Case Report: Identification of a Novel Variant (m.8909T>C) of Human Mitochondrial ATP6 Gene and Its Functional Consequences on Yeast ATP Synthase

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Kidney Analyses
2.2. Patient Consent, Ethical Committees, and Adhesion to Biosecurity and Institutional Safety Procedures
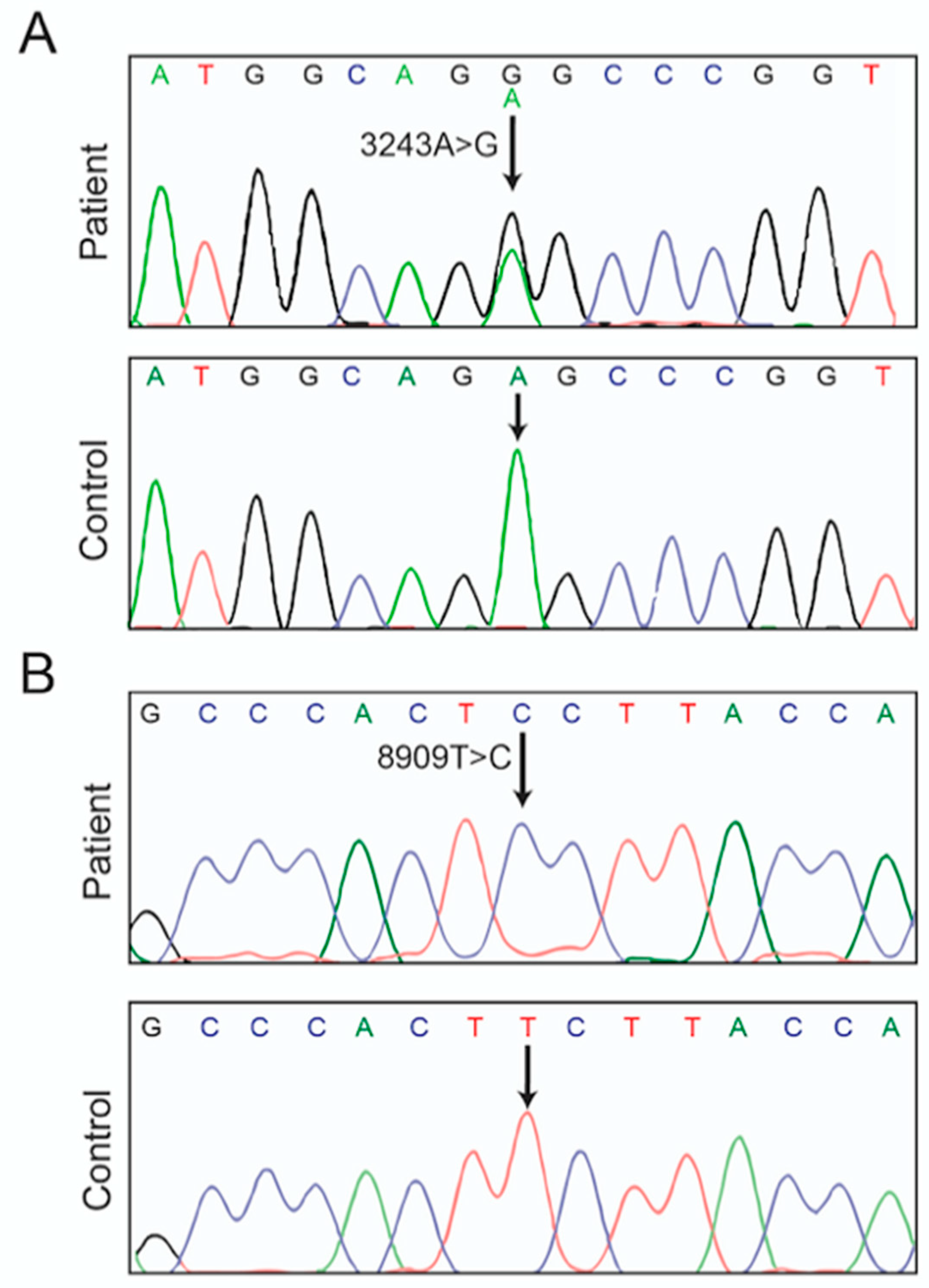
2.3. mtDNA Characterization
2.4. Media for Growing Yeast
2.5. Construction of S. cerevisiae Strain RKY108 (aF145S)
2.6. Yeast-Based Drug Assay
2.7. Biochemical Investigation of Mitochondria
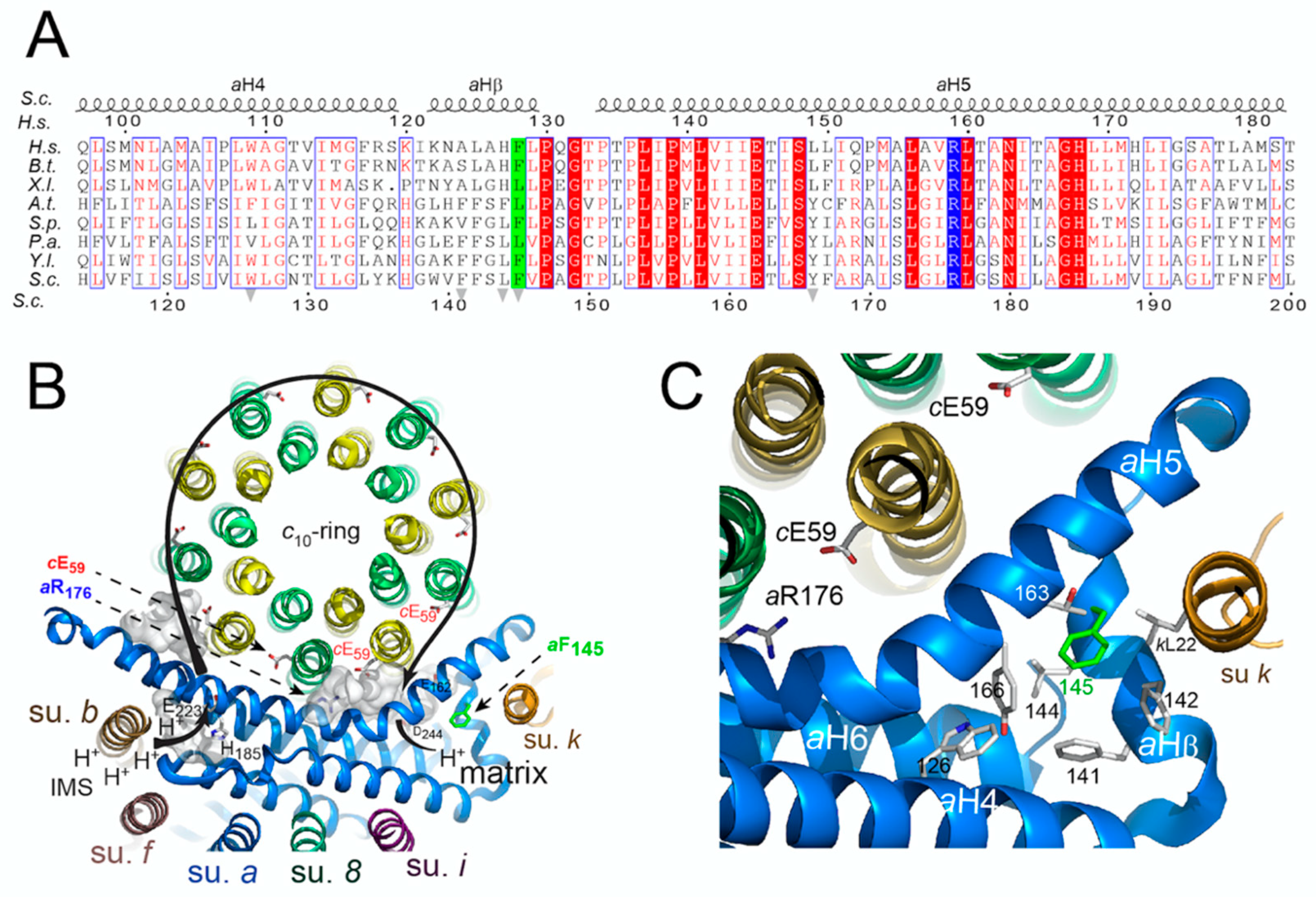
2.8. Amino-Acid Alignments and Subunit a Topology
2.9. Statistical Analyses
3. Results
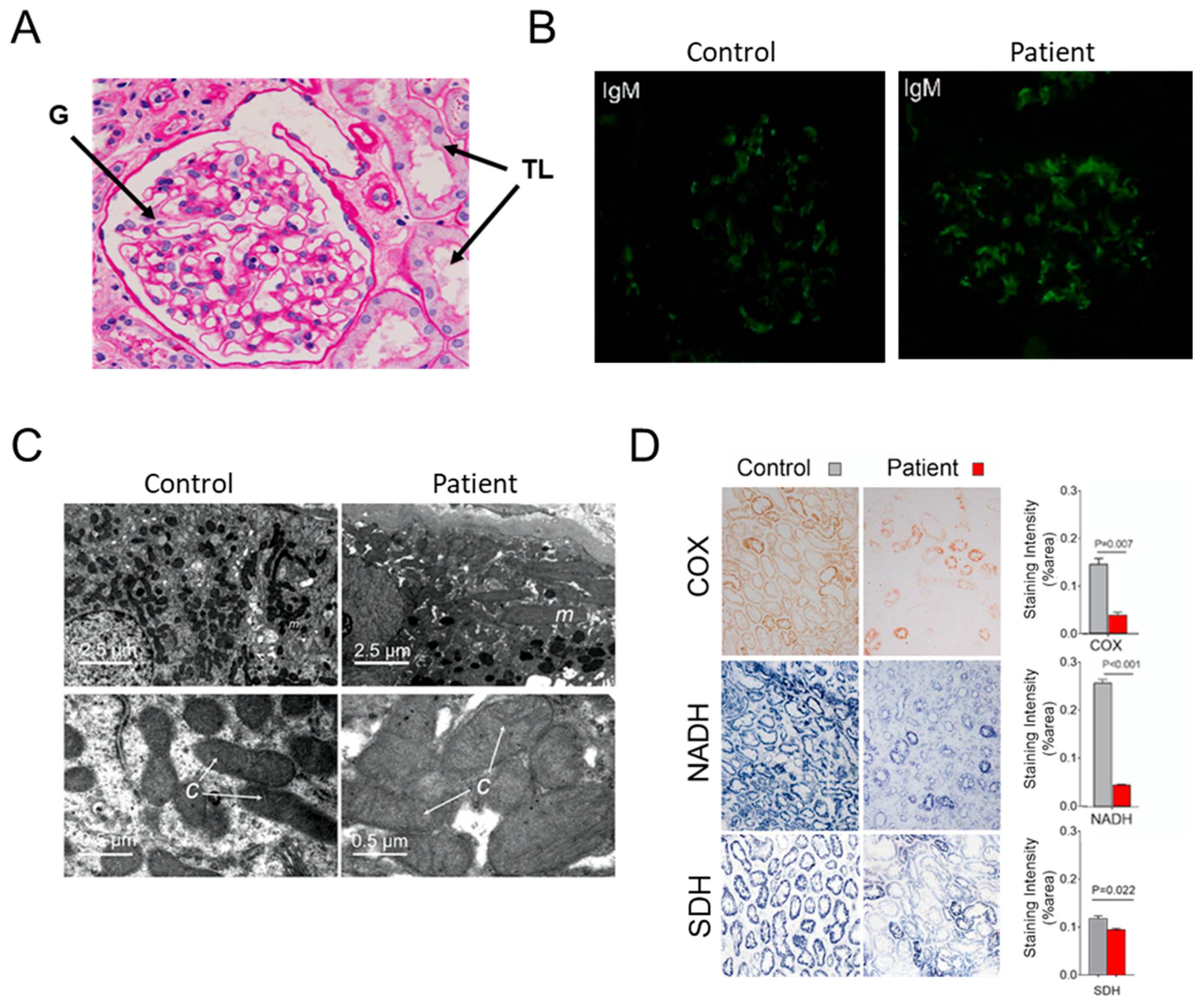
3.1. Case Report
3.2. Hints for Mitochondrial Dysfunction
3.3. Consequences of the m.8909T>C Mutation on Yeast ATP Synthase
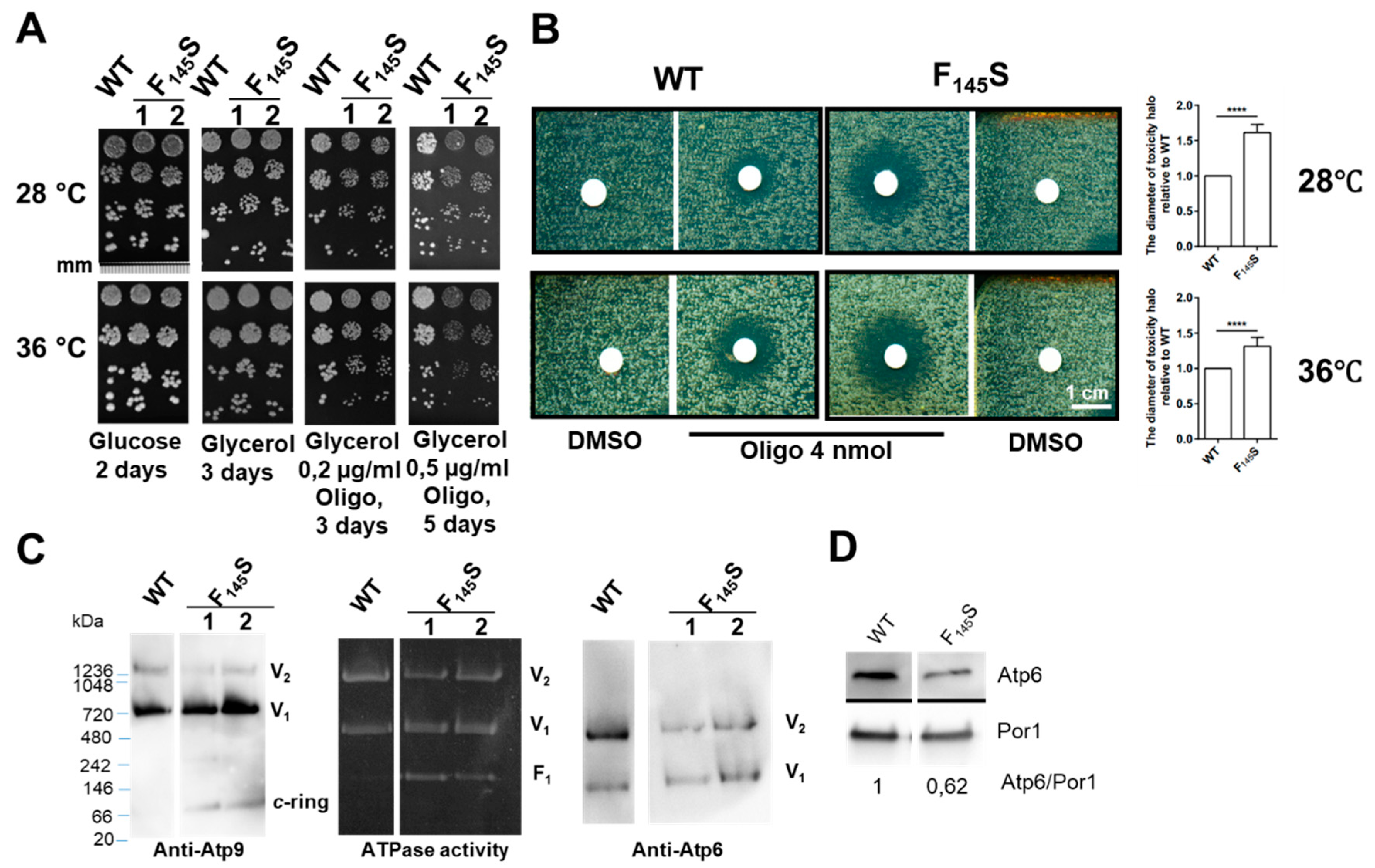
3.3.1. Influence of the aF145S Mutation on Yeast Respiratory Growth
3.3.2. Influence of the aF145S Mutation on Mitochondrial Respiration and ATP Synthesis
3.3.3. Influence of the aF145S Mutation on ATP Synthase Assembly/Stability
3.4. Topology of the Phenylalanine Residue Targeted by the m.8909T>C Mutation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Ethical Approval Number
Abbreviations
| Alb | Albumin |
| ADP | Adenosine di phosphate |
| ATP | Adenosine tri phosphate |
| BN-PAGE | Blue-native polyacrylamide gel electrophoresis |
| BUN | Blood urea nitrogen |
| CCCP | Cyanide m-chlorophenylhydrazone |
| Chol | Cholesterol |
| CIV | Cytochrome c oxidase |
| COX | Complex IV |
| CI-CIV | Complex I–Complex IV |
| CV | ATP synthase |
| DMSO | Dimethylsulfoxide |
| EGTA | Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| ESRD | End-stage renal disease |
| F | Female |
| FO | Domain FO of ATP synthase |
| F1 | Domain F1 of ATP synthase |
| FSGS | FSGS, Focal segmental glomerulosclerosis |
| Glo | Globulin |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| IgM | Immunoglobulin M |
| M | Male |
| MELAS | Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes |
| MPGNs | Mesangial proliferative glomerulonephritis |
| mtDNA | Mitochondrial DNA |
| NADH | Nicotinamide adenine dinucleotide |
| Oligo | Oligomycin |
| OXPHOS | Oxidative phosphorylation |
| Pi | Inorganic phosphate |
| PVDF | Polyvinylidene difluoride |
| ROS | Reactive oxygen species |
| Scr | Serum creatinine |
| SDH | Complex II dehydrogenase activities |
| SDS-PAGE | Sodium dodecyl sulfate–polyacrylamide gel electrophoresis |
| SIDS | Sudden infant death syndrome |
| TMPD | N,N,N,N,-tetramethyl-p-phenylenediamine |
| TG | Triglyceride |
| WB | Western-blot |
| WT | Wild-type |
References
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Carelli, V. Mitochondrial disorders. Curr. Opin. Neurol. 2007, 20, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 2010, 51, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, S.; Salviati, L.; Nishigaki, Y.; Walker, W.F.; Hernandez-Rosa, E.; Trevisson, E.; Delplace, S.; Desnuelle, C.; Shanske, S.; Hirano, M.; et al. A functionally dominant mitochondrial DNA mutation. Hum. Mol. Genet. 2008, 17, 1814–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Fu, Q.; Zhou, X.; Qu, J.; Tong, Y.; Guan, M.X. Mitochondrial variants may influence the phenotypic manifestation of Leber’s hereditary optic neuropathy-associated ND4 G11778A mutation. J. Genet. Genom. 2008, 35, 649–655. [Google Scholar] [CrossRef]
- Swalwell, H.; Blakely, E.L.; Sutton, R.; Tonska, K.; Elstner, M.; He, L.; Taivassalo, T.; Burns, D.K.; Turnbull, D.M.; Haller, R.G.; et al. A homoplasmic mtDNA variant can influence the phenotype of the pathogenic m.7472Cins MTTS1 mutation: Are two mutations better than one? Eur. J. Hum. Genet. 2008, 16, 1265–1274. [Google Scholar] [CrossRef]
- Bonnefoy, N.; Fox, T.D. Genetic transformation of Saccharomyces cerevisiae mitochondria. Methods Cell Biol. 2001, 65, 381–396. [Google Scholar]
- Okamoto, K.; Perlman, P.S.; Butow, R.A. The sorting of mitochondrial DNA and mitochondrial proteins in zygotes: Preferential transmission of mitochondrial DNA to the medial bud. J. Cell Biol. 1998, 142, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Rak, M.; Tetaud, E.; Duvezin-Caubet, S.; Ezkurdia, N.; Bietenhader, M.; Rytka, J.; di Rago, J.P. A yeast model of the neurogenic ataxia retinitis pigmentosa (NARP) T8993G mutation in the mitochondrial ATP synthase-6 gene. J. Biol. Chem. 2007, 282, 34039–34047. [Google Scholar] [CrossRef] [Green Version]
- Kucharczyk, R.; Rak, M.; di Rago, J.P. Biochemical consequences in yeast of the human mitochondrial DNA 8993T>C mutation in the ATPase6 gene found in NARP/MILS patients. Biochim. Biophys. Acta 2009, 1793, 817–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucharczyk, R.; Giraud, M.F.; Brethes, D.; Wysocka-Kapcinska, M.; Ezkurdia, N.; Salin, B.; Velours, J.; Camougrand, N.; Haraux, F.; di Rago, J.P. Defining the pathogenesis of human mtDNA mutations using a yeast model: The case of T8851C. Int. J. Biochem. Cell Biol. 2013, 45, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Kucharczyk, R.; Ezkurdia, N.; Couplan, E.; Procaccio, V.; Ackerman, S.H.; Blondel, M.; di Rago, J.P. Consequences of the pathogenic T9176C mutation of human mitochondrial DNA on yeast mitochondrial ATP synthase. Biochim. Biophys. Acta 2010, 1797, 1105–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabala, A.M.; Lasserre, J.P.; Ackerman, S.H.; di Rago, J.P.; Kucharczyk, R. Defining the impact on yeast ATP synthase of two pathogenic human mitochondrial DNA mutations, T9185C and T9191C. Biochimie 2014, 100, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Kucharczyk, R.; Salin, B.; di Rago, J.P. Introducing the human Leigh syndrome mutation T9176G into Saccharomyces cerevisiae mitochondrial DNA leads to severe defects in the incorporation of Atp6p into the ATP synthase and in the mitochondrial morphology. Hum. Mol. Genet. 2009, 18, 2889–2898. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.; Niedzwiecka, K.; Zhao, W.; Xu, S.; Liang, S.; Zhu, X.; Xie, H.; Tribouillard-Tanvier, D.; Giraud, M.F.; Zeng, C.; et al. Identification of G8969>A in mitochondrial ATP6 gene that severely compromises ATP synthase function in a patient with IgA nephropathy. Sci. Rep. 2016, 6, 36313. [Google Scholar] [CrossRef] [Green Version]
- Morava, E.; Rodenburg, R.J.; Hol, F.; de Vries, M.; Janssen, A.; van den Heuvel, L.; Nijtmans, L.; Smeitink, J. Clinical and biochemical characteristics in patients with a high mutant load of the mitochondrial T8993G/C mutations. Am. J. Med. Genet. A 2006, 140, 863–868. [Google Scholar] [CrossRef]
- Baracca, A.; Barogi, S.; Carelli, V.; Lenaz, G.; Solaini, G. Catalytic activities of mitochondrial ATP synthase in patients with mitochondrial DNA T8993G mutation in the ATPase 6 gene encoding subunit a. J. Biol. Chem. 2000, 275, 4177–4182. [Google Scholar] [CrossRef] [Green Version]
- Baracca, A.; Sgarbi, G.; Mattiazzi, M.; Casalena, G.; Pagnotta, E.; Valentino, M.L.; Moggio, M.; Lenaz, G.; Carelli, V.; Solaini, G. Biochemical phenotypes associated with the mitochondrial ATP6 gene mutations at nt8993. Biochim. Biophys. Acta 2007, 1767, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Carrozzo, R.; Murray, J.; Santorelli, F.M.; Capaldi, R.A. The T9176G mutation of human mtDNA gives a fully assembled but inactive ATP synthase when modeled in Escherichia coli. FEBS Lett. 2000, 486, 297–299. [Google Scholar] [CrossRef] [Green Version]
- Carrozzo, R.; Rizza, T.; Lucioli, S.; Pierini, R.; Bertini, E.; Santorelli, F.M. A mitochondrial ATPase 6 mutation is associated with Leigh syndrome in a family and affects proton flow and adenosine triphosphate output when modeled in Escherichia coli. Acta Paediatr. Suppl. 2004, 93, 65–67. [Google Scholar] [CrossRef]
- Cortes-Hernandez, P.; Vazquez-Memije, M.E.; Garcia, J.J. ATP6 homoplasmic mutations inhibit and destabilize the human F1F0-ATP synthase without preventing enzyme assembly and oligomerization. J. Biol. Chem. 2007, 282, 1051–1058. [Google Scholar] [CrossRef] [Green Version]
- Dionisi-Vici, C.; Seneca, S.; Zeviani, M.; Fariello, G.; Rimoldi, M.; Bertini, E.; De Meirleir, L. Fulminant Leigh syndrome and sudden unexpected death in a family with the T9176C mutation of the mitochondrial ATPase 6 gene. J. Inherit. Metab. Dis. 1998, 21, 2–8. [Google Scholar] [CrossRef]
- Houstek, J.; Pickova, A.; Vojtiskova, A.; Mracek, T.; Pecina, P.; Jesina, P. Mitochondrial diseases and genetic defects of ATP synthase. Biochim. Biophys. Acta 2006, 1757, 1400–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiazzi, M.; Vijayvergiya, C.; Gajewski, C.D.; DeVivo, D.C.; Lenaz, G.; Wiedmann, M.; Manfredi, G. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum. Mol. Genet. 2004, 13, 869–879. [Google Scholar] [CrossRef] [PubMed]
- De Meirleir, L.; Seneca, S.; Lissens, W.; Schoentjes, E.; Desprechins, B. Bilateral striatal necrosis with a novel point mutation in the mitochondrial ATPase 6 gene. Pediatr. Neurol. 1995, 13, 242–246. [Google Scholar] [CrossRef]
- Hench, J.; Bratic Hench, I.; Pujol, C.; Ipsen, S.; Brodesser, S.; Mourier, A.; Tolnay, M.; Frank, S.; Trifunovic, A. A tissue-specific approach to the analysis of metabolic changes in Caenorhabditis elegans. PLoS ONE 2011, 6, e28417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef] [Green Version]
- Kiernan, J.A. Indigogenic substrates for detection and localization of enzymes. Biotech. Histochem. Off. Publ. Biol. Stain Comm. 2007, 82, 73–103. [Google Scholar] [CrossRef]
- Rak, M.; Tetaud, E.; Godard, F.; Sagot, I.; Salin, B.; Duvezin-Caubet, S.; Slonimski, P.P.; Rytka, J.; di Rago, J.P. Yeast cells lacking the mitochondrial gene encoding the ATP synthase subunit 6 exhibit a selective loss of complex IV and unusual mitochondrial morphology. J. Biol. Chem. 2007, 282, 10853–10864. [Google Scholar] [CrossRef] [Green Version]
- Steele, D.F.; Butler, C.A.; Fox, T.D. Expression of a recoded nuclear gene inserted into yeast mitochondrial DNA is limited by mRNA-specific translational activation. Proc. Natl. Acad. Sci. USA 1996, 93, 5253–5257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerin, B.; Labbe, P.; Somlo, M. Preparation of yeast mitochondria (Saccharomyces cerevisiae) with good P/O and respiratory control ratios. Methods Enzymol. 1979, 55, 149–159. [Google Scholar] [PubMed]
- Somlo, M. Induction and repression of mitochondrial ATPase in yeast. Eur. J. Biochem. 1968, 5, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brethes, D.; di Rago, J.P.; Velours, J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Guo, H.; Bueler, S.A.; Rubinstein, J.L. Atomic model for the dimeric FO region of mitochondrial ATP synthase. Science 2017, 358, 936–940. [Google Scholar] [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System; Version 0.99; Schrödinger, LLC. DeLano Scientific: San Carlos, CA, USA, 2002.
- Majamaa, K.; Moilanen, J.S.; Uimonen, S.; Remes, A.M.; Salmela, P.I.; Karppa, M.; Majamaa-Voltti, K.A.; Rusanen, H.; Sorri, M.; Peuhkurinen, K.J.; et al. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: Prevalence of the mutation in an adult population. Am. J. Hum. Genet. 1998, 63, 447–454. [Google Scholar] [CrossRef] [Green Version]
- Fornuskova, D.; Brantova, O.; Tesarova, M.; Stiburek, L.; Honzik, T.; Wenchich, L.; Tietzeova, E.; Hansikova, H.; Zeman, J. The impact of mitochondrial tRNA mutations on the amount of ATP synthase differs in the brain compared to other tissues. Biochim. Biophys. Acta 2008, 1782, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Sasarman, F.; Antonicka, H.; Shoubridge, E.A. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum. Mol. Genet. 2008, 17, 3697–3707. [Google Scholar] [CrossRef] [Green Version]
- McMillan, R.P.; Stewart, S.; Budnick, J.A.; Caswell, C.C.; Hulver, M.W.; Mukherjee, K.; Srivastava, S. Quantitative Variation in m.3243A>G Mutation Produce Discrete Changes in Energy Metabolism. Sci. Rep. 2019, 9, 5752. [Google Scholar] [CrossRef] [Green Version]
- Michon, T.; Galante, M.; Velours, J. NH2-terminal sequence of the isolated yeast ATP synthase subunit 6 reveals post-translational cleavage. Eur. J. Biochem. 1988, 172, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Symersky, J.; Osowski, D.; Walters, D.E.; Mueller, D.M. Oligomycin frames a common drug-binding site in the ATP synthase. Proc. Natl. Acad. Sci. USA 2012, 109, 13961–13965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, A.; Uh, M.; Mueller, D.M. Level of ATP synthase activity required for yeast Saccharomyces cerevisiae to grow on glycerol media. FEBS Lett. 1994, 343, 160–164. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre-Legendre, L.; Vaillier, J.; Benabdelhak, H.; Velours, J.; Slonimski, P.P.; di Rago, J.P. Identification of a nuclear gene (FMC1) required for the assembly/stability of yeast mitochondrial F(1)-ATPase in heat stress conditions. J. Biol. Chem. 2001, 276, 6789–6796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Rak, M.; Tetaud, E.; Godard, F.; Sardin, E.; Bouhier, M.; Gombeau, K.; Caetano-Anollés, D.; Salin, B.; Chen, H.; et al. Deregulating mitochondrial metabolite and ion transport has beneficial effects in yeast and human cellular models for NARP syndrome. Hum. Mol. Genet. 2019. [Google Scholar] [CrossRef]
- Morales-Rios, E.; Montgomery, M.G.; Leslie, A.G.; Walker, J.E. Structure of ATP synthase from Paracoccus denitrificans determined by X-ray crystallography at 4.0 A resolution. Proc. Natl. Acad. Sci. USA 2015, 112, 13231–13236. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.; Rohou, A.; Schep, D.G.; Bason, J.V.; Montgomery, M.G.; Walker, J.E.; Grigorieff, N.; Rubinstein, J.L. Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM. Elife 2015, 4. [Google Scholar] [CrossRef]
- Allegretti, M.; Klusch, N.; Mills, D.J.; Vonck, J.; Kuhlbrandt, W.; Davies, K.M. Horizontal membrane-intrinsic alpha-helices in the stator a-subunit of an F-type ATP synthase. Nature 2015, 521, 237–240. [Google Scholar] [CrossRef]
- Tzagoloff, A.; Barrientos, A.; Neupert, W.; Herrmann, J.M. Atp10p assists assembly of Atp6p into the F0 unit of the yeast mitochondrial ATPase. J. Biol. Chem. 2004, 279, 19775–19780. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Kucharczyk, R.; di Rago, J.P.; Tzagoloff, A. The leader peptide of yeast Atp6p is required for efficient interaction with the Atp9p ring of the mitochondrial ATPase. J. Biol. Chem. 2007, 282, 36167–36176. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.; Parey, K.; Bublitz, M.; Mills, D.J.; Zickermann, V.; Vonck, J.; Kuhlbrandt, W.; Meier, T. Structure of a Complete ATP Synthase Dimer Reveals the Molecular Basis of Inner Mitochondrial Membrane Morphology. Mol. Cell 2016, 63, 445–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, A.; Vonck, J.; Mills, D.J.; Meier, T.; Kuhlbrandt, W. Structure, mechanism, and regulation of the chloroplast ATP synthase. Science 2018, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.P.; Luo, M.; Zhou, W.; Symersky, J.; Bai, D.; Chambers, M.G.; Faraldo-Gomez, J.D.; Liao, M.; Mueller, D.M. High-resolution cryo-EM analysis of the yeast ATP synthase in a lipid membrane. Science 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vik, S.B.; Antonio, B.J. A mechanism of proton translocation by F1F0 ATP synthases suggested by double mutants of the a subunit. J. Biol. Chem. 1994, 269, 30364–30369. [Google Scholar] [PubMed]
- Junge, W.; Lill, H.; Engelbrecht, S. ATP synthase: An electrochemical transducer with rotatory mechanics. Trends Biochem. Sci. 1997, 22, 420–423. [Google Scholar] [CrossRef]
- Pogoryelov, D.; Krah, A.; Langer, J.D.; Yildiz, O.; Faraldo-Gomez, J.D.; Meier, T. Microscopic rotary mechanism of ion translocation in the F(o) complex of ATP synthases. Nat. Chem. Biol. 2010, 6, 891–899. [Google Scholar] [CrossRef]
- Davidson, M.M.; Walker, W.F.; Hernandez-Rosa, E. The m.3243A>G mtDNA mutation is pathogenic in an in vitro model of the human blood brain barrier. Mitochondrion 2009, 9, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Seidowsky, A.; Hoffmann, M.; Glowacki, F.; Dhaenens, C.M.; Devaux, J.P.; de Sainte Foy, C.L.; Provot, F.; Gheerbrant, J.D.; Hummel, A.; Hazzan, M.; et al. Renal involvement in MELAS syndrome−a series of 5 cases and review of the literature. Clin. Nephrol. 2013, 80, 456–463. [Google Scholar] [CrossRef]
- Kurogouchi, F.; Oguchi, T.; Mawatari, E.; Yamaura, S.; Hora, K.; Takei, M.; Sekijima, Y.; Ikeda, S.; Kiyosawa, K. A case of mitochondrial cytopathy with a typical point mutation for MELAS, presenting with severe focal-segmental glomerulosclerosis as main clinical manifestation. Am. J. Nephrol. 1998, 18, 551–556. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshinari, M.; Doi, Y.; Yoshizumi, H.; Katafuchi, R.; Yokomizo, Y.; Nishiyama, K.; Wakisaka, M.; Fujishima, M. Renal complications in patients with diabetes mellitus associated with an A to G mutation of mitochondrial DNA at the 3243 position of leucine tRNA. Diabetes Res. Clin. Pract. 1999, 44, 183–189. [Google Scholar] [CrossRef]
- Löwik, M.M.; Hol, F.A.; Steenbergen, E.J.; Wetzels, J.F.; van den Heuvel, L.P. Mitochondrial tRNALeu(UUR) mutation in a patient with steroid-resistant nephrotic syndrome and focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2005, 20, 336–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guéry, B.; Choukroun, G.; Noël, L.H.; Clavel, P.; Rötig, A.; Lebon, S.; Rustin, P.; Bellané-Chantelot, C.; Mougenot, B.; Grünfeld, J.P.; et al. The spectrum of systemic involvement in adults presenting with renal lesion and mitochondrial tRNA (Leu) gene mutation. J. Am. Soc. Nephrol. 2003, 14, 2099–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, H.; Mori, Y.; Kayamori, K.; Igari, T.; Ito, E.; Akashi, T.; Noguchi, Y.; Kitamura, K.; Okado, T.; Terada, Y.; et al. A familial case of mitochondrial disease resembling Alport syndrome. Clin. Exp. Nephrol. 2008, 12, 159–163. [Google Scholar] [CrossRef]
- Suzuki, T.; Fujino, T.; Sugiyama, M.; Ishida, M. A case of mitochondrial encephalomyopathy (MELAS). Nihon Jinzo Gakkai Shi 1996, 38, 109–114. [Google Scholar]
- Mima, A.; Shiota, F.; Matsubara, T.; Iehara, N.; Akagi, T.; Abe, H.; Nagai, K.; Matsuura, M.; Murakami, T.; Kishi, S.; et al. An autopsy case of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) with intestinal bleeding in chronic renal failure. Ren. Fail. 2011, 33, 622–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, N.; Shimada, T.; Ishibashi, Y.; Yoshitomi, H.; Oyake, N.; Murakami, Y.; Nishino, I.; Nonaka, I.; Goto, Y.; Kitamura, J. Marked left ventricular hypertrophy in a patient with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Int. J. Cardiol. 2008, 129, e77–e80. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Hsieh, R.H.; Peng, N.J.; Lai, P.H.; Lee, C.F.; Lo, Y.K.; Wei, Y.H. A follow-up study in a Taiwanese family with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes syndrome. J. Formos. Med. Assoc. 2007, 106, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Jansen, J.J.; Maassen, J.A.; van der Woude, F.J.; Lemmink, H.A.; van den Ouweland, J.M.; t’ Hart, L.M.; Smeets, H.J.; Bruijn, J.A.; Lemkes, H.H. Mutation in mitochondrial tRNA(Leu(UUR)) gene associated with progressive kidney disease. J. Am. Soc. Nephrol. 1997, 8, 1118–1124. [Google Scholar]
- Ireland, J.; Rossetti, S.; Haugen, E.; Ireland, J.; Michels, V.; Harris, P. Mitochondrial causes of renal insufficiency and hearing loss. Kidney Int. 2004, 65, 2444–2445. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, K.; Tomida, C.; Umeyama, K.; Urakami, K.; Ishizu, T.; Hirayama, K.; Gotoh, M.; Iitsuka, T.; Takemura, K.; Kikuchi, H.; et al. Prevalence of Japanese dialysis patients with an A-to-G mutation at nucleotide 3243 of the mitochondrial tRNA(Leu(UUR)) gene. Nephrol. Dial. Transplant. 2000, 15, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, O.; Vilarinho, L.; Almeida, F.; Ferreira, F.; Guardado, J.; Ferreira, M.; Lourenço, A.; Medeiros, R.; Almeida, J. Cardiomyopathy and kidney disease in a patient with maternally inherited diabetes and deafness caused by the 3243A>G mutation of mitochondrial DNA. Cardiology 2010, 115, 71–74. [Google Scholar] [CrossRef]
- Manouvrier, S.; Rötig, A.; Hannebique, G.; Gheerbrandt, J.D.; Royer-Legrain, G.; Munnich, A.; Parent, M.; Grünfeld, J.P.; Largilliere, C.; Lombes, A.; et al. Point mutation of the mitochondrial tRNA(Leu) gene (A 3243 G) in maternally inherited hypertrophic cardiomyopathy, diabetes mellitus, renal failure, and sensorineural deafness. J. Med. Genet. 1995, 32, 654–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujita, Y.; Kunitomo, T.; Fujii, M.; Furukawa, S.; Otsuki, H.; Fujino, K.; Hamamoto, T.; Tabata, T.; Matsumura, K.; Sasaki, T.; et al. A surviving case of mitochondrial cardiomyopathy diagnosed from the symptoms of multiple organ dysfunction syndrome. Int. J. Cardiol. 2008, 128, e43–e45. [Google Scholar] [CrossRef] [PubMed]
- Bergamin, C.S.; Rolim, L.C.; Dib, S.A.; Moisés, R.S. Unusual occurrence of intestinal pseudo obstruction in a patient with maternally inherited diabetes and deafness (MIDD) and favorable outcome with coenzyme Q10. Arq. Bras. Endocrinol. Metabol. 2008, 52, 1345–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihara, M.; Tanaka, H.; Yashiro, M.; Nishimura, Y. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) with chronic renal failure: Report of mother-child cases. Rinsho Shinkeigaku. 1996, 36, 1069–1073. [Google Scholar] [PubMed]
- Ueda, Y.; Ando, A.; Nagata, T.; Yanagida, H.; Yagi, K.; Sugimoto, K.; Okada, M.; Takemura, T. A boy with mitochondrial disease: asymptomatic proteinuria without neuromyopathy. Pediatr. Nephrol. 2004, 19, 107–110. [Google Scholar] [CrossRef]
- Cheong, H.I.; Chae, J.H.; Kim, J.S.; Park, H.W.; Ha, I.S.; Hwang, Y.S.; Lee, H.S.; Choi, Y. Hereditary glomerulopathy associated with a mitochondrial tRNA(Leu) gene mutation. Pediatr. Nephrol. 1999, 13, 477–480. [Google Scholar] [CrossRef]
- Yorifuji, T.; Kawai, M.; Momoi, T.; Sasaki, H.; Furusho, K.; Muroi, J.; Shimizu, K.; Takahashi, Y.; Matsumura, M.; Nambu, M.; et al. Nephropathy and growth hormone deficiency in a patient with mitochondrial tRNA(Leu(UUR)) mutation. J. Med. Genet. 1996, 33, 621–622. [Google Scholar] [CrossRef] [Green Version]
- Hotta, O.; Inoue, C.N.; Miyabayashi, S.; Furuta, T.; Takeuchi, A.; Taguma, Y. Clinical and pathologic features of focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR) gene mutation. Kidney Int. 2001, 59, 1236–1243. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.Y.; Wei, R.B.; Wang, Y.D.; Zhang, X.G.; Tang, L.; Chen, X.M. Focal segmental glomerulosclerosis associated with maternally inherited diabetes and deafness: Clinical pathological analysis. Indian J. Pathol. Microbiol. 2013, 56, 272–275. [Google Scholar] [CrossRef]
- Thyagarajan, D.; Shanske, S.; Vazquez-Memije, M.; De Vivo, D.; DiMauro, S. A novel mitochondrial ATPase 6 point mutation in familial bilateral striatal necrosis. Ann. Neurol. 1995, 38, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.M.; Santorelli, F.M.; Shanske, S.; Gelb, B.D.; Fyfe, B.; Wolfe, D.; Willner, J.P. Leigh syndrome and hypertrophic cardiomyopathy in an infant with a mitochondrial DNA point mutation (T8993G). Am. J. Med. Genet. 1994, 50, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Santorelli, F.M.; Schlessel, J.S.; Slonim, A.E.; DiMauro, S. Novel mutation in the mitochondrial DNA tRNA glycine gene associated with sudden unexpected death. Pediatr. Neurol. 1996, 15, 145–149. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward | Reverse | Product Length (bp) | |
|---|---|---|---|
| For detection of point mutations | |||
| 1 | CCCACAGTTTATGTAGCTTACC | GTACTATATCTATTGCGCCAGG | 1215 |
| 2 | ACTACCAGACAACCTTAGCC | AACATCGAGGTCGTAAACCC | 1293 |
| 3 | CTTCACCAGTCAAAGCGAAC | AGAAGTAGGGTCTTGGTGAC | 1242 |
| 4 | CGAACTAGTCTCAGGCTTCAAC | TCGTGGTGCTGGAGTTTAAG | 1228 |
| 5 | ACGTAAGCCTTCTCCTCACT | TCGTTACCTAGAAGGTTGCC | 1138 |
| 6 | CCGACCGTTGACTATTCTCT | GATGGCAAATACAGCTCCTA | 1160 |
| 7 | GCAAACTCATCACTAGACATCG | AGCTTTACAGTGGGCTCTAG | 1329 |
| 8 | ACCACAGTTTCATGCCCATC | TGGCCTTGGTATGTGCTTTC | 1223 |
| 9 | CACTTCCACTCCATAACGCT | GTTGAGGGTTATGAGAGTAGC | 1308 |
| 10 | TACCAAATGCCCCTCATTTA | GTAATGAGGATGTAAGCCCG | 1272 |
| 11 | TTCAATCAGCCACATAGCCC | GATGAAACCGATATCGCCGA | 1260 |
| 12 | GAGGGCGTAGGAATTATATCC | GTCAGGTTAGGTCTAGGAGG | 1240 |
| 13 | CATACTCGGATTCTACCCTAG | TGTAATTACTGTGGCCCCTC | 1280 |
| 14 | TCGGCATTATCCTCCTGCTT | GTGCTATGTACGGTAAATGGC | 1250 |
| 15 | TGACTCACCCATCAACAACC | ATAGAAAGGCTAGGACCAAACC | 1179 |
| For detection of mtDNA deletion | |||
| 1 | GCACCCTATGTCGCAGTATCTGTCTTTG | GGACGAGAAGGGATTTGACTGTAATGTGC | 16,255 |
| 2 | CACTTCCACTCCATAACGCTCCTCATACT | GGGCTATTGGTTGAATGAGTAGGCTGATG | 16,250 |
| For detection of mtDNA copy number | |||
| COX1 | TTCGCCGACCGTTGACTATTCTCT | AAGATTATTACAAATGCATGGGC | 197 |
| 18S | GTCTGTGATGCCCTTAGATG | AGCTTATGACCCGCACTTAC | 177 |
| For pyrosequencing of m.3243A>G | |||
| Amplification | AAGGACAAGAGAAATAAGGC | ATGAGGAGTAGGAGGTTGG | 207 |
| Sequencing | TTTTATGCGATTACCG | ||
| Strain | Nuclear Genotype | mtDNA | Reference |
|---|---|---|---|
| DFS160 | MATα leu2Δ ura3-52 ade2-101 arg8::URA3 kar1-1 | ρo | [31] |
| NB40-3C | MATa lys2 leu2-3,112 ura3-52 his3ΔHindIII arg8::hisG | ρ+ cox2-62 | [31] |
| MR6 | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 CAN1 arg8::HIS3 | ρ+ | [30] |
| MR10 | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 CAN1 arg8::HIS3 | ρ+ atp6::ARG8m | [30] |
| RKY109 | MATa leu2Δura3-52 ade2-101 arg8::URA3 kar1-1 | ρ− atp6-F145S | This study |
| RKY108 | MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 CAN1 arg8::HIS3 | ρ atp6-F145S | This study |
| The Time of First Kidney Biopsy | |
|---|---|
| Age/years | 14 |
| Sex (M/F) | F |
| Ethnicity | Han Chinese |
| Course/mouth | 8 |
| Urinary protein/g·24 h−1 | 3.74 |
| BUN/mg·dL−1 | 20.6 |
| Scr/mg·dL−1 | 1.23 |
| Alb/g·L−1 | 27.5 |
| Glo/g·L−1 | 16.8 |
| TG/mmol·L−1 | 3.99 |
| Chol/mmol·L−1 | 8.6 |
| At uremia stage | |
| BUN/mg·dL−1 | 58.1 |
| Scr/mg·dL−1 | 9.27 |
| Gene | Nucleotide Changes | Frequency a | Amino Acid Change Function Annotation | Type |
|---|---|---|---|---|
| D-Loop | m.263A>G | 1861/1867 | non-coding | polymorphic |
| m.499G>A * | 2106/2144 | non-coding | polymorphic | |
| m.16217T>C * | 103/1867 | non-coding | polymorphic | |
| m.16261C>T | 111/1867 | non-coding | polymorphic | |
| m.16136T>C * | 24/1867 | non-coding | polymorphic | |
| 12S rRNA | m.750A>G | 2682/2704 | non-coding | polymorphic |
| m.827A>G * | 54/2704 | non-coding | polymorphic | |
| m.1438A>G | 2620/2704 | non-coding | polymorphic | |
| 16S rRNA | m.2706A>G | 2178/2704 | non-coding | polymorphic |
| TL1 | m.3243A>G | 0/2704 | MELAS | Mutation |
| ND2 | m.4769A>G | 2674/2704 | Met100Met | polymorphic |
| m.4820G>A * | 45/2704 | Glu117Glu | polymorphic | |
| m.5063T>C | 3/2704 | Pro198Pro | polymorphic | |
| COXI | m.6023G>A * | 34/2704 | Glu40Glu | polymorphic |
| m.6413T>C * | 23/2704 | Asn170Asn | polymorphic | |
| m.7028C>T | 2199/2704 | Ala375Ala | polymorphic | |
| ATP6 | m.8860A>G | 2698/2704 | Thr112Ala | polymorphic |
| m.8909T>C | 0/2704 | Phe128Ser | Mutation | |
| ND4L | m.10646G>A | 13/2704 | Val59Val | polymorphic |
| ND4 | m.11254T>C | 1/2704 | Ile165Ile | polymorphic |
| m.11719G>A | 2100/2704 | Gly320Gly | polymorphic | |
| ND5 | m.13590G>A * | 110/2704 | Leu418Leu | polymorphic |
| m.13779A>G | 0/2704 | Thr481Thr | polymorphic | |
| CYTB | m.14766T>C | 610/2704 | Thr7Ile | polymorphic |
| m.15326A>G | 2687/2704 | Thr194Ala | polymorphic | |
| m.15535C>T * | 48/2704 | Asn263Asn | polymorphic | |
| m.15688C>T | 0/2704 | Ser314Ser | polymorphic | |
| m.15758A>G | 29/2704 | Ile338Val | polymorphic |
| Strain | Respiration Rate nmoL O min−1 mg−1 | ATP Synthesis Rate nmoL ATP min−1 mg−1 | |||
|---|---|---|---|---|---|
| NADH | NADH + ADP | NADH + CCCP | −oligo | +oligo | |
| WT | 249 ± 21 | 654 ± 46 | 1028 ± 21 | 1393 ± 113 | 236 +/− 60 |
| aF145S | 178 ± 2 * | 474 ± 5 * | 811 ± 9 * | 988 ± 101 * | 110 +/− 49 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Q.; Kucharczyk, R.; Zhao, W.; Dautant, A.; Xu, S.; Niedzwiecka, K.; Su, X.; Giraud, M.-F.; Gombeau, K.; Zhang, M.; et al. Case Report: Identification of a Novel Variant (m.8909T>C) of Human Mitochondrial ATP6 Gene and Its Functional Consequences on Yeast ATP Synthase. Life 2020, 10, 215. https://doi.org/10.3390/life10090215
Ding Q, Kucharczyk R, Zhao W, Dautant A, Xu S, Niedzwiecka K, Su X, Giraud M-F, Gombeau K, Zhang M, et al. Case Report: Identification of a Novel Variant (m.8909T>C) of Human Mitochondrial ATP6 Gene and Its Functional Consequences on Yeast ATP Synthase. Life. 2020; 10(9):215. https://doi.org/10.3390/life10090215
Chicago/Turabian StyleDing, Qiuju, Róża Kucharczyk, Weiwei Zhao, Alain Dautant, Shutian Xu, Katarzyna Niedzwiecka, Xin Su, Marie-France Giraud, Kewin Gombeau, Mingchao Zhang, and et al. 2020. "Case Report: Identification of a Novel Variant (m.8909T>C) of Human Mitochondrial ATP6 Gene and Its Functional Consequences on Yeast ATP Synthase" Life 10, no. 9: 215. https://doi.org/10.3390/life10090215
APA StyleDing, Q., Kucharczyk, R., Zhao, W., Dautant, A., Xu, S., Niedzwiecka, K., Su, X., Giraud, M. -F., Gombeau, K., Zhang, M., Xie, H., Zeng, C., Bouhier, M., di Rago, J. -P., Liu, Z., Tribouillard-Tanvier, D., & Chen, H. (2020). Case Report: Identification of a Novel Variant (m.8909T>C) of Human Mitochondrial ATP6 Gene and Its Functional Consequences on Yeast ATP Synthase. Life, 10(9), 215. https://doi.org/10.3390/life10090215