result is interesting because it reveals a π interaction between the aromatic ring and the O atoms layer of the mineral surface in the former adsorption complexes where the adsorbate is placed parallel to the surface. This interaction is absent in this model with the perpendicular orientation of 5-ASA, justifying its lower adsorption energy.
3.2. Adsorption of the Conformer F2 of 5-ASA in the Halloysite
Analogous to the above conformer, the F2 conformer of the 5-ASA molecule was placed in a parallel disposition with respect to the internal mineral surface with a crossing orientation, where the axis formed by the amino and hydroxyl substituents in the 5-ASA molecule is oriented perpendicularly with respect to the cylinder axis,
c axis, of the halloysite nanotube. After the optimization of this adsorption complex the molecular orientation is similar to that of the conformer F1 (
Figure 6) and main adsorbate-surface interatomic distances are:
d(C=O…HOAl) = 1.97 Å,
d(CC(H)O…HOAl) = 2.07 Å,
d(CCOH…O(H)Al) = 2.24 Å, and
d(CN…HOHAl) = 2.14 Å. In this adsorption complex, the amino H atoms break the coplanarity of the 5-ASA molecule but are oriented against the surface, in contrast to the abovementioned adsorption models. The intramolecular hydrogen bond of 5-ASA is maintained with
d(CCOH…O=CO) = 1.95 Å, this distance being longer than that in F1 and in the isolated molecule. The interaction between the negatively charged amino N atom and the positively charged H atoms of mineral surface is stronger, whereas those of the 5-ASA carboxylic and hydroxyl groups are weaker than in above adsorption complexes. The adsorption energy is −29.61 kcal/mol (
Table 2) in this adsorption model, mainly due to the electrostatic interactions and the π interaction between the aromatic ring and surface. This adsorption energy shows that this adsorption is also energetically favorable, even more so than in F1. The difference of the total energy of the adsorption complexes between F1 and F2 conformers with the same orientation with respect to surface is only 3.38 kcal/mol more stable for F2 than F1.
On the other hand, the adsorption of the F2 conformer of 5-ASA can be placed in a parallel disposition with respect to the surface as well as in a parallel orientation with respect to the
c axis of the nanotube (
Figure 7). This orientation was maintained during the optimization process. The intramolecular hydrogen bond of the adsorbate molecule is maintained with
d(CCOH…O=CO) = 1.89 Å, being shorter than that in the former F2 adsorption complex but longer than that in the F1 complex. The main adsorbate-surface interactions are
d(C=O…HOAl) = 2.33 Å,
d(CC(H)O…HOAl) = 2.20 Å,
d(OCOH…O(H)Al) = 1.83 Å, and
d(CN…HOHAl) = 2.10 Å, being similar to the former model of F2. In this case, the main interaction is between the carboxylic H atom and the surface O atom. This interaction breaks the coplanarity of the carboxylic H atom, with the aromatic ring approaching the surface. The adsorption energy of this complex is −29.05 kcal/mol (
Table 2), being slightly smaller than the former one with F2 and greater than that with F1. However, considering the total energy of the adsorption complexes between F1 and F2 conformers with the same orientation with respect to surface, the difference is only 0.19 kcal/mol more stable for F2 than F1.
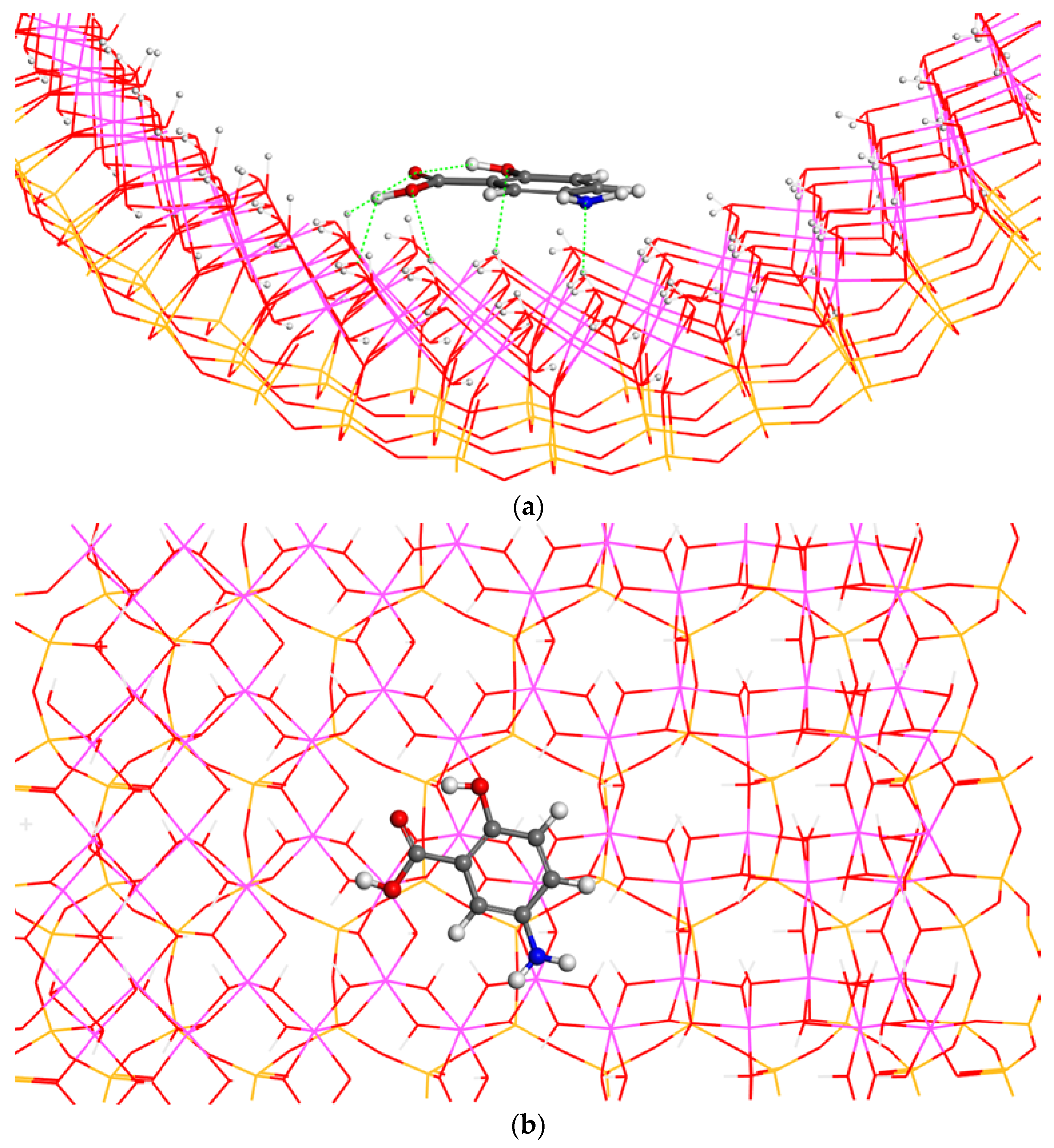
In addition, the F2 conformer of 5-ASA can be placed perpendicularly to the internal halloysite surface with the carboxylic group oriented to the surface. This orientation was maintained during the optimization (
Figure 8). The adsorption energy is −18.67 kcal/mol. This energy is higher than that achieved with the conformer F1 of 5-ASA with the same relative orientation with respect to the mineral surface, but lower than those of F2 with a parallel orientation of adsorbate with respect to the surface (
Table 2). Moreover, the intramolecular hydrogen bond of the 5-ASA molecule is maintained with
d(OC=O…HOCC) = 1.88 Å. The main adsorbate-surface interatomic interactions are
d(C=O…HOAl) = 1.99 Å,
d(O=C–(H)O…HOAl) = 2.27 Å, and
d(O=COH…O(H)Al) = 1.51 Å. The carboxylic H atom forms a very strong hydrogen bond with the surface O atoms.
3.3. Adsorption of the Conformer F1 of 5-ASA in the Halloysite Nanotube in the Presence of Water Molecules
A similar study was conducted with the presence of water molecules. After the optimization of the halloysite filled with water in the internal and external zones, the water molecules placed outside the nanotube are at an average distance of
d(OH…OSi) = 1.7–2.0 Å to the basal tetrahedral O atoms. However, the internal water molecules placed inside the nanotube are closer to the aluminol surface with an average distance of
d(OH…OH) = 1.6–1.7 Å. These distances are shorter than those with the external siloxane surface, indicating that the aluminol surface is more hydrophilic than the siloxane surface (
Figure 9). For the adsorption study, one 5-ASA molecule was placed inside a halloysite nanotube filled with water molecules. Two relative positions of the 5-ASA molecule were explored, one placed parallel to the mineral surface near the O–H groups of the aluminol surface of the halloysite (F1Wsurf) and another one placed in the middle of the nanotube in a parallel orientation to the
c axis of the nanotube (F1Wcenter). After the optimization both adsorbates remained in the same positions (
Figure 9). In both adsorption complexes the intramolecular H bond of the 5-ASA molecule remained similar to
d(CCOH…O(H)CO) = 1.70 Å. In F1Wsurf, the adsorbate-surface interactions are similar to the first model of F1 along some hydrogen bonds between the water molecules and the 5-ASA molecule. Nevertheless, no water molecule entered the space between the 5-ASA molecule and the mineral surface. In the F1Wcenter complex, the main interactions are between 5-ASA and the water molecules. The adsorption energies of the complexes are −31.50 and −29.58 kcal/mol for the F1Wsurf and F1Wcenter complexes, respectively (
Table 2).
The adsorption energy of the 5-ASA in halloysite with water is also energetically favorable. The adsorption energy in F1Wsurf is higher than that in F1Wcenter, meaning that the 5-ASA is likely to be adsorbed in the halloysite internal surface. On the other hand, the adsorption energy in F1Wsurf is higher than the adsorption of the F1 conformer of 5-ASA without water molecules, due to the interaction of solvating water molecules with 5-ASA (
Table 2).
4. Conclusions
In all adsorption models of 5-ASA and the internal surface of halloysite, the adsorption is energetically favorable. The adsorption energy is independent of the conformer type of 5-ASA, and both conformers exhibit the same behavior. The presence of water favors the adsorption of 5-ASA, forming hydrogen bonds with the 5-ASA molecule.
The 5-ASA molecule tends to be adsorbed in a parallel disposition with respect to the mineral surface, whereas the occurrence of a perpendicular orientation of the molecule with respect to the surface is highly improbable. The main interactions between 5-ASA and the halloysite surface are the π interaction between the aromatic ring of 5-ASA and the mineral surface, electrostatic interactions between the positively charged atoms and the negatively charged O atoms of the polar substituents of 5-ASA and the mineral surface, and some weak hydrogen bonds.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}